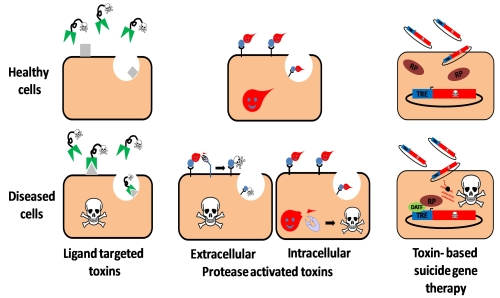
Toxin-Based Therapeutic Approaches


Abstract
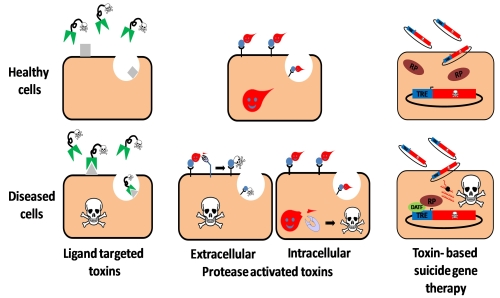
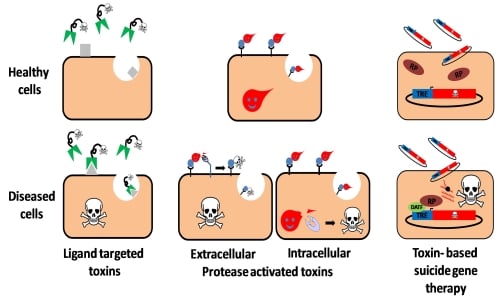
:
1. Introduction
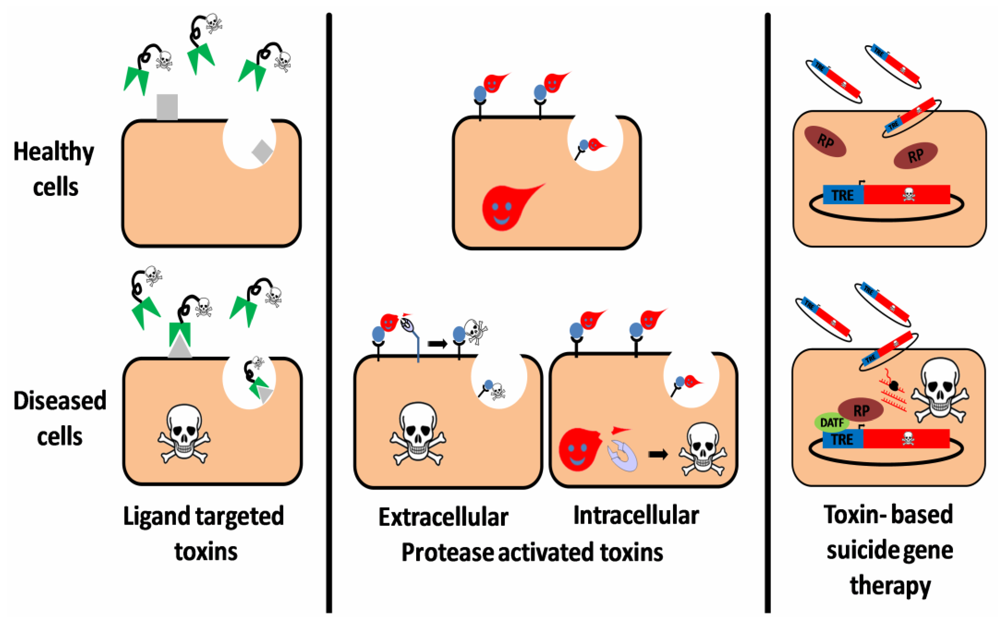
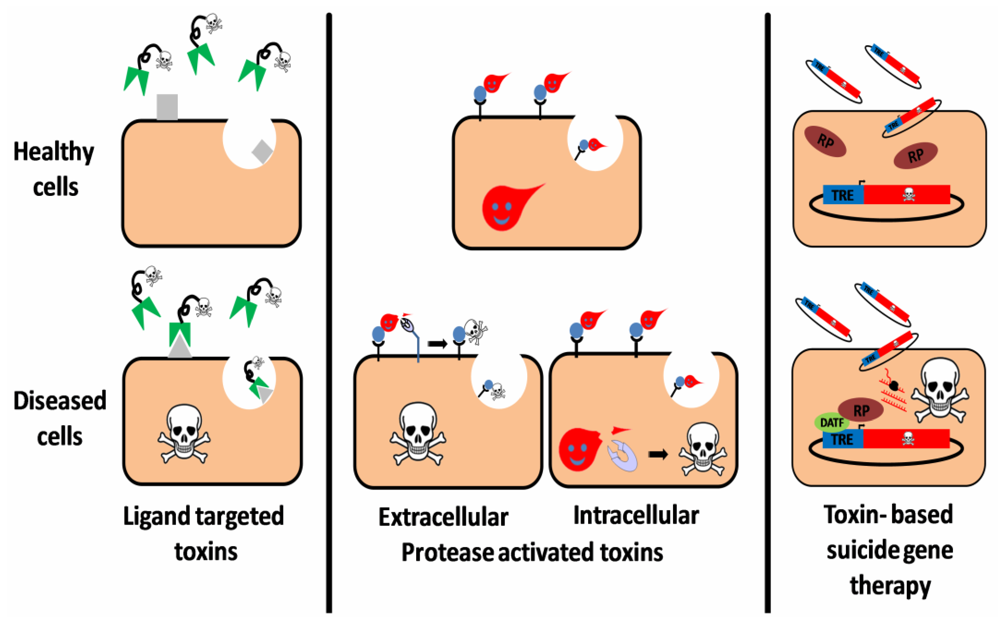
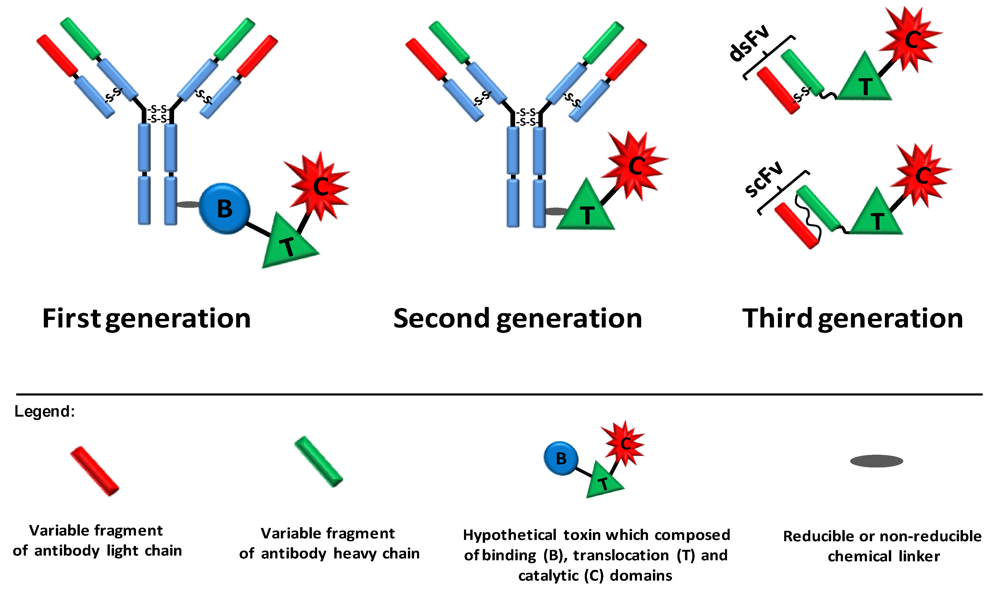
2. Ligand Targeted Toxins—Immunotoxins
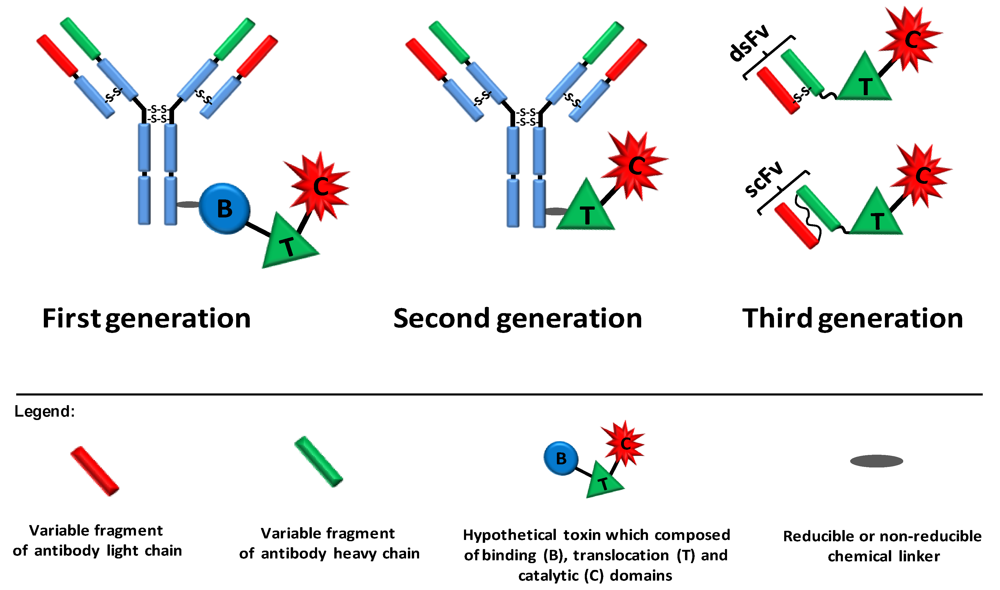

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Construct Name | Targeting Moiety | Toxic Moiety | Toxin Source | Target | Indication | Clinical Trial Phase | References |
|---|---|---|---|---|---|---|---|
| DAB389IL2 (Denileukin Diftitox) | IL˒ | DAB389 | DT | IL˒R | CTCL, NHL, CLL, NSCLC, GVHD, psoriasis, melanoma, ovarian, breast, kidney cancers | I,II,III,IV *FDA approvedFor CTCLtreatment | [17,18,19,20,21,22,23,24,25] |
| DAB486IL2 | IL-2 | DAB486 | DT | IL-2R | NHL, HD, CLL, CTCL, KS, RA | I/II | [26,27,28,29,30,31] |
| Tf-CRM107 (TransMID) | Transferrin | CRM107 | DT | TfR | Brain and CNS tumors | I, III | [32,33,34] |
| DT388-GM-CSF | GM-CSF | DT388 | DT | GM-CSFR | AML | I | [35] |
| DAB389EGF | EGF | DAB389 | DT | EGFR | EGFR-expressing carcinoma | I/II | [11] |
| A-dmDT390-bisFV (UCHT1) | bisFv | DT390 | DT | CD3ε | T-cell lymphoma/leukemia | I/II | [36,37] |
| DT388-IL3 | VariantIL-3 | DT388 | DT | IL-3R | AML, MDS | I/II | [38] |
| OVB3-PE | MAb | Full length PE | PE | Ovarian antigen | Ovarian cancer | I | [39] |
| ERB-38 | dsFv | PE38 | PE | erbB2/ HER2 | Breast, esophageal cancers | I | [40] |
| SS1(dsFv)PE38 (SS1P) | dsFv | PE38 | PE | Mesothelin | Mesothelioma, ovarian, pancreatic cancers | I | [41,42] |
| B3(Fv)-PE38 (LMB-7) | scFv | PE38 | PE | Lewis Y | Adenocarcinoma | I | [10] |
| LMB-1 | MAb | PE38 | PE | Lewis Y | Adenocarcinoma | I | [43] |
| RFB4(dsFv)-PE38 (BL22/CAT3888) | dsFv | PE38 | PE | CD22 | NHL, CLL, HCL, ALL | I,II | [44,45,46,47] |
| LMB-2 | scFv | PE38 | PE | CD25 | Leukemia, lymphoma | II | [48] |
| scFv(FRP5)-ETA | scFv | PE40 | PE | erbB2 /HER2 | Melanoma, Breast, colon cancers | I | [49,50] |
| TP40 | TGFα | Modified PE40 | PE | EGFR | Bladder cancer | I | [51] |
| TP38 | TGFα | PE38 | PE | EGFR | Glioblastoma | II | [52,53,54] |
| BR96sFv-PE40 (SGN-10) | scFv | PE40 | PE | Lewis Y | Adenocarcinoma | I | [55] |
| B3(dsFv)-PE38 (LMB-9) | dsFv | PE38 | PE | Lewis Y | Adenocarcinoma | I | [10] |
| IL4(38-37) PE38KDEL (NBI-3001) | Circularly permuted IL-4 | Modified PE38 | PE | IL-4R | Brain, CNS, kidney, lung, breast cancers | I,II | [56,57,58] |
| Mutated RFB4(dsFv)-PE38 (HA22/CAT-8015) | dsFv | PE38 | PE | CD22 | HCL, ALL, NHL CLL, PLL, SLL | I | [59,60] |
| IL13-PE38QQR (cinterdekin besudotox) | IL-13 | Modified PE38 | PE | IL-13R | Glioma | I/II, III | [61,62,63,64] |
| RFB4-Fab'-dgA | Fab' | Deglycosylated RTA | Ricin | CD22 | B-NHL | I | [65] |
| RFB4-dgA (IMTOX-22) | MAb | Deglycosylated RTA | Ricin | CD22 | B-NHL, CLL | I | [66,67] |
| HD37-dgA (IMTOX-19) | MAb | Deglycosylated RTA | Ricin | CD19 | NHL | I | [68,69] |
| RFB4-dgA + HD37-dgA (Combotox) | MAb | Deglycosylated RTA | Ricin | CD22, CD19 | NHL, ALL | I | [70,71] |
| RFT5-dgA (IMTOX-25) | MAb | Deglycosylated RTA | Ricin | CD25 | HD, CTCL, melanoma, GVHD | I,II | [72,73,74,75,76] |
| Ki-4.dgA | MAb | Deglycosylated RTA | Ricin | CD30 | HD, NHL | I | [75,77] |
| Anti-B4-bR | MAb | Blocked ricin | Ricin | CD19 | B-NHL | II | [78,79,80,81] |
| Anti-CEA-bR | MAb | Blocked ricin | Ricin | CEA | Colorectal cancer | I/II | [82] |
| N901-bR | MAb | Blocked ricin | Ricin | CD56 | SCLC | I | [83,84,85] |
| Anti-CD7-dgA (DA7) | MAb | Deglycosylated RTA | Ricin | CD7 | T-NHL | I | [86] |
| Anti-CD3-dgA +Anti-CD7-dgA | MAb | Deglycosylated RTA | Ricin | CD3, CD7 | GVHD | I/II | [87] |
| CD5-IC, CD5 Plus | MAb | RTA | Ricin | CD5 | RA, SLE, diabetes mellitus | I,II | [88,89,90,91,92] |
| H65-RTA | MAb | RTA | Ricin | CD5 | CTCL, GVHD | I, I/II | [93,94,95] |
| T101-RTA | MAb | RTA | Ricin | CD5 | CLL | I | [96,97,98] |
| MDX-RA | MAb | RTA | Ricin | Human lens epithelial antigen | Posterior capsule opacification (secondary cataract) | III | [99,100,101] |
| XomaZyme-Mel(XMMME-001-RTA) | MAb | RTA | Ricin | Melanoma antigen | Melanoma | I/II | [102,103,104,105,106,107] |
| XomaZyme-791(79IT/36-RTA) | MAb | RTA | Ricin | 72kDa TAA | Colorectal cancer | I | [108,109,110] |
| 454A12-rRA | MAb | RTA | Ricin | TfR | Leptomeningeal neoplasia | I | [111] |
| 260F9-rRTA | MAb | RTA | Ricin | 55 kDa breast cancer antigen | Breast cancer | I | [112,113] |
| B43-PAP | MAb | PAP | PAP | CD19 | ALL | I | [114] |
| TXU-PAP | MAb | PAP | PAP | CD7 | HIV-1 infection | I | [115] |
| BER-H2-Sap6 | MAb | Saporin | Saporin | CD30 | HD | I | [116] |
| HUM-195/rGel | MAb | Gelonin | Gelonin | CD33 | AML, CML | I | [117] |
| BDI-1-MD | MAb | Momordin | Momordin | Bladder carcinoma antigen | Bladder cancer | I | [118] |
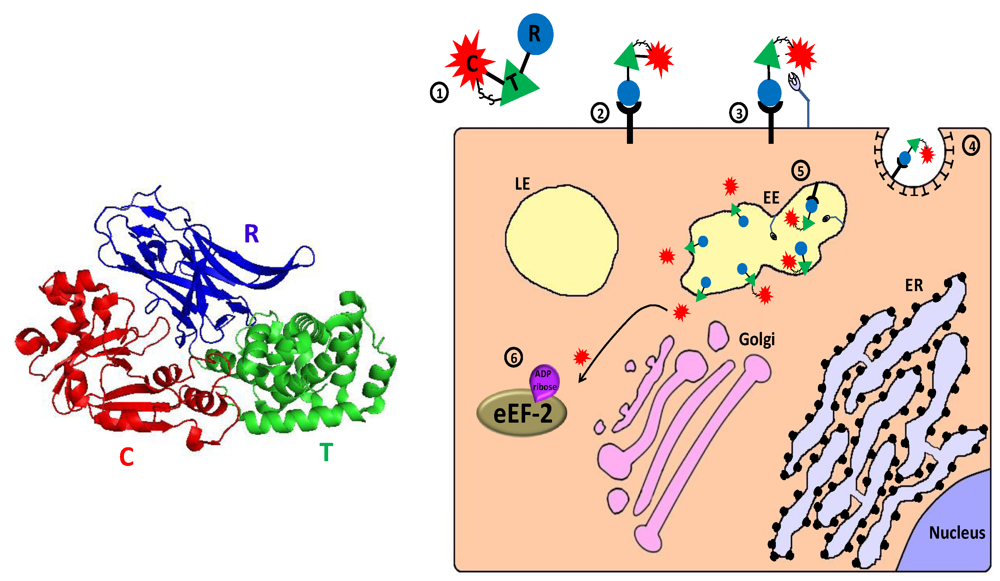
2.1. Diphtheria Toxin Based Immunotoxins
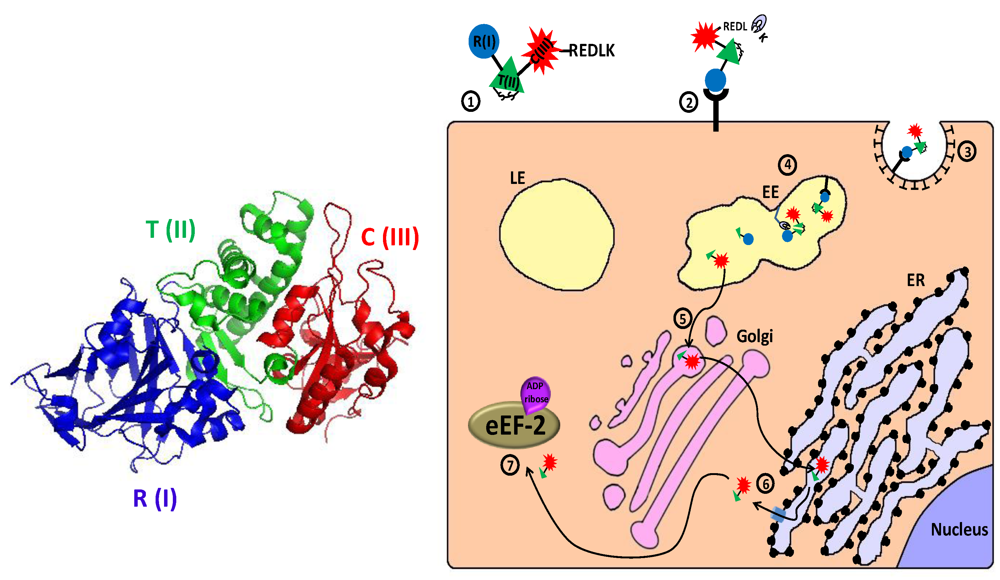
2.1.1. Diphtheria Toxin—Mechanism of Action
2.1.2. Targeting IL-2 Receptor: Denileukin Diftitox (Ontak)
2.1.3. Targeting Granulocyte-Macrophage Colony Stimulating Factor Receptor: DT388-GM-CSF
2.1.4. Targeting Transferrin Receptor: Tf-CRM107 (TransMID)
2.2. Pseudomonas Exotoxin A Based Immunotoxins
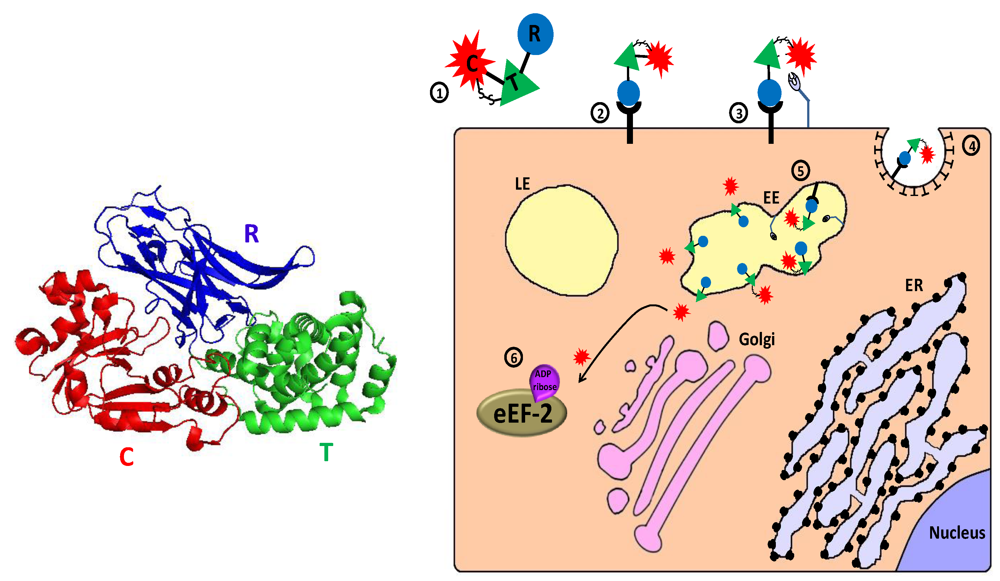
2.2.1. Pseudomonas Exotoxin A—Mechanism of Action
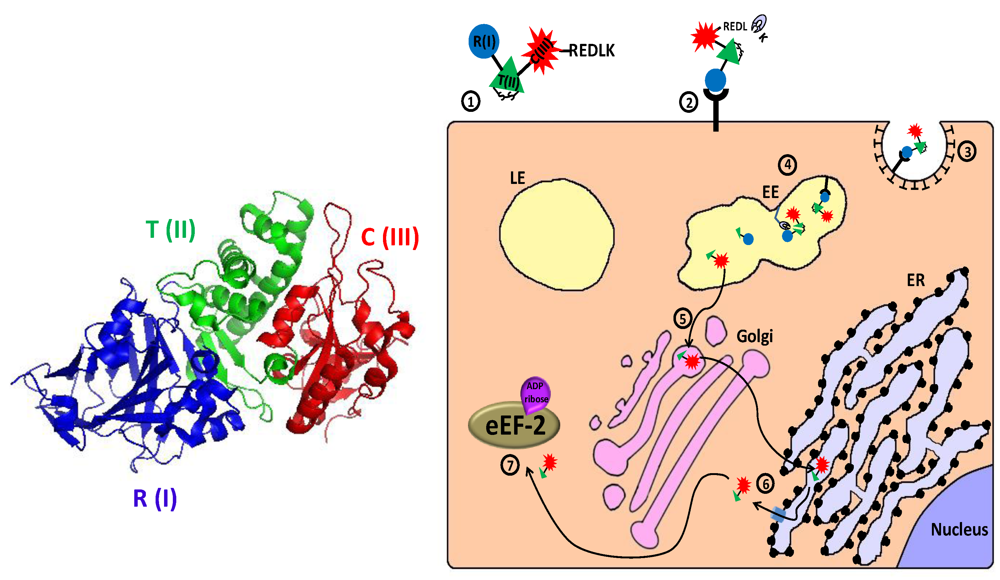
2.2.2. Targeting the CD25 Subunit of IL2-Receptor: LMB-2
2.2.3. Targeting CD22: BL22
2.2.4. Targeting the LeY Antigen: LMB-1
2.3. Ribosome Inactivating Proteins Based Immunotoxins
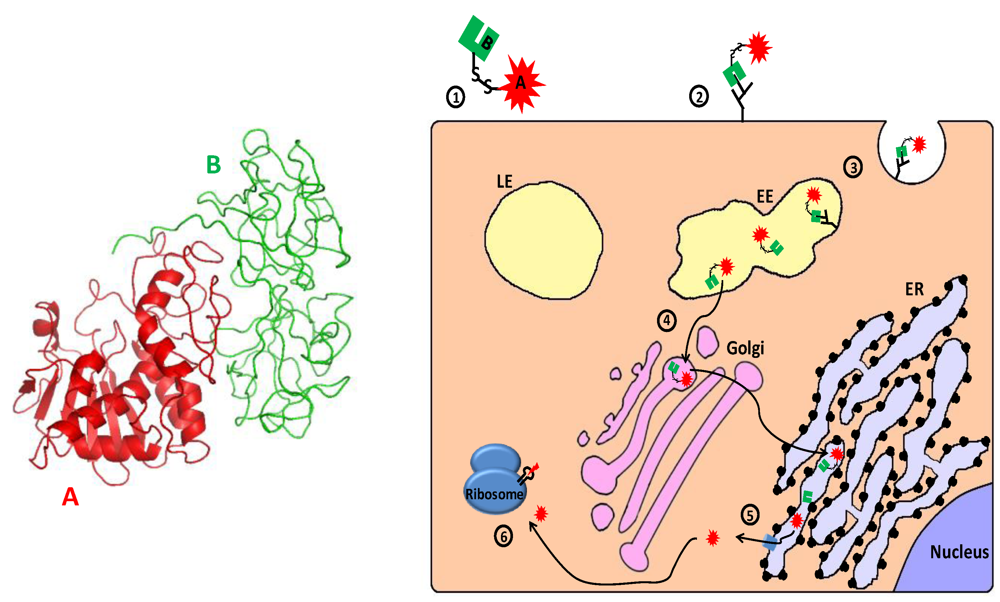
2.3.1. Ribosome Inactivating Proteins—Mechanism of Action
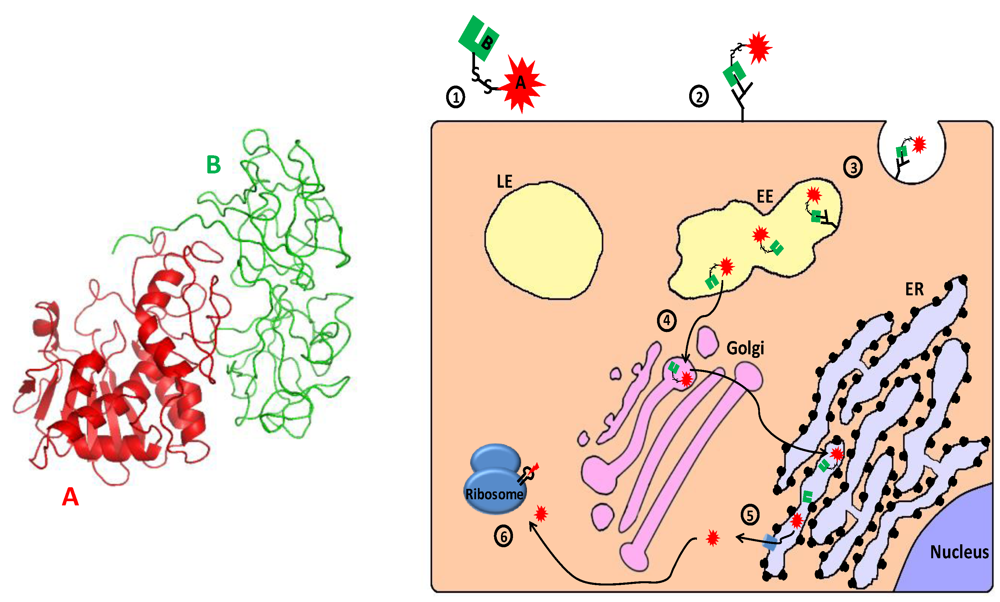
2.3.2. Targeting CD25 and CD30: RFT5-dgA and ki-4.dgA.
2.3.3. Targeting CD22 and CD19: RFB4-dgA and HD37-dgA
3. Toxin Based Suicide Gene Therapy
| Construct Name | Transcription Regulatory Element | Toxin | Delivery Vector | Disease | Clinical Trial Phase | Reference |
|---|---|---|---|---|---|---|
| Ad5-PSE/PSA-DT-A | PSA * | DTA | Adenovirus | Prostate cancer | Preclinical phase | [339] |
| Ad-PSA/FLP +Ad-RSV/FRT2neo/DT-A | PSA * | DTA | Adenovirus | Prostate cancer | Preclinical phase | [340] |
| C32-PSA/DT-A | PSA * | DTA | Cationic polymer | Prostate cancer | Preclinical phase | [341] |
| pTHA-47, pTHA-49 | hCG (α or β subunits) * | DTA | Naked DNA-electroporation | Ovarian cancer | Preclinica lphase | [342] |
| pHE-4/DT-A,117-MSLN/DT-A | HE4, MSLN * | DTA | Cationic polymer | Ovarian cancer | Preclinica lphase | [343] |
| DTA-H19 (BC-819) | H19 * | DTA | Naked DNA, Cationic polymer | Ovarian, bladder, pancreatic cancers | I/II, II | [344,345,346] |
| DTA-TER, DTA-TERT | hTER, hTERT * | DTA | Naked DNA- CaPO4 precipitate | Bladder cancer | Preclinical phase | [347] |
| HIV-DT-A | HIV Tat and Rev cis-acting responsive sequences | DTA | Retrovirus, cationic liposomes | HIV-1 infection | Preclinical phase | [348,349,350] |
| pNL-DTΔN-GFP-RRE-SA | HIV Rev cis-acting responsive sequence | Attenuated DTA variant | Non-integrating lentivirus | HIV-1 infection | Preclinical phase | [351] |
| pA3-6PED | PAX3 DNA responsive sequences | DTA | Cationic liposomes | ARMS | Preclinical phase | [352] |
| petbz.ES.DT-A, pA.E-Sel.DT-A | E-selectin * | DTA | Naked DNA-electroporation | Activated endothelial cells (Angiogenesis) | Preclinical phase | [353] |
| GH-loxP-DT + CMV-Cre / GH-Cre | Growth hormone (GH) * | DTA | Adenovorus | Pituitary Tumor | Preclinical phase | [354] |
| BV-CG/ITR-DTA | GFAP * + CMV enhancer + ITR of AAV | DTA | Baculovirus | glioma | Preclinical phase | [355] |
| G1CEAPEANa, G1CEADTANa | CEA * | PEA, DTA | Cationic liposomes | Colorectal carcinoma | Preclinical phase | [356] |
| pRad51-DTA | Rad51 * | DTA | Various transfection methods | Variety of cancer cells | Preclinical phase | [357] |
| pAF-DTA, pAF5.1DTA | AFP * | DTA | Cationic liposomes | HCC | Preclinical phase | [358,359] |
| pTHA45, pTHA17 | Immunoglobulin heavy/κ-light chain * | DTA | Naked DNA-electroporation | B-Lymphoid Cells | Preclinical phase | [360] |
| pTyrIII/DT-A, pMIA III/DT-A | MIA, tyrosinase, * | DTA | Cationic lipids | Melanoma | Preclinical phase | [361] |
| retro-1.3MBPppe, retro-1.3MBPpri | MBP * | PE/RTA | Retrovirus | Glioblastoma | Preclinical phase | [362] |
| pMSLN/DT-A | MSLN * | DTA | Cationic polymer | Pancreatic cancer | Preclinical phase | [363] |
| V3 | Hsp70B' * + HSEs | DTA, attenuated DTA variants | Cationic liposomes | Pancreatic cancer | Preclinical phase | [364] |
| pLTR-DT | p34 responsive sequences (BLV-LTR) | DTA | Cationic liposomes | BVL infected cells | Preclinical phase (veterinary use) | [365,366] |
3.1. Targeting Prostate Cancer
3.2. Targeting Ovarian Cancer
3.3. Targeting Bladder Cancer
3.4. Targeting Viral Infected Cells
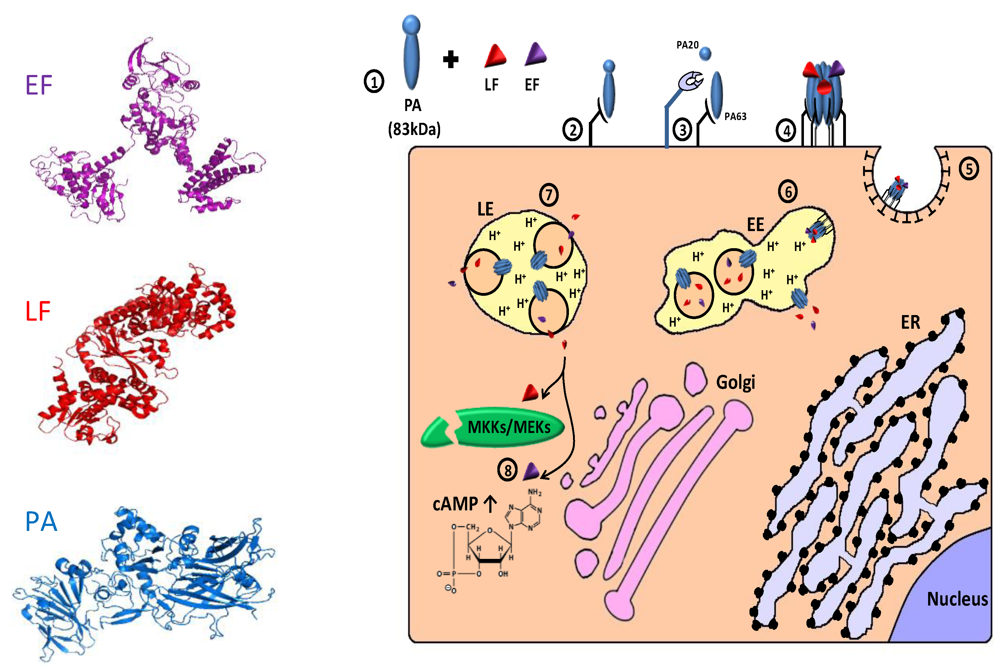
4. Protease Activated Toxins
4.1. Extracellular Protease Activated Toxins
| Construct Name | Activating Protease | Protease Localization | Components | Toxin Source | Activation Mode | Target | References |
|---|---|---|---|---|---|---|---|
| PA-L1/L2 + FP59 | MMPs (mainly MMP2 and MMP9) | Extracellular | PA (modified) +FP59 | Anthrax + PE | Binding and translocation of the toxic moiety | MMPs expressing tumor cells | [421] |
| PA-L1 + LF | MMPs (mainly MMP2 and MMP9) | Extracellular | PA (modified) +LF | Anthrax | Binding and translocation of the toxic moiety | Tumor vasculature; MMPs expressing tumor cells with V600E B-Raf mutation | [422,423,424] |
| PrAg-U2 + FP59 | uPA | Extracellular | PA (modified) +FP59 | Anthrax + PE | Binding and translocation of the toxic moiety | Tumor cells with receptor-associated uPA activity | [425,426,427,428,429] |
| PrAg-L1-I210A + PrAg-U2 R200A + FP59 | MMPs + uPA (both required) | Extracellular | PA (modified) +FP59 | Anthrax + PE | Binding and translocation of the toxic moiety | MMPs expressing tumor cells with receptor-associated uPA activity | [430] |
| UFT3 | PSA | Extracellular and intracellular | Ubiquitin (mutant), saporin | Saporin | Toxin stabilization | Prostate cancer cells | [431] |
| DTU2GMCSF | uPA | Extracellular | DT388 (modified), GM-CSF | DT | Translocation of the toxic moiety | AML cells (the toxin is targeted also by fusion to GM-CSF) | [432] |
| FLD/MM, FLD/YV | HIV-1 protease | Intracellular | PA + LFN-DTA | Anthrax + DT | Toxin stabilization | HIV-1 infected cells | [433] |
| TAT-Pro-HIV-p2/NC, TAT-Pro-HIV-MA/CA | HIV-1 protease | Intracellular | HIV-1 TAT transduction peptide, Maize RIP (modified) | Maize RIP | Enhancement in the enzymatic activity of the toxic moiety | HIV-1 infected cells | [434] |
4.1.1. Targeting Matrix Metalloproteinases (MMPs) Overexpressing Tumor Cells
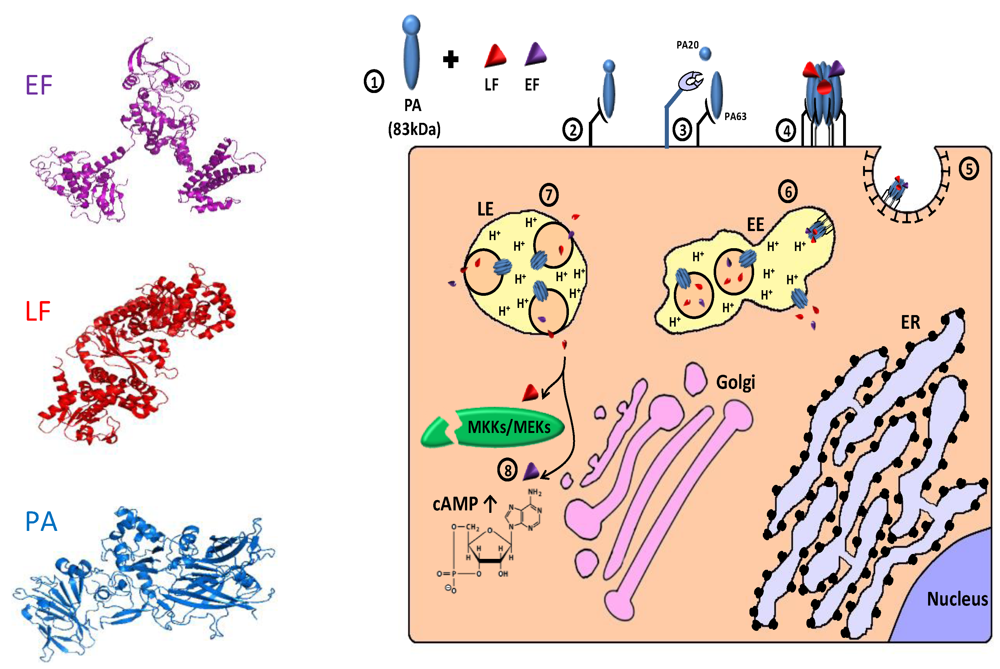
4.1.2. Targeting Malignant Cells Overexpressing the Urokinase Plasminogen Activator System
4.2. Intracellular Protease Activated Toxins
4.2.1. Targeting HIV Infected Cells
5. Concluding Remarks
References
- Kostrzewa, R.; Segura-Aguilar, J. Botulinum neurotoxin: Evolution from poison, to research tool—onto medicinal therapeutic and future pharmaceutical panacea. Neurotox. Res. 2007, 12, 275–290. [Google Scholar]
- Mahajan, S.T.; Brubaker, L. Botulinum toxin: From life-threatening disease to novel medical therapy. Am. J. Obstet. Gynecol. 2007, 196, 7–15. [Google Scholar]
- Erbguth, F.J. From poison to remedy: The chequered history of botulinum toxin. J. Neur. Transm. 2008, 115, 559–565. [Google Scholar]
- Truong, D.D.; Stenner, A.; Reichel, G. Current Clinical Applications of Botulinum Toxin. Curr. Pharm. Design 2009, 15, 3671–3680. [Google Scholar]
- Winau, F.; Westphal, O.; Winau, R. Paul Ehrlich—In search of the magic bullet. Microb. Infect. 2004, 6, 786–789. [Google Scholar]
- Bosch, F.; Rosich, L. The contributions of Paul Ehrlich to pharmacology: A tribute on the occasion of the centenary of his Nobel Prize. Pharmacology 2008, 82, 171–179. [Google Scholar]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich's magic bullet concept: 100 years of progress. Nat. Rev. Canc. 2008, 8, 473–480. [Google Scholar]
- Moolten, F.L.; Cooperband, S.R. Selective destruction of target cells by diphtheria toxin conjugated to antibody directed against antigens on the cells. Science 1970, 169, 68–70. [Google Scholar]
- Moolten, F.; Zajdel, S.; Cooperband, S. Immunotherapy of experimental animal tumors with antitumor antibodies conjugated to diphtheria toxin or ricin. Ann. NY Acad. Sci. 1976, 277, 690–699. [Google Scholar]
- Pastan, I.; Hassan, R.; FitzGerald, D.J.; Kreitman, R.J. Immunotoxin treatment of cancer. Annu. Rev. Med. 2007, 58, 221–237. [Google Scholar]
- Kreitman, R.J. Immunotoxins for targeted cancer therapy. AAPS J. 2006, 8, E532–E551. [Google Scholar]
- Brumlik, M.J.; Daniel, B.J.; Waehler, R.; Curiel, D.T.; Giles, F.J.; Curiel, T.J. Trends in immunoconjugate and ligand-receptor based targeting development for cancer therapy. Expert Opin. Drug Deliv. 2008, 5, 87–103. [Google Scholar]
- Potala, S.; Sahoo, S.K.; Verma, R.S. Targeted therapy of cancer using diphtheria toxin-derived immunotoxins. Drug Discov. Today 2008, 13, 807–815. [Google Scholar]
- Fuchs, H.; Bachran, C. Targeted tumor therapies at a glance. Curr. Drug Targets 2009, 10, 89–93. [Google Scholar]
- Kreitman, R.J. Recombinant immunotoxins containing truncated bacterial toxins for the treatment of hematologic malignancies. BioDrugs 2009, 23, 1–13. [Google Scholar]
- Fracasso, G.; Stirpe, F.; Colombatti, M. Ribosome-Inactivating Protein-Containing Conjugates for Therapeutic Use. In Toxic Plant Proteins; Lord, J.M., Hartley, M.R., Eds.; Springer: Berlin/Heidelberg, Germany, 2010; Volume 18, pp. 225–263. [Google Scholar]
- Bagel, J.; Garland, W.T.; Breneman, D.; Holick, M.; Littlejohn, T.W.; Crosby, D.; Faust, H.; Fivenson, D.; Nichols, J. Administration of DAB389IL-2 to patients with recalcitrant psoriasis: A double-blind, phase II multicenter trial. J. Am. Acad. Dermatol. 1998, 38, 938–944. [Google Scholar]
- Olsen, E.; Duvic, M.; Frankel, A.; Kim, Y.; Martin, A.; Vonderheid, E.; Jegasothy, B.; Wood, G.; Gordon, M.; Heald, P.; Oseroff, A.; Pinter-Brown, L.; Bowen, G.; Kuzel, T.; Fivenson, D.; Foss, F.; Glode, M.; Molina, A.; Knobler, E.; Stewart, S.; Cooper, K.; Stevens, S.; Craig, F.; Reuben, J.; Bacha, P.; Nichols, J. Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphoma. J. Clin. Oncol. 2001, 19, 376–388. [Google Scholar]
- Frankel, A.E.; Fleming, D.R.; Hall, P.D.; Powell, B.L.; Black, J.H.; Leftwich, C.; Gartenhaus, R. A phase II study of DT fusion protein denileukin diftitox in patients with fludarabine-refractory chronic lymphocytic leukemia. Clin. Canc. Res. 2003, 9, 3555–3561. [Google Scholar]
- Dang, N.H.; Hagemeister, F.B.; Pro, B.; McLaughlin, P.; Romaguera, J.E.; Jones, D.; Samuels, B.; Samaniego, F.; Younes, A.; Wang, M.; Goy, A.; Rodriguez, M.A.; Walker, P.L.; Arredondo, Y.; Tong, A.T.; Fayad, L. Phase II study of denileukin diftitox for relapsed/refractory B-Cell non-Hodgkin's lymphoma. J. Clin. Oncol. 2004, 22, 4095–4102. [Google Scholar]
- Shaughnessy, P.J.; Bachier, C.; Grimley, M.; Freytes, C.O.; Callander, N.S.; Essell, J.H.; Flomenberg, N.; Selby, G.; Lemaistre, C.F. Denileukin diftitox for the treatment of steroid-resistant acute graft-versus-host disease. Biol. Blood Marrow Transplant. 2005, 11, 188–193. [Google Scholar]
- Frankel, A.E.; Surendranathan, A.; Black, J.H.; White, A.; Ganjoo, K.; Cripe, L.D. Phase II clinical studies of denileukin diftitox diphtheria toxin fusion protein in patients with previously treated chronic lymphocytic leukemia. Cancer 2006, 106, 2158–2164. [Google Scholar]
- Dang, N.H.; Pro, B.; Hagemeister, F.B.; Samaniego, F.; Jones, D.; Samuels, B.I.; Rodriguez, M.A.; Goy, A.; Romaguera, J.E.; McLaughlin, P.; Tong, A.T.; Turturro, F.; Walker, P.L.; Fayad, L. Phase II trial of denileukin diftitox for relapsed/refractory T-cell non-Hodgkin lymphoma. Br. J. Haematol. 2007, 136, 439–447. [Google Scholar]
- Gerena-Lewis, M.; Crawford, J.; Bonomi, P.; Maddox, A.M.; Hainsworth, J.; McCune, D.E.; Shukla, R.; Zeigler, H.; Hurtubise, P.; Chowdhury, T.R.; Fletcher, B.; Dyehouse, K.; Ghalie, R.; Jazieh, A.R. A Phase II trial of Denileukin Diftitox in patients with previously treated advanced non-small cell lung cancer. Am. J. Clin. Oncol. 2009, 32, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Prince, H.M.; Duvic, M.; Martin, A.; Sterry, W.; Assaf, C.; Sun, Y.; Straus, D.; Acosta, M.; Negro-Vilar, A. Phase III placebo-controlled trial of denileukin diftitox for patients with cutaneous T-cell lymphoma. J.Clin. Oncol. 2010, 28, 1870–1877. [Google Scholar]
- LeMaistre, C.F.; Meneghetti, C.; Rosenblum, M.; Reuben, J.; Parker, K.; Shaw, J.; Deisseroth, A.; Woodworth, T.; Parkinson, D.R. Phase I trial of an interleukin-2 (IL-2) fusion toxin (DAB486IL-2) in hematologic malignancies expressing the IL-2 receptor. Blood 1992, 79, 2547–2554. [Google Scholar]
- Kuzel, T.M.; Rosen, S.T.; Gordon, L.I.; Winter, J.; Samuelson, E.; Kaul, K.; Roenigk, H.H.; Nylen, P.; Woodworth, T. Phase I trial of the diphtheria toxin/interleukin-2 fusion protein DAB486IL-2: Efficacy in mycosis fungoides and other non-Hodgkin's lymphomas. Leuk. Lymphoma 1993, 11, 369–377. [Google Scholar]
- LeMaistre, C.F.; Craig, F.E.; Meneghetti, C.; McMullin, B.; Parker, K.; Reuben, J.; Boldt, D.H.; Rosenblum, M.; Woodworth, T. Phase I trial of a 90-minute infusion of the fusion toxin DAB486IL-2 in hematological cancers. Canc. Res. 1993, 53, 3930–3934. [Google Scholar]
- Platanias, L.C.; Ratain, M.J.; O'Brien, S.; Larson, R.A.; Vardiman, J.W.; Shaw, J.P.; Williams, S.F.; Baron, J.M.; Parker, K.; Woodworth, T.G. Phase I trial of a genetically engineered interleukin-2 fusion toxin (DAB486IL-2) as a 6 hour intravenous infusion in patients with hematologic malignancies. Leuk. Lymphoma 1994, 14, 257–262. [Google Scholar]
- Tepler, I.; Schwartz, G.; Parker, K.; Charette, J.; Kadin, M.E.; Woodworth, T.G.; Schnipper, L.E. Phase I trial of an interleukin-2 fusion toxin (DAB486IL-2) in hematologic malignancies: Complete response in a patient with Hodgkin's disease refractory to chemotherapy. Cancer 1994, 73, 1276–1285. [Google Scholar]
- Moreland, L.W.; Sewell, K.L.; Trentham, D.E.; Bucy, R.P.; Sullivan, W.F.; Schrohenloher, R.E.; Shmerling, R.H.; Parker, K.C.; Swartz, W.G.; Woodworth, T.G.; et al. Interleukin-2 diphtheria fusion protein (DAB486IL-2) in refractory rheumatoid arthritis. A double-blind, placebo-controlled trial with open-label extension. Arthritis Rheum. 1995, 38, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Laske, D.W.; Youle, R.J.; Oldfield, E.H. Tumor regression with regional distribution of the targeted toxin TF-CRM107 in patients with malignant brain tumors. Nat. Med. 1997, 3, 1362–1368. [Google Scholar]
- Weaver, M.; Laske, D.W. Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) for therapy of malignant gliomas. J. Neurooncol. 2003, 65, 3–13. [Google Scholar]
- Debinski, W. Molecular targeting with recombinant cytotoxins for the treatment of brain tumors. Drug Dev. Res. 2008, 69, 407–414. [Google Scholar]
- Frankel, A.E.; Powell, B.L.; Hall, P.D.; Case, L.D.; Kreitman, R.J. Phase I trial of a novel diphtheria toxin/granulocyte macrophage colony-stimulating factor fusion protein (DT388GMCSF) for refractory or relapsed acute myeloid leukemia. Clin. Canc. Res. 2002, 8, 1004–1013. [Google Scholar]
- Frankel, A.E.; Zuckero, S.L.; Mankin, A.A.; Grable, M.; Mitchell, K.; Lee, Y.J.; Neville, D.M.; Woo, J.H. Anti-CD3 recombinant diphtheria immunotoxin therapy of cutaneous T cell lymphoma. Curr. Drug Targets 2009, 10, 104–109. [Google Scholar]
- Woo, J.H.; Lee, Y.J.; Neville, D.M.; Frankel, A.E. Pharmacology of anti-CD3 diphtheria immunotoxin in CD3 positive T-cell lymphoma trials. Meth. Mol. Biol. 2010, 651, 157–175. [Google Scholar]
- Testa, U.; Riccioni, R.; Biffoni, M.; Diverio, D.; Lo-Coco, F.; Foa, R.; Peschle, C.; Frankel, A.E. Diphtheria toxin fused to variant human interleukin-3 induces cytotoxicity of blasts from patients with acute myeloid leukemia according to the level of interleukin-3 receptor expression. Blood 2005, 106, 2527–2529. [Google Scholar]
- Pai, L.; Bookman, M.; Ozols, R.; Young, R.; Smith, J., 2d; Longo, D.; Gould, B.; Frankel, A.; McClay, E.; Howell, S. Clinical evaluation of intraperitoneal Pseudomonas exotoxin immunoconjugate OVB3-PE in patients with ovarian cancer. J. Clin. Oncol. 1991, 9, 2095–2103. [Google Scholar] [PubMed]
- Pai-Scherf, L.H.; Villa, J.; Pearson, D.; Watson, T.; Liu, E.; Willingham, M.C.; Pastan, I. Hepatotoxicity in Cancer Patients Receiving erb-38, a Recombinant Immunotoxin That Targets the erbB2 Receptor. Clin. Canc. Res. 1999, 5, 2311–2315. [Google Scholar]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin. Canc. Res. 2007, 13, 5144–5149. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Hassan, R.; Fitzgerald, D.J.; Pastan, I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin. Canc. Res. 2009, 15, 5274–5279. [Google Scholar]
- Pai, L.H.; Wittes, R.; Setser, A.; Willingham, M.C.; Pastan, I. Treatment of advanced solid tumors with immunotoxin LMB-1: An antibody linked to Pseudomonas exotoxin. Nat. Med. 1996, 2, 350–353. [Google Scholar]
- Kreitman, R.J.; Wilson, W.H.; Bergeron, K.; Raggio, M.; Stetler-Stevenson, M.; FitzGerald, D.J.; Pastan, I. Efficacy of the Anti-CD22 Recombinant Immunotoxin BL22 in Chemotherapy-Resistant Hairy-Cell Leukemia. New Engl. J. Med. 2001, 345, 241–247. [Google Scholar]
- Kreitman, R.J.; Squires, D.R.; Stetler-Stevenson, M.; Noel, P.; FitzGerald, D.J.P.; Wilson, W.H.; Pastan, I. Phase I Trial of Recombinant Immunotoxin RFB4(dsFv)-PE38 (BL22) in Patients With B-Cell Malignancies. J. Clin. Oncol. 2005, 23, 6719–6729. [Google Scholar]
- Kreitman, R.J.; Stetler-Stevenson, M.; Margulies, I.; Noel, P.; FitzGerald, D.J.P.; Wilson, W.H.; Pastan, I. Phase II Trial of Recombinant Immunotoxin RFB4(dsFv)-PE38 (BL22) in Patients With Hairy Cell Leukemia. J. Clin. Oncol. 2009, 27, 2983–2990. [Google Scholar]
- Wayne, A.S.; Kreitman, R.J.; Findley, H.W.; Lew, G.; Delbrook, C.; Steinberg, S.M.; Stetler-Stevenson, M.; FitzGerald, D.J.; Pastan, I. Anti-CD22 Immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-Positive Hematologic Malignancies of Childhood: Preclinical Studies and Phase I Clinical Trial. Clin. Canc. Res. 2010, 16, 1894–1903. [Google Scholar]
- Kreitman, R.J.; Wilson, W.H.; White, J.D.; Stetler-Stevenson, M.; Jaffe, E.S.; Giardina, S.; Waldmann, T.A.; Pastan, I. Phase I Trial of Recombinant Immunotoxin Anti-Tac(Fv)-PE38 (LMB-2) in Patients With Hematologic Malignancies. J. Clin. Oncol. 2000, 18, 1622–1636. [Google Scholar]
- Azemar, M.; Djahansouzi, S.; Jäger, E.; Solbach, C.; Schmidt, M.; Maurer, A.B.; Mross, K.; Unger, C.; Minckwitz, G.v.; Dall, P.; Groner, B.; Wels, W.S. Regression of Cutaneous Tumor Lesions in Patients Intratumorally Injected with a Recombinant Single-chain Antibody-toxin Targeted to ErbB2/HER2. Breast Canc. Res. Treat. 2003, 82, 155–164. [Google Scholar]
- von Minckwitz, G.; Harder, S.; Hovelmann, S.; Jager, E.; Al-Batran, S.E.; Loibl, S.; Atmaca, A.; Cimpoiasu, C.; Neumann, A.; Abera, A.; Knuth, A.; Kaufmann, M.; Jager, D.; Maurer, A.B.; Wels, W.S. Phase I clinical study of the recombinant antibody toxin scFv(FRP5)-ETA specific for the ErbB2/HER2 receptor in patients with advanced solid malignomas. Breast Canc. Res. 2005, 7, R617–R626. [Google Scholar]
- Goldberg, M.R.; Heimbrook, D.C.; Russo, P.; Sarosdy, M.F.; Greenberg, R.E.; Giantonio, B.J.; Linehan, W.M.; Walther, M.; Fisher, H.A.; Messing, E.; et al. Phase I clinical study of the recombinant oncotoxin TP40 in superficial bladder cancer. Clin. Canc. Res. 1995, 1, 57–61. [Google Scholar]
- Sampson, J.H.; Akabani, G.; Archer, G.E.; Bigner, D.D.; Berger, M.S.; Friedman, A.H.; Friedman, H.S.; Herndon, J.E., 2nd; Kunwar, S.; Marcus, S.; McLendon, R.E.; Paolino, A.; Penne, K.; Provenzale, J.; Quinn, J.; Reardon, D.A.; Rich, J.; Stenzel, T.; Tourt-Uhlig, S.; Wikstrand, C.; Wong, T.; Williams, R.; Yuan, F.; Zalutsky, M.R.; Pastan, I. Progress report of a Phase I study of the intracerebral microinfusion of a recombinant chimeric protein composed of transforming growth factor (TGF)-alpha and a mutated form of the Pseudomonas exotoxin termed PE-38 (TP-38) for the treatment of malignant brain tumors. J. Neurooncol. 2003, 65, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.H.; Reardon, D.A.; Friedman, A.H.; Friedman, H.S.; Coleman, R.E.; McLendon, R.E.; Pastan, I.; Bigner, D.D. Sustained radiographic and clinical response in patient with bifrontalrecurrent glioblastoma multiforme with intracerebral infusion of therecombinant targeted toxin TP-38: Case study. Neuro. Oncol. 2005, 7, 90–96. [Google Scholar]
- Sampson, J.H.; Akabani, G.; Archer, G.E.; Berger, M.S.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; Greