Host-Toxin Interactions Involving EspC and Pet, Two Serine Protease Autotransporters of the Enterobacteriaceae

Abstract
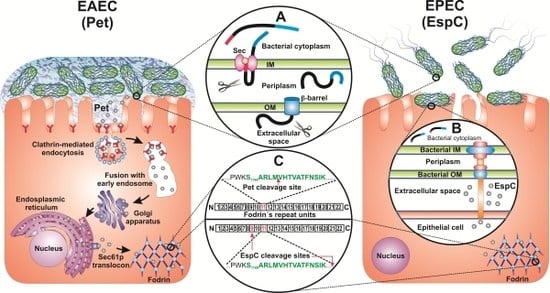
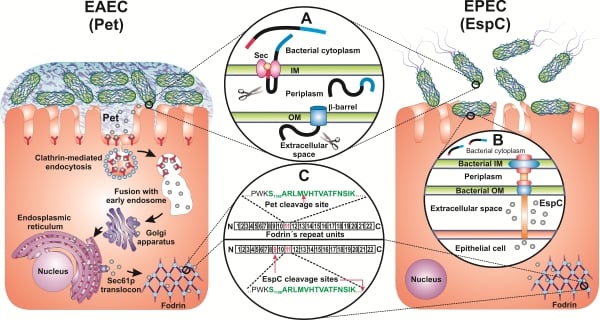
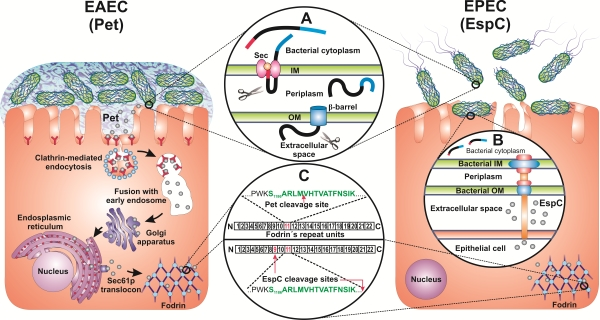
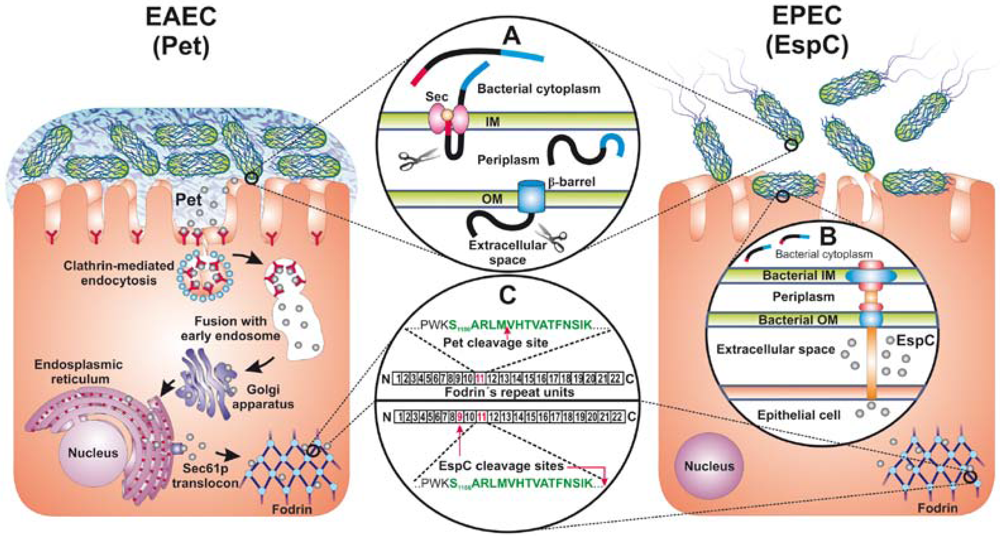
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. EAEC and EPEC Infections
3. Structure of Pet and EspC
4. Pet and EspC Activity on Epithelial Cells
5. Delivery of Pet and EspC to the Host Cell Cytosol
6. Effect of Pet and EspC on Fodrin and the Actin Cytoskeleton
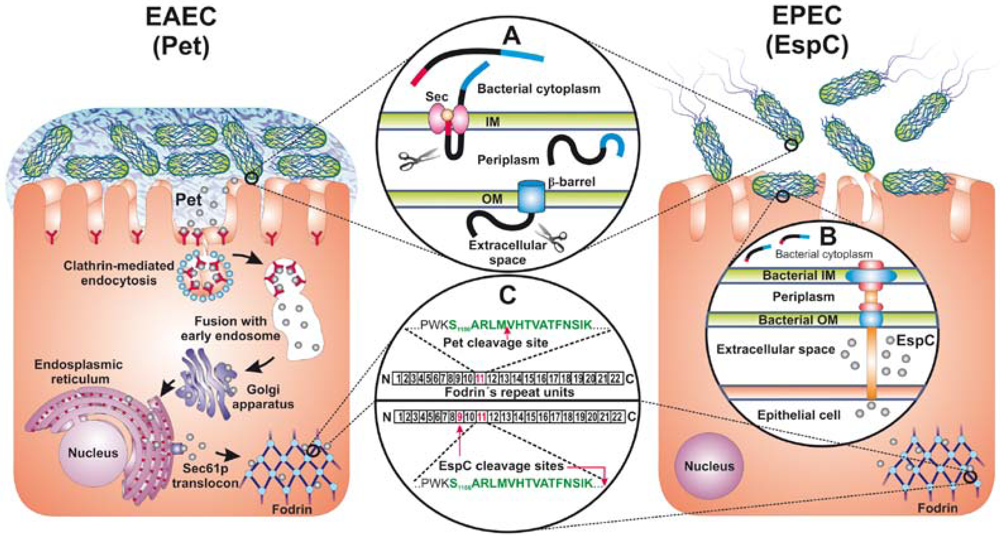
7. Summary
Acknowledgements
References
- Nataro, J.P.; Kaper, J.B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 1998, 11, 142–201. [Google Scholar] [PubMed]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar] [CrossRef] [PubMed]
- Harrington, S.M.; Dudley, E.G.; Nataro, J.P. Pathogenesis of enteroaggregative Escherichia coli infection. FEMS Microbiol. Lett. 2006, 254, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.B.; Mohanty, A.; DuPont, H.L.; Okhuysen, P.C.; Chiang, T. A review of an emerging enteric pathogen: Enteroaggregative Escherichia coli. J. Med. Microbiol. 2006, 55, 1303–1311. [Google Scholar] [CrossRef] [PubMed]
- Hicks, S.; Candy, D.C.; Phillips, A.D. Adhesion of enteroaggregative Escherichia coli to formalin-fixed intestinal and ureteric epithelia from children. J. Med. Microbiol. 1996, 44, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Nataro, J.P.; Hicks, S.; Phillips, A.D.; Vial, P.A.; Sears, C.L. T84 cells in culture as a model for enteroaggregative Escherichia coli pathogenesis. Infect. Immun. 1996, 64, 4761–4768. [Google Scholar] [PubMed]
- Vial, P.A.; Robins-Browne, R.; Lior, H.; Prado, V.; Kaper, J.B.; Nataro, J.P.; Maneval, D.; Elsayed, A.; Levine, M.M. Characterization of enteroadherent-aggregative Escherichia coli, a putative agent of diarrheal disease. J. Infect. Dis. 1988, 158, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Eslava, C.; Villaseca, J.; Morales, R.; Navarro, A.; Cravioto, A. Identification of a protein with toxigenic activity produced by enteroaggregative Escherichia coli. 93rd General Meeting of the American Society for Microbiology, Washington, DC, USA; 1993; p. 44. [Google Scholar]
- Hicks, S.; Candy, D.C.; Phillips, A.D. Adhesion of enteroaggregative Escherichia coli to pediatric intestinal mucosa in vitro. Infect. Immun. 1996, 64, 4751–4760. [Google Scholar] [PubMed]
- Tzipori, S.; Montanaro, J.; Robins-Browne, R.M.; Vial, P.; Gibson, R.; Levine, M.M. Studies with enteroaggregative Escherichia coli in the gnotobiotic piglet gastroenteritis model. Infect. Immun. 1992, 60, 5302–5306. [Google Scholar] [PubMed]
- Nataro, J.P.; Kaper, J.B.; Robins-Browne, R.; Prado, V.; Vial, P.; Levine, M.M. Patterns of adherence of diarrheagenic Escherichia coli to HEp-2 cells. Pediatr. Infect. Dis. J. 1987, 6, 829–831. [Google Scholar] [CrossRef] [PubMed]
- Nataro, J.P.; Deng, Y.; Maneval, D.R.; German, A.L.; Martin, W.C.; Levine, M.M. Aggregative adherence fimbriae I of enteroaggregative Escherichia coli mediate adherence to HEp-2 cells and hemagglutination of human erythrocytes. Infect. Immun. 1992, 60, 2297–2304. [Google Scholar] [PubMed]
- Moreira, C.G.; Carneiro, S.M.; Nataro, J.P.; Trabulsi, L.R.; Elias, W.P. Role of type I fimbriae in the aggregative adhesion pattern of enteroaggregative Escherichia coli. FEMS Microbiol. Lett. 2003, 226, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Eslava, C.; Navarro-Garcia, F.; Czeczulin, J.R.; Henderson, I.R.; Cravioto, A.; Nataro, J.P. Pet, an autotransporter enterotoxin from enteroaggregative Escherichia coli. Infect. Immun. 1998, 66, 3155–3163. [Google Scholar] [PubMed]
- Knutton, S.; Lloyd, D.R.; McNeish, A.S. Adhesion of enteropathogenic Escherichia coli to human intestinal enterocytes and cultured human intestinal mucosa. Infect. Immun. 1987, 55, 69–77. [Google Scholar]
- Taylor, C.J.; Hart, A.; Batt, R.M.; McDougall, C.; McLean, L. Ultrastructural and biochemical changes in human jejunal mucosa associated with enteropathogenic Escherichia coli (0111) infection. J. Pediatr. Gastroenterol. Nutr. 1986, 5, 70–73. [Google Scholar] [CrossRef]
- Tzipori, S.; Robins-Browne, R.M.; Gonis, G.; Hayes, J.; Withers, M.; McCartney, E. Enteropathogenic Escherichia coli enteritis: Evaluation of the gnotobiotic piglet as a model of human infection. Gut 1985, 26, 570–578. [Google Scholar] [CrossRef]
- Jerse, A.E.; Yu, J.; Tall, B.D.; Kaper, J.B. A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7839–7843. [Google Scholar]
- Moon, H.W.; Whipp, S.C.; Argenzio, R.A.; Levine, M.M.; Giannella, R.A. Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infect. Immun. 1983, 41, 1340–1351. [Google Scholar]
- Jarvis, K.G.; Giron, J.A.; Jerse, A.E.; McDaniel, T.K.; Donnenberg, M.S.; Kaper, J.B. Enteropathogenic Escherichia coli contains a putative type III secretion system necessary for the export of proteins involved in attaching and effacing lesion formation. Proc. Natl. Acad. Sci. USA 1995, 92, 7996–8000. [Google Scholar]
- Elliott, S.J.; Wainwright, L.A.; McDaniel, T.K.; Jarvis, K.G.; Deng, Y.K.; Lai, L.C.; McNamara, B.P.; Donnenberg, M.S.; Kaper, J.B. The complete sequence of the locus of enterocyte effacement (LEE) from enteropathogenic Escherichia coli E2348/69. Mol. Microbiol. 1998, 28, 1–4. [Google Scholar]
- McDaniel, T.K.; Kaper, J.B. A cloned pathogenicity island from enteropathogenic Escherichia coli confers the attaching and effacing phenotype on E. coli K-12. Mol. Microbiol. 1997, 23, 399–407. [Google Scholar] [PubMed]
- Stein, M.; Kenny, B.; Stein, M.A.; Finlay, B.B. Characterization of EspC, a 110-kilodalton protein secreted by enteropathogenic Escherichia coli which is homologous to members of the immunoglobulin A protease-like family of secreted proteins. J. Bacteriol. 1996, 178, 6546–6554. [Google Scholar]
- Mellies, J.L.; Navarro-Garcia, F.; Okeke, I.; Frederickson, J.; Nataro, J.P.; Kaper, J.B. espC pathogenicity island of enteropathogenic Escherichia coli encodes an enterotoxin. Infect. Immun. 2001, 69, 315–324. [Google Scholar]
- Mellies, J.L.; Elliott, S.J.; Sperandio, V.; Donnenberg, M.S.; Kaper, J.B. The Per regulon of enteropathogenic Escherichia coli: identification of a regulatory cascade and a novel transcriptional activator, the locus of enterocyte effacement (LEE)-encoded regulator (Ler). Mol. Microbiol. 1999, 33, 296–306. [Google Scholar] [CrossRef]
- Elliott, S.J.; Sperandio, V.; Giron, J.A.; Shin, S.; Mellies, J.L.; Wainwright, L.; Hutcheson, S.W.; McDaniel, T.K.; Kaper, J.B. The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infect. Immun. 2000, 68, 6115–6126. [Google Scholar]
- Navarro-Garcia, F. Serine protease autotransporters of Enterobacteriaceae. In Handbook of Proteolytic Enzymes: Cysteine, Serine and Threonine Peptidases, 2nd; Barrett A.J.;, Rawlings, N.D.; Woessner, J.F., Eds.; Elsevier Academic: Oxford, UK, 2004; Volume 2, pp. 1761–1765. [Google Scholar]
- Yen, Y.T.; Kostakioti, M.; Henderson, I.R.; Stathopoulos, C. Common themes and variations in serine protease autotransporters. Trends Microbiol. 2008, 16, 370–379. [Google Scholar]
- Henderson, I.R.; Navarro-Garcia, F.; Desvaux, M.; Fernandez, R.C.; Ala'Aldeen, D. Type V protein secretion pathway: The autotransporter story. Microbiol. Mol. Biol. Rev. 2004, 68, 692–744. [Google Scholar] [CrossRef]
- Dautin, N.; Bernstein, H.D. Protein secretion in gram-negative bacteria via the autotransporter pathway. Annu. Rev. Microbiol. 2007, 61, 89–112. [Google Scholar] [CrossRef]
- Vidal, J.E.; Navarro-Garcia, F. EspC translocation into epithelial cells by enteropathogenic Escherichia coli requires a concerted participation of type V and III secretion systems. Cell Microbiol. 2008, 10, 1975–1986. [Google Scholar] [CrossRef]
- Navarro-Garcia, F.; Sears, C.; Eslava, C.; Cravioto, A.; Nataro, J.P. Cytoskeletal effects induced by pet, the serine protease enterotoxin of enteroaggregative Escherichia coli. Infect. Immun. 1999, 67, 2184–2192. [Google Scholar]
- Nemec, K.N.; Scaglione, P.; Navarro-Garcia, F.; Huerta, J.; Tatulian, S.A.; Teter, K. A host-specific factor is necessary for efficient folding of the autotransporter plasmid-encoded toxin. Biochimie 2010, 92, 171–177. [Google Scholar]
- Jain, S.; Goldberg, M.B. Requirement for YaeT in the outer membrane assembly of autotransporter proteins. J. Bacteriol. 2007, 189, 5393–5398. [Google Scholar] [CrossRef]
- Purdy, G.E.; Fisher, C.R.; Payne, S.M. IcsA surface presentation in Shigella flexneri requires the periplasmic chaperones DegP, Skp, and SurA. J. Bacteriol. 2007, 189, 5566–5573. [Google Scholar]
- Purdy, G.E.; Hong, M.; Payne, S.M. Shigella flexneri DegP facilitates IcsA surface expression and is required for efficient intercellular spread. Infect. Immun. 2002, 70, 6355–6364. [Google Scholar] [CrossRef]
- Ruiz-Perez, F.; Henderson, I.R.; Leyton, D.L.; Rossiter, A.E.; Zhang, Y.; Nataro, J.P. Roles of periplasmic chaperone proteins in the biogenesis of serine protease autotransporters of Enterobacteriaceae. J. Bacteriol. 2009, 191, 6571–6583. [Google Scholar]
- Wagner, J.K.; Heindl, J.E.; Gray, A.N.; Jain, S.; Goldberg, M.B. Contribution of the periplasmic chaperone Skp to efficient presentation of the autotransporter IcsA on the surface of Shigella flexneri. J. Bacteriol. 2009, 191, 815–821. [Google Scholar]
- Dutta, P.R.; Cappello, R.; Navarro-Garcia, F.; Nataro, J.P. Functional comparison of serine protease autotransporters of Enterobacteriaceae. Infect. Immun. 2002, 70, 7105–7113. [Google Scholar] [CrossRef]
- Henderson, I.R.; Czeczulin, J.; Eslava, C.; Noriega, F.; Nataro, J.P. Characterization of pic, a secreted protease of Shigella flexneri and enteroaggregative Escherichia coli. Infect. Immun. 1999, 67, 5587–5596. [Google Scholar]
- Harrington, S.M.; Sheikh, J.; Henderson, I.R.; Ruiz-Perez, F.; Cohen, P.S.; Nataro, J.P. The Pic protease of enteroaggregative Escherichia coli promotes intestinal colonization and growth in the presence of mucin. Infect. Immun. 2009, 77, 2465–2473. [Google Scholar]
- Navarro-Garcia, F.; Eslava, C.; Villaseca, J.M.; Lopez-Revilla, R.; Czeczulin, J.R.; Srinivas, S.; Nataro, J.P.; Cravioto, A. In vitro effects of a high-molecular-weight heat-labile enterotoxin from enteroaggregative Escherichia coli. Infect. Immun. 1998, 66, 3149–3154. [Google Scholar]
- Henderson, I.R.; Hicks, S.; Navarro-Garcia, F.; Elias, W.P.; Philips, A.D.; Nataro, J.P. Involvement of the enteroaggregative Escherichia coli plasmid-encoded toxin in causing human intestinal damage. Infect. Immun. 1999, 67, 5338–5344. [Google Scholar]
- Kenny, B.; Finlay, B.B. Protein secretion by enteropathogenic Escherichia coli is essential for transducing signals to epithelial cells. Proc. Natl. Acad. Sci. USA 1995, 92, 7991–7995. [Google Scholar] [CrossRef]
- Navarro-Garcia, F.; Canizalez-Roman, A.; Sui, B.Q.; Nataro, J.P.; Azamar, Y. The serine protease motif of EspC from enteropathogenic Escherichia coli produces epithelial damage by a mechanism different from that of Pet toxin from enteroaggregative E. coli. Infect. Immun. 2004, 72, 3609–3621. [Google Scholar] [CrossRef]
- Navarro-Garcia, F.; Canizalez-Roman, A.; Luna, J.; Sears, C.; Nataro, J.P. Plasmid-encoded toxin of enteroaggregative Escherichia coli is internalized by epithelial cells. Infect. Immun. 2001, 69, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Garcia, F.; Canizalez-Roman, A.; Vidal, J.E.; Salazar, M.I. Intoxication of epithelial cells by plasmid-encoded toxin requires clathrin-mediated endocytosis. Microbiology 2007, 153, 2828–2838. [Google Scholar]
- Vidal, J.E.; Navarro-Garcia, F. Efficient translocation of EspC into epithelial cells depends on enteropathogenic Escherichia coli and host cell contact. Infect. Immun. 2006, 74, 2293–2303. [Google Scholar] [CrossRef]
- Navarro-Garcia, F.; Canizalez-Roman, A.; Burlingame, K.E.; Teter, K.; Vidal, J.E. Pet, a non-AB toxin, is transported and translocated into epithelial cells by a retrograde trafficking pathway. Infect. Immun. 2007, 75, 2101–2109. [Google Scholar]
- Lord, J.M.; Roberts, L.M.; Lencer, W.I. Entry of protein toxins into mammalian cells by crossing the endoplasmic reticulum membrane: Co-opting basic mechanisms of endoplasmic reticulum-associated degradation. Curr. Top Microbiol. Immunol. 2005, 300, 149–168. [Google Scholar] [CrossRef]
- Vembar, S.S.; Brodsky, J.L. One step at a time: Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell. Biol. 2008, 9, 944–957. [Google Scholar] [CrossRef]
- Hazes, B.; Read, R.J. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 1997, 36, 11051–11054. [Google Scholar] [CrossRef]
- Scaglione, P.; Nemec, K.N.; Burlingame, K.E.; Grabon, A.; Huerta, J.; Navarro-Garcia, F.; Tatulian, S.A.; Teter, K. Structural characteristics of the plasmid-encoded toxin from enteroaggregative Escherichia coli. Biochemistry 2008, 47, 9582–9591. [Google Scholar]
- Argent, R.H.; Parrott, A.M.; Day, P.J.; Roberts, L.M.; Stockley, P.G.; Lord, J.M.; Radford, S.E. Ribosome-mediated folding of partially unfolded ricin A-chain. J. Biol. Chem. 2000, 275, 9263–9269. [Google Scholar]
- Pande, A.H.; Moe, D.; Jamnadas, M.; Tatulian, S.A.; Teter, K. The pertussis toxin S1 subunit is a thermally unstable protein susceptible to degradation by the 20S proteasome. Biochemistry 2006, 45, 13734–13740. [Google Scholar]
- Pande, A.H.; Scaglione, P.; Taylor, M.; Nemec, K.N.; Tuthill, S.; Moe, D.; Holmes, R.K.; Tatulian, S.A.; Teter, K. Conformational instability of the cholera toxin A1 polypeptide. J. Mol. Biol. 2007, 374, 1114–1128. [Google Scholar]
- Mayerhofer, P.U.; Cook, J.P.; Wahlman, J.; Pinheiro, T.T.; Moore, K.A.; Lord, J.M.; Johnson, A.E.; Roberts, L.M. Ricin A chain insertion into endoplasmic reticulum membranes is triggered by a temperature increase to 37 °C. J. Biol. Chem. 2009, 284, 10232–10242. [Google Scholar]
- Betancourt-Sanchez, M.; Navarro-Garcia, F. Pet secretion, internalization and induction of cell death during infection of epithelial cells by enteroaggregative Escherichia coli. Microbiology 2009, 155, 2895–2906. [Google Scholar] [CrossRef]
- Canizalez-Roman, A.; Navarro-Garcia, F. Fodrin CaM-binding domain cleavage by Pet from enteroaggregative Escherichia coli leads to actin cytoskeletal disruption. Mol. Microbiol. 2003, 48, 947–958. [Google Scholar] [CrossRef]
- Baines, A.J. Evolution of spectrin function in cytoskeletal and membrane networks. Biochem. Soc. Trans. 2009, 37, 796–803. [Google Scholar] [CrossRef]
- Chakrabarti, A.; Kelkar, D.A.; Chattopadhyay, A. Spectrin organization and dynamics: New insights. Biosci. Rep. 2006, 26, 369–386. [Google Scholar] [CrossRef]
- Kizhatil, K.; Yoon, W.; Mohler, P.J.; Davis, L.H.; Hoffman, J.A.; Bennett, V. Ankyrin-G and beta2-spectrin collaborate in biogenesis of lateral membrane of human bronchial epithelial cells. J. Biol. Chem. 2007, 282, 2029–2037. [Google Scholar]
- Nath, R.; Raser, K.J.; Stafford, D.; Hajimohammadreza, I.; Posner, A.; Allen, H.; Talanian, R.V.; Yuen, P.; Gilbertsen, R.B.; Wang, K.K. Non-erythroid alpha-spectrin breakdown by calpain and interleukin 1 beta-converting-enzyme-like protease(s) in apoptotic cells: Contributory roles of both protease families in neuronal apoptosis. Biochem. J. 1996, 319, 683–690. [Google Scholar]
- Wang, K.K.; Posmantur, R.; Nath, R.; McGinnis, K.; Whitton, M.; Talanian, R.V.; Glantz, S.B.; Morrow, J.S. Simultaneous degradation of alphaII- and betaII-spectrin by caspase 3 (CPP32) in apoptotic cells. J. Biol. Chem. 1998, 273, 22490–22497. [Google Scholar]
- Villaseca, J.M.; Navarro-Garcia, F.; Mendoza-Hernandez, G.; Nataro, J.P.; Cravioto, A.; Eslava, C. Pet toxin from enteroaggregative Escherichia coli produces cellular damage associated with fodrin disruption. Infect. Immun. 2000, 68, 5920–5927. [Google Scholar]
- Djabali, K. Cytoskeletal proteins connecting intermediate filaments to cytoplasmic and nuclear periphery. Histol. Histopathol. 1999, 14, 501–509. [Google Scholar]
- Harris, A.S.; Morrow, J.S. Calmodulin and calcium-dependent protease I coordinately regulate the interaction of fodrin with actin. Proc. Natl. Acad. Sci. USA 1990, 87, 3009–3013. [Google Scholar]
- Wang, K.K.W. Calpain and caspase: Can you tell the difference? Trends Neurosci. 2000, 23, 20–26. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Navarro-Garcia, F.; Sonnested, M.; Teter, K. Host-Toxin Interactions Involving EspC and Pet, Two Serine Protease Autotransporters of the Enterobacteriaceae. Toxins 2010, 2, 1134-1147. https://doi.org/10.3390/toxins2051134
Navarro-Garcia F, Sonnested M, Teter K. Host-Toxin Interactions Involving EspC and Pet, Two Serine Protease Autotransporters of the Enterobacteriaceae. Toxins. 2010; 2(5):1134-1147. https://doi.org/10.3390/toxins2051134
Chicago/Turabian StyleNavarro-Garcia, Fernando, Michael Sonnested, and Ken Teter. 2010. "Host-Toxin Interactions Involving EspC and Pet, Two Serine Protease Autotransporters of the Enterobacteriaceae" Toxins 2, no. 5: 1134-1147. https://doi.org/10.3390/toxins2051134