Emergence of Anthrax Edema Toxin as a Master Manipulator of Macrophage and B Cell Functions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Effects of ET on Essential Macrophage Functions
2.1. Global Gene Changes Induced by ET
2.2. Intracellular Signaling of ET
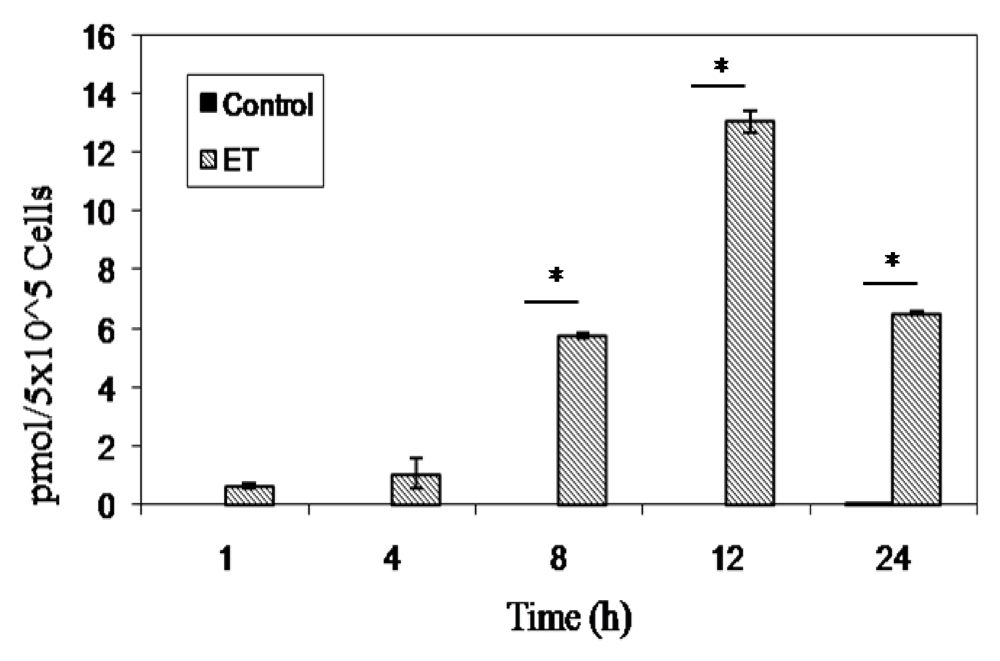
2.3. ET-Induced cAMP Production and Cytotoxicity Is Cell-Dependent
2.4. ET Alters the Motility and Phagocytic Activity of Macrophages
2.5. Macrophage Cytokine Modulation by ET
2.6. Synergistic Effects of LT and ET
3. B Cells: An Unknown Target of ET
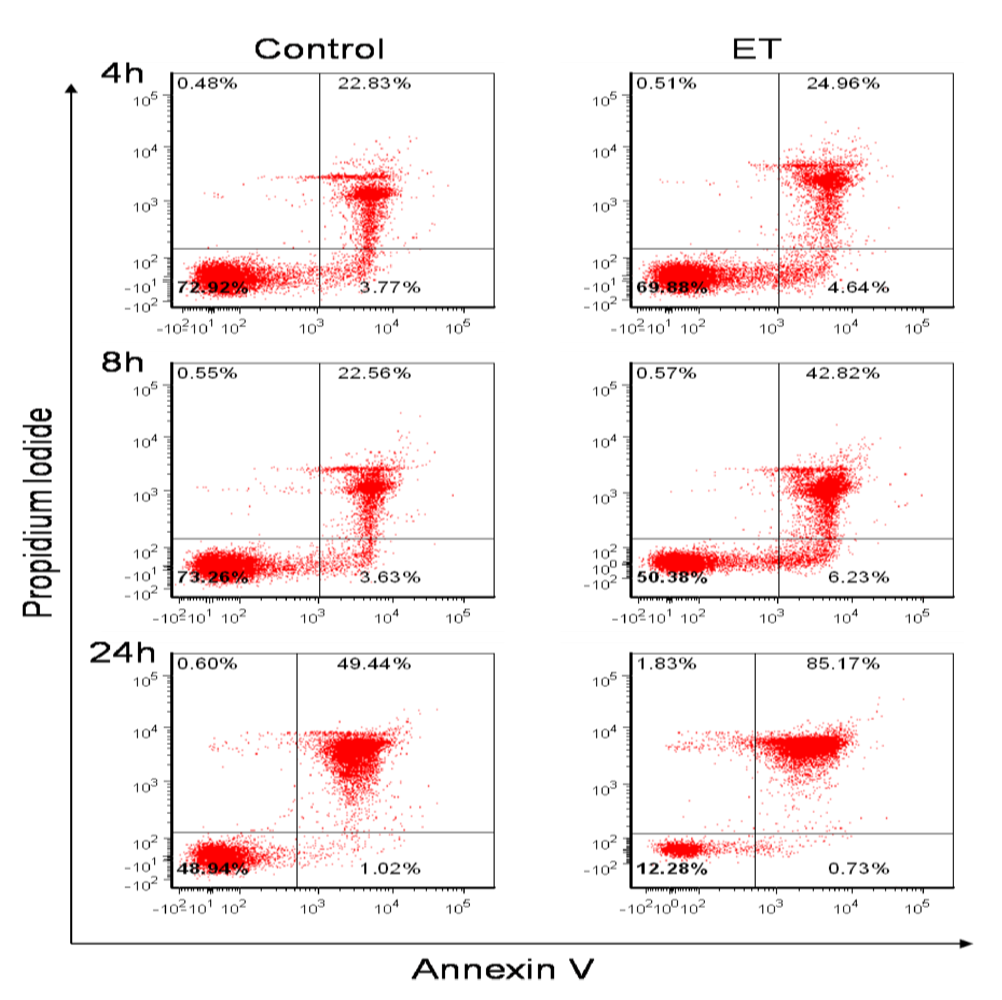
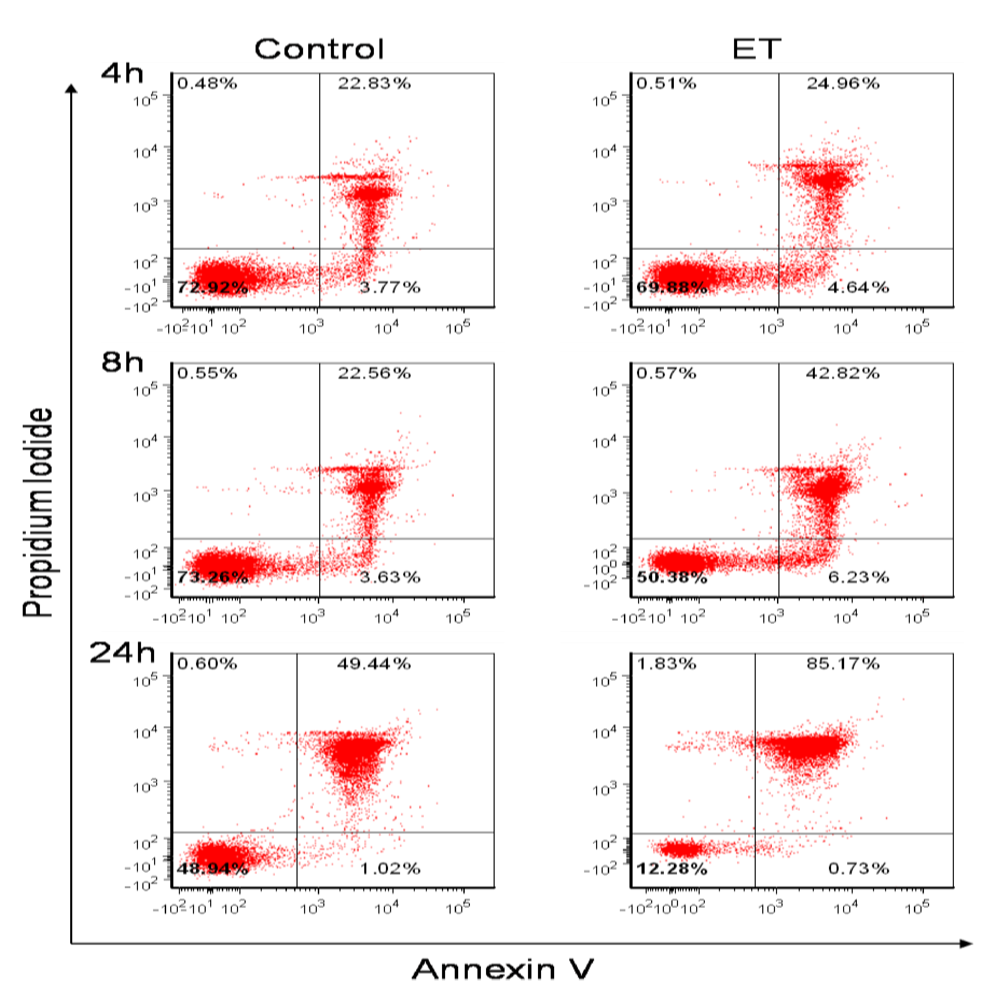
3.1. B Cells Are Susceptible to ET Activity
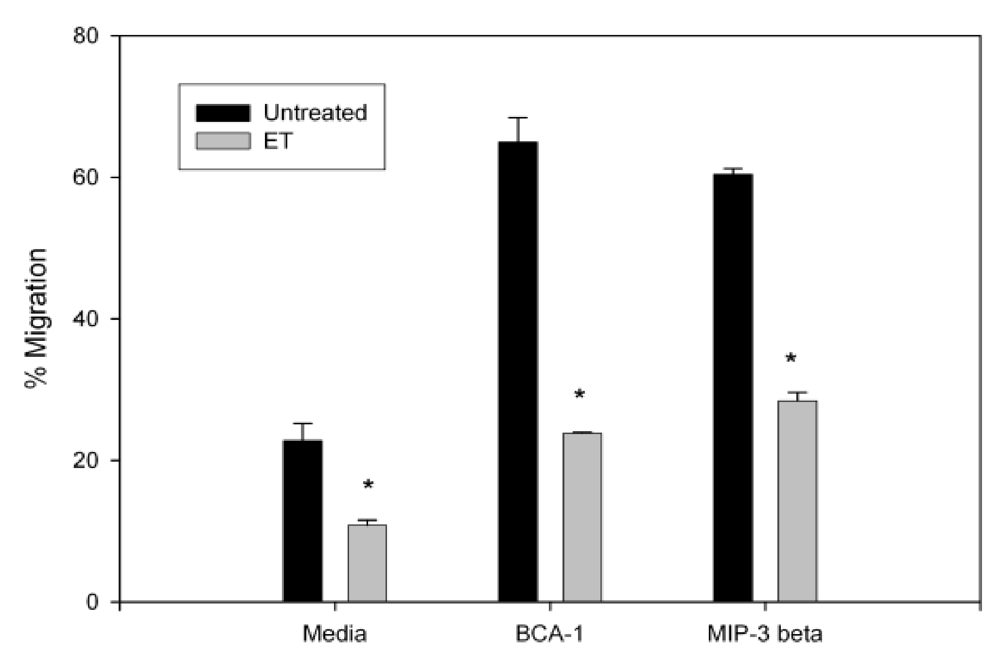
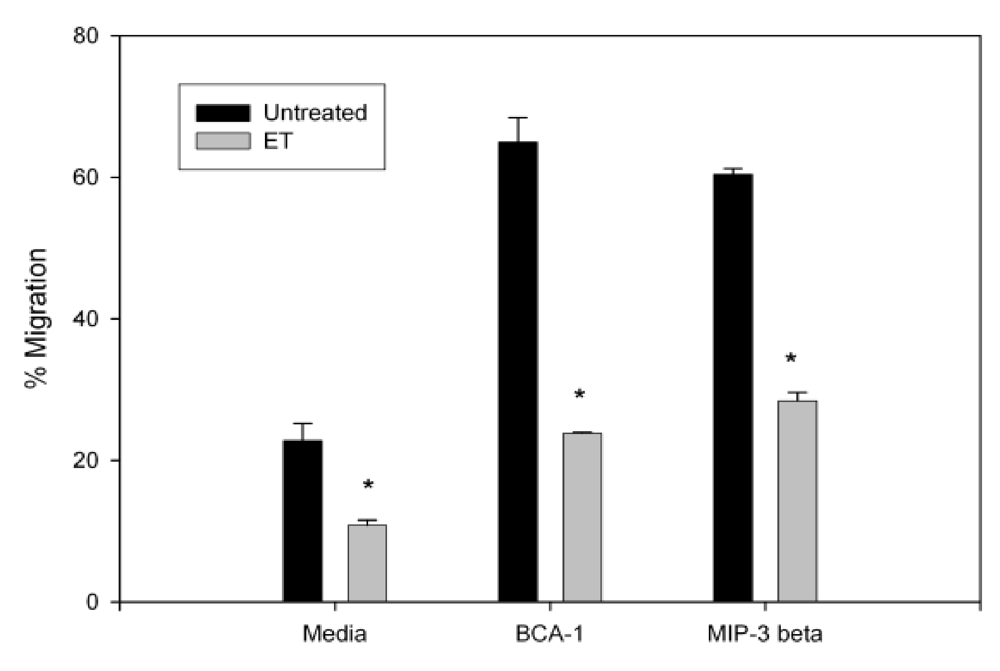
3.2. Anthrax ET Inhibits B Cell Migration
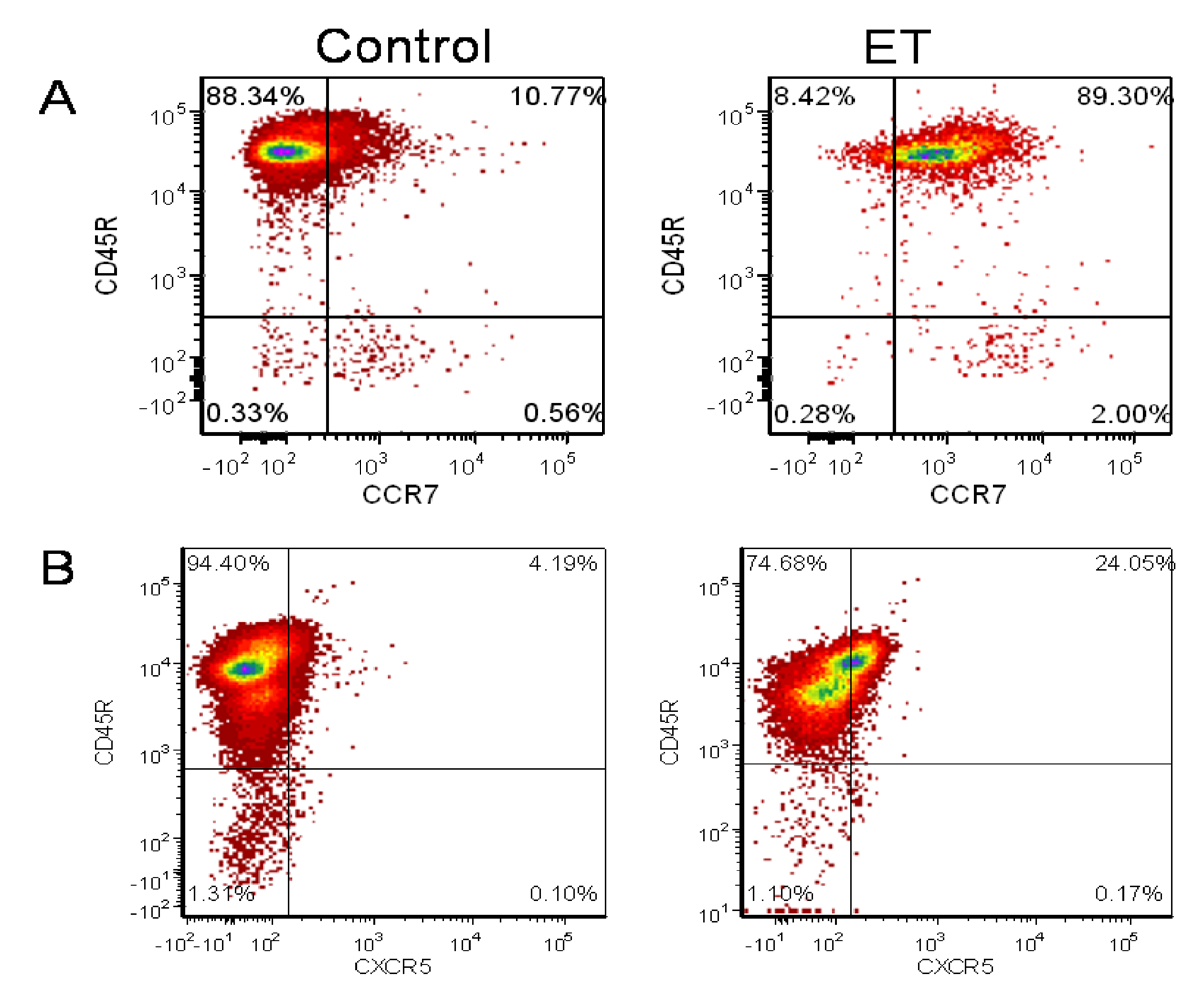
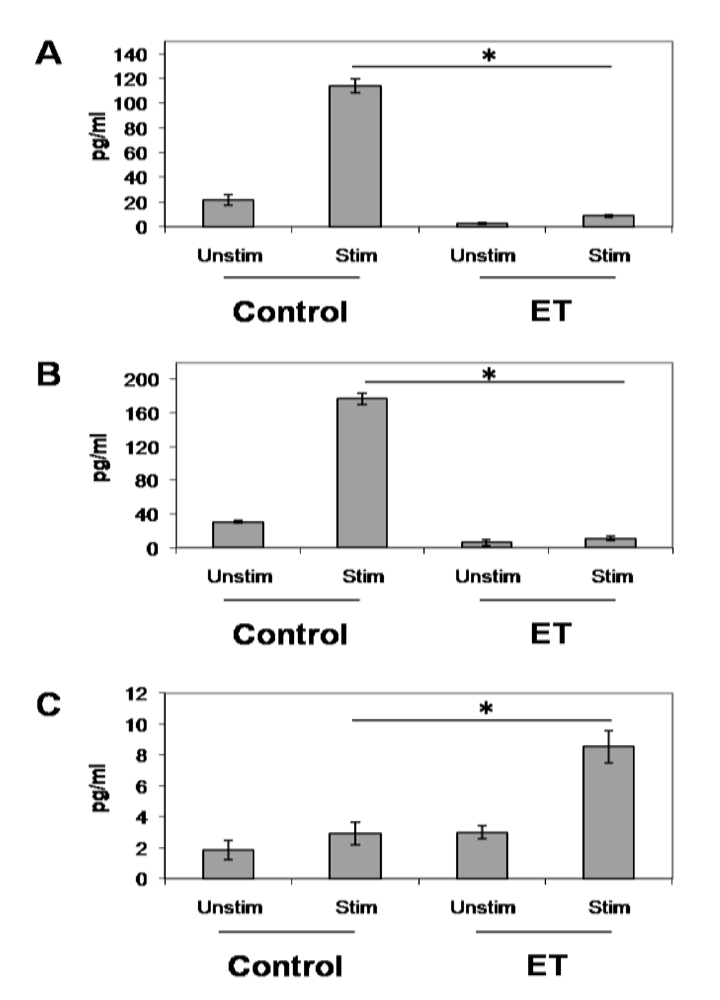
3.3. ET-Induced Modulation of B Cell Cytokine Production
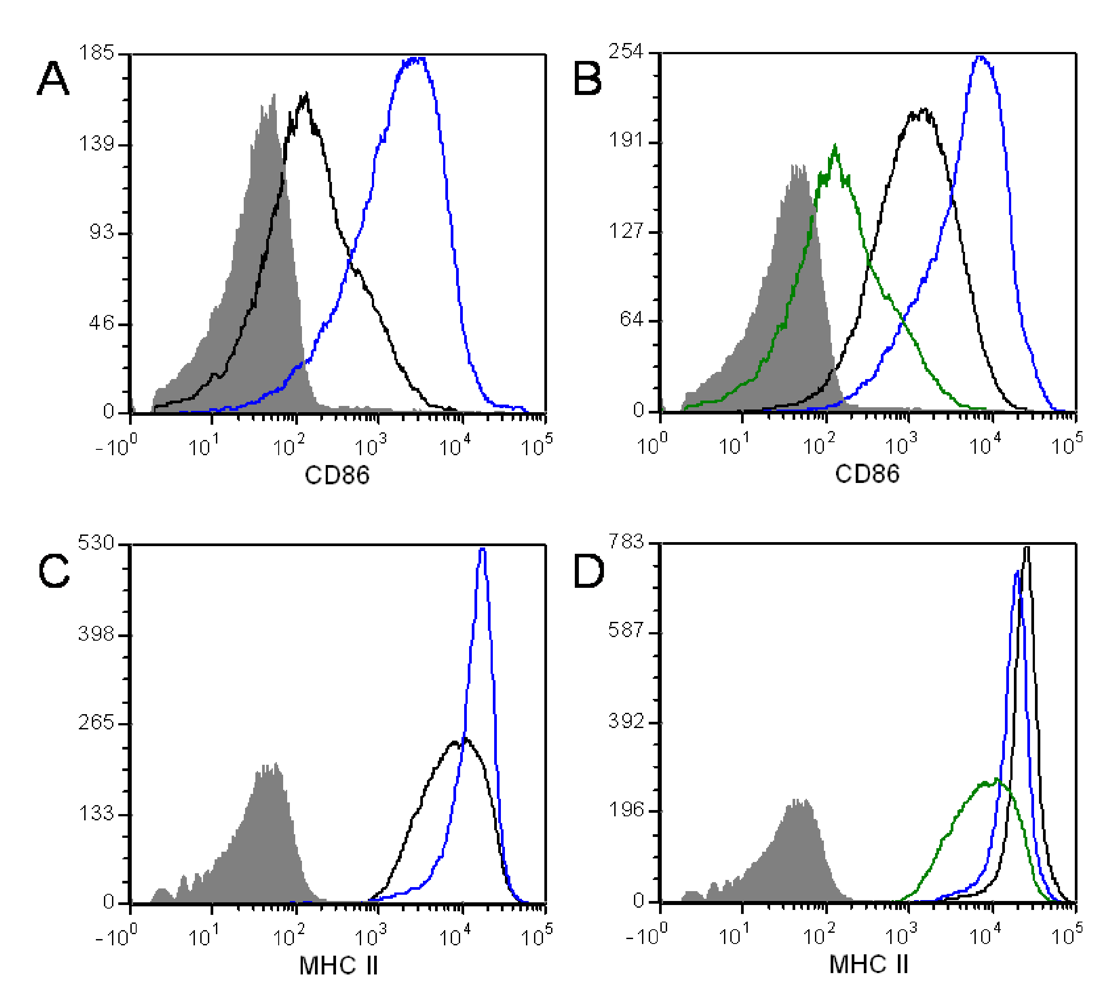
3.4. ET Alters B Cell Surface Marker Expression
4. Conclusions
Acknowledgements
References
- Scobie, H.; Rainey, G.; Bradley, K.; Young, J. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar]
- Bradley, K.; Mogridge, J.; Mourez, M.; Collier, R.; Young, J. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar]
- Cataldi, A.; Labruyere, E.; Mock, M. Construction and characterization of a protective antigen-deficient Bacillus anthracis strain. Mol. Microbiol. 1990, 4, 1111–1117. [Google Scholar]
- Pezard, C.; Duflot, E.; Mock, M. Construction of Bacillus anthracis mutant strains producing a single toxin component. J. Gen. Microbiol. 1993, 139, 2459–2463. [Google Scholar]
- Heninger, S.; Drysdale, M.; Lovchik, J.; Hutt, J.; Lipscomb, M.F.; Koehler, T.M.; Lyons, C.R. Toxin-deficient mutants of Bacillus anthracis are lethal in a murine model for pulmonary anthrax. Infect. Immun. 2006, 74, 6067–6074. [Google Scholar]
- Welkos, S.L.; Vietri, N.J.; Gibbs, P.H. Non-toxigenic derivatives of the Ames strain of Bacillus anthracis are fully virulent for mice: role of plasmid pX02 and chromosome in strain-dependent virulence. Microb. Pathog. 1993, 14, 381–388. [Google Scholar]
- Brachman, P.; Friedlander, A. Anthrax. In Vaccines, 3rd; Plotkin, S., Orenstein, W., Eds.; WB Saunders Co: Philadelphia, PA, USA, 1999; pp. 629–637. [Google Scholar]
- Lebowich, R.J.; McKillip, B.G.; Conboy, J.R. Cutaneous anthrax: a pathological study with clinical correlation. Am. J. Clin. Pathol. 1943, 13, 505–515. [Google Scholar]
- Beatty, M.E.; Ashford, D.A.; Griffin, P.M.; Tauxe, R.V.; Sobel, J. Gastrointestinal anthrax: Review of the literature. Arch. Intern. Med. 2003, 163, 2527–2531. [Google Scholar]
- Glomski, I.; Piris-Gimenez, A.; Huerre, M.; Mock, M.; Goossens, P. Primary involvement of pharynx and Peyer’s patch in inhalational and intestinal anthrax. PLoS Pathog. 2007, 3, e76. [Google Scholar]
- Chakrabarty, K.; Wu, W.; Booth, J.; Duggan, E.; Coggeshall, K.; Metcalf, J. Bacillus anthracis spores stimulate cytokine and chemokine innate immune responses in human alveolar macrophages through multiple mitogen-activated protein kinase pathways. Infect. Immun. 2006, 74, 4430–4438. [Google Scholar]
- Cleret, A.; Quesnel-Hellmann, A.; Vallon-Eberhard, A.; Verrier, B.; Jung, S.; Vidal, D.; Mathieu, J.; Tournier, J. Lung dendritic cells rapidly mediate anthrax spore entry through the pulmonary route. J. Immunol. 2007, 178, 7994–8001. [Google Scholar]
- Vitale, G.; Bernardi, L.; Napolitano, G.; Mock, M.; Montecucco, C. Susceptibility of mitogen- activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000, 352, 739–745. [Google Scholar]
- Kirby, J. Anthrax lethal toxin induces human endothelial cell apoptosis. Infect. Immun. 2004, 72, 430–439. [Google Scholar]
- Friedlander, A.; Bhatnagar, R.; Leppla, S.; Johnson, L.; Singh, Y. Characterization of macrophage sensitivity and resistance to anthrax lethal toxin. Infect. Immun. 1993, 61, 245–252. [Google Scholar]
- Lin, C.; Kao, Y.; Liu, W.; Huang, H.; Chen, K.; Wang, T.; Lin, H. Cytotoxic effects of anthrax lethal toxin on macrophage-like cell lines J774A.1. Curr. Microbiol. 1996, 33, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Alileche, A.; Serfass, E.; Muehlbauer, S.; Porcelli, S.; Brojatsch, J. Anthrax lethal toxin-mediated killing of human and murine dendritic cells impairs the adaptive immune response. PLoS Pathog. 2005, 1, 150–158. [Google Scholar]
- Popov, S.; Villasmil, R.; Bernardi, J.; Grene, E.; Cardwell, J.; Wu, A.; Alibek, D.; Bailey, C.; Alibek, K. Lethal toxin of Bacillus anthracis causes apoptosis of macrophages. Biochem. Biophys. Res. Commun. 2002, 293, 349–355. [Google Scholar]
- Molin, F.D.; Fasanella, A.; Simonato, M.; Garofolo, G.; Montecucco, C.; Tonello, F. Ratio of lethal and edema factors in rabbit systemic anthrax. Toxicon 2008, 52, 824–828. [Google Scholar]
- Leppla, S. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar]
- Drum, C.L.; Yan, S.Z.; Bard, J.; Shen, Y.Q.; Lu, D.; Soelaiman, S.; Grabarek, Z.; Bohm, A.; Tang, W.J. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 2002, 415, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Leppla, S. Bacillus anthracis calmodulin-dependent adenylate cyclase: chemical and enzymatic properties and interactions with eukaryotic cells. In Advances in Cyclic Nucleotide and Protein Phosphorylation Research; Greengard, P., Robinson, B.A., Eds.; Raven Press: New York, NY, USA, 1984; pp. 189–198. [Google Scholar]
- Tang, W.; Krupinski, J.; Gilman, A. Expression and characterization of calmodulin-activated (type 1) adenylyl cyclases. J. Biol. Chem. 1991, 266, 8595–8603. [Google Scholar]
- Ahuja, N.; Kumar, P.; Bhatnagar, R. The adenylate cyclase toxins. Crit. Rev. Microbiol. 2004, 30, 187–196. [Google Scholar]
- Crawford, M.A.; Aylott, C.V.; Bourdeau, R.W.; Bokoch, G.M. Bacillus anthracis toxins inhibit human neutrophil NADPH oxidase activity. J. Immunol. 2006, 176, 7557–7565. [Google Scholar] [PubMed]
- Hong, J.; Doebele, R.; Lingen, M.; Quilliam, L.; Tang, W.; Rosner, M. Anthrax edema toxin inhibits endothelial cell chemotaxis via Epac and Rap1. J. Biol. Chem. 2007, 282, 19781–19787. [Google Scholar]
- Comer, J.E.; Chopra, A.K.; Peterson, J.W.; Konig, R. Direct inhibition of T-lymphocyte activation by anthrax toxins in vivo. Infect. Immun. 2005, 73, 8275–8281. [Google Scholar]
- Rossi Paccani, S.; Tonello, F.; Ghittoni, R.; Natale, M.; Muraro, L.; d’elios, M.M.; Tang, W.J.; Montecucco, C.; Baldari, C.T. Anthrax toxins suppress T-lymphocyte activation by disrupting antigen receptor signaling. J. Exp. Med. 2005, 201, 325–331. [Google Scholar]
- Tournier, J.N.; Quesnel-Hellmann, A.; Mathieu, J.; Montecucco, C.; Tang, W.J.; Mock, M.; Vidal, D.R.; Goossens, P.L. Anthrax edema toxin cooperates with lethal toxin to impair cytokine secretion during infection of dendritic cells. J. Immunol. 2005, 174, 4934–4941. [Google Scholar]
- Szarowicz, S.E.; During, R.L.; Li, W.; Quinn, C.P.; Tang, W.J.; Southwick, F.S. Bacillus anthracis edema toxin impairs neutrophil actin-based motility. Infect. Immun. 2009, 77, 2455–2464. [Google Scholar]
- Puhar, A.; Molin, F.D.; Horvath, S.; Ladants, D.; Montecucco, C. Anthrax edema toxin modulates PKA- and CREB-dependent signaling in two phases. PLoS One 2008, 3, e3564. [Google Scholar]
- Paccani, S.R.; Tonello, F.; Patrussi, L.; Capitani, N.; Simonato, M.; Montecucco, C.; Baldari, C. Anthrax toxins inhibit immune cell chemotaxis by perturbing chemokine receptor signaling. Cell. Microbiol. 2007, 9, 924–929. [Google Scholar]
- Kim, C.; Wilcox-Adelman, S; Sano, Y.; Tang, W.; Collier, R.J.; Park, J. Anti-inflammatory cAMP signaling and cell migration genes co-opted by the anthrax bacillus. Proc. Natl. Acad. Sci. USA 2008, 105, 6150–6155. [Google Scholar]
- Maldonado-Arocho, F.J.; Fulcher, J.A.; Lee, B.; Bradley, K.A. Anthrax oedema toxin induces anthrax toxin receptor expression in monocyte-derived cells. Mol. Microbiol. 2006, 61, 324–337. [Google Scholar]
- Xu, L.; Frucht, D. Bacillus anthracis: a multi-faceted role for anthrax lethal toxin in thwarting host immune defenses. Int. J. Biochem. Cell. Biol. 2007, 39, 20–24. [Google Scholar]
- Turk, B.E. Manipulation of host signaling pathways by anthrax toxins. Biochem. J. 2007, 402, 405–417. [Google Scholar]
- Cote, C.K.; Rea, K.M.; Norris, S.L.; van Rooijen, N.; Welkos, S.L. The use of a model of in vivo macrophage depletion to study the role of macrophages during infection with Bacillus anthracis spores. Microb. Pathog. 2004, 37, 169–175. [Google Scholar]
- Cote, C.; Van Rooijen, N.; Welkos, S. Roles of macrophages and neutrophils in the early host response to Bacillus anthracis spores in a mouse model of infection. Infect. Immun. 2006, 74, 469–480. [Google Scholar]
- Cote, C.; DiMezzo, T.; Banks, D.; France, B.; Bradley, K.; Welkos, S. Early interactions between fully virulent Bacillus anthracis and macrophages that influence the balance between spore clearance and development of a lethal infection. Microb. Infect. 2008, 10, 613–619. [Google Scholar]
- Comer, J.; Galindo, C.; Zhang, F.; Wenglikowski, A.; Bush, K.; Garner, H.; Peterson, J.; Chopra, A. Murine macrophage transcriptional and functional responses to Bacillus anthracis edema toxin. Microb. Pathog. 2006, 41, 96–110. [Google Scholar]
- Banks, D.; Barnajian, B.; Maldonado-Arocho, F.; Sanchez, A.; Bradley, K. Anthrax toxin receptor 2 mediates Bacillus anthracis killing of macrophages following spore challenge. Cell. Microbiol. 2005, 7, 1173–1185. [Google Scholar]
- Park, J.M.; Greten, F.R.; Wong, A.; Westrick, R.J.; Arthur, J.S.C.; Otsu, K.; Hoffmann, A.; Montminy, M.; Karin, M. Signaling pathways and genes that inhibit pathogen-induced macrophage apoptosis-CREB and NF-kappaβ as key regulators. Immunity 2005, 23, 319–329. [Google Scholar]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar]
- Yeager, L.A.; Chopra, A.K.; Peterson, J.W. Bacillus anthracis edema toxin suppresses human macrophage phagocytosis and cytoskeletal remodeling via the Protein Kinase A and Exchange Protein Activated by cyclic AMP pathways. Infect. Immun. 2009, 77, 2530–2543. [Google Scholar]
- Kumar, P.; Ahuja, N.; Bhatnagar, R. Anthrax edema toxin requires influx of calcium for inducing cyclic AMP toxicity in target cells. Infect. Immun. 2002, 70, 4997–5007. [Google Scholar]
- Voth, D.; Hamm, E.; Nguyen, L.; Tucker, A.; Salles, I.; Ortiz-Leduc, O.; Ballard, J. Bacillus anthracis oedema toxin as a cause of tissue necrosis and cell type-specific cytotoxicity. Cell. Microbiol. 2005, 7, 1139–1149. [Google Scholar]
- Firoved, A.M.; Miller, G.F.; Moayeri, M.; Kakkar, R.; Shen, Y.; Wiggins, J.F.; McNally, E.M.; Tang, W.J.; Leppla, S.H. Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am. J. Pathol. 2005, 167, 1309–1320. [Google Scholar]
- O’Brien, J.; Friedlander, A.; Dreier, T.; Ezzell, J.; Leppla, S. Effects of anthrax toxin components on human neutrophils. Infect. Immun. 1985, 47, 306–310. [Google Scholar]
- Shen, Y.; Zhukovskaya, N.; Zimmers, M.; Soelaiman, S.; Bergson, P.; Wang, C.; Gibbs, C.; Tang, W. Selective inhibition of anthrax edema factor by adefovir, a drug for chronic hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 3242–3247. [Google Scholar]
- Bailly, S.; Ferrua, B.; Fay, M.; Gougerot, P.M. Differential regulation of IL-6, IL-1 alpha, IL-1 beta and TNF alpha production in LPS-stimulated human monocytes: role of cyclic AMP. Infect. Immun. 1994, 62, 4432–4439. [Google Scholar]
- Hart, P.H.; Whitty, G.A.; Piccoli, S.; Hamilton, J.A. Control by IFN-γ and PGE2 of TNFα and IL-1 production by human monocytes. Immunology 1989, 66, 376–383. [Google Scholar]
- Hoover, D.; Firedlander, A.; Rogers, L.; Yoon, I. Anthrax edema toxin differentially regulates LPS-induced monocyte production of tumor necrosis factor alpha and interleukin-6 by increasing intracellular cAMP. Infect. Immun. 1994, 62, 4432–4439. [Google Scholar]
- Bermudez, L.E.; Young, L.S. Tumor necrosis factor, alone or in combination with IL-2, but not IFN-gamma, is associated with macrophage killing of Mycobacterium avium complex. J. Immunol. 1988, 140, 3006–3013. [Google Scholar]
- Cross, A.S.; Sadoff, J.C.; Kelly, N.; Bernton, E.; Gemski, P. Pretreatment with recombinant murine tumor necrosis factor alpha/cachectin and murine interleukin 1 alpha protects mice from lethal bacterial infection. J. Exp. Med. 1989, 169, 2021–2027. [Google Scholar]
- Pezard, C.; Berche, P.; Mock, M. Contribution of individual toxin components to virulence of Bacillus anthracis. Infect. Immun. 1991, 59, 3472–3477. [Google Scholar]
- Fang, H.; Xu, L.; Chen, T.Y.; Cyr, J.M.; Frucht, D.M. Anthrax lethal toxin has direct and potent inhibitory effects on B cell proliferation and immunoglobulin production. J. Immunol. 2006, 176, 6155–6161. [Google Scholar]
- Lomo, J.; Blomhoff, H.K.; Beiske, K.; Stokke, T.; Smeland, E.B. TGF-beta 1 and cyclic AMP promote apoptosis in resting human B lymphocytes. J. Immunol. 1995, 154, 1634–1643. [Google Scholar]
- Okada, T.; Cyster, J.G. B cell migration and interactions in the early phase of antibody responses. Curr. Opin. Immunol. 2006, 18, 278–285. [Google Scholar]
- Reif, K.; Ekland, E.H.; Ohl, L.; Nakano, H.; Lipp, M.; Forster, R.; Cyster, J.G. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature 2002, 416, 94–99. [Google Scholar]
- Okada, T.; Ngo, V.N.; Ekland, E.H.; Forster, R.; Lipp, M.; Littman, D.R.; Cyster, J.G. Chemokine requirements for B cell entry to lymph nodes and Peyer's patches. J. Exp. Med. 2002, 196, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Krzysiek, R.; Lefevre, E.A.; Zou, W.; Foussat, A.; Bernard, J.; Portier, A.; Galanaud, P.; Richard, Y. Antigen receptor engagement selectively induces macrophage inflammatory protein-1 alpha (MIP-1 alpha) and MIP-1 beta chemokine production in human B cells. J. Immunol. 1999, 162, 4455–4463. [Google Scholar]
- Zhang, Y.; Lin, J.X.; Vilcek, J. Synthesis of interleukin 6 (interferon-beta 2/B cell stimulatory factor 2) in human fibroblasts is triggered by an increase in intracellular cyclic AMP. J. Biol. Chem. 1988, 263, 6177–6182. [Google Scholar]
- Orlikowsky, T.W.; Dannecker, G.E.; Spring, B.; Eichner, M.; Hoffmann, M.K.; Poets, C.F. Effect of dexamethasone on B7 regulation and T cell activation in neonates and adults. Pediatr. Res. 2005, 57, 656–661. [Google Scholar]
- Wortis, H.H.; Teutsch, M.; Higer, M.; Zheng, J.; Parker, D.C. B-cell activation by crosslinking of surface IgM or ligation of CD40 involves alternative signal pathways and results in different B-cell phenotypes. Proc. Natl. Acad. Sci. USA 1995, 92, 3348–3352. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gnade, B.T.; Moen, S.T.; Chopra, A.K.; Peterson, J.W.; Yeager, L.A. Emergence of Anthrax Edema Toxin as a Master Manipulator of Macrophage and B Cell Functions. Toxins 2010, 2, 1881-1897. https://doi.org/10.3390/toxins2071881
Gnade BT, Moen ST, Chopra AK, Peterson JW, Yeager LA. Emergence of Anthrax Edema Toxin as a Master Manipulator of Macrophage and B Cell Functions. Toxins. 2010; 2(7):1881-1897. https://doi.org/10.3390/toxins2071881
Chicago/Turabian StyleGnade, Bryan T., Scott T. Moen, Ashok K. Chopra, Johnny W. Peterson, and Linsey A. Yeager. 2010. "Emergence of Anthrax Edema Toxin as a Master Manipulator of Macrophage and B Cell Functions" Toxins 2, no. 7: 1881-1897. https://doi.org/10.3390/toxins2071881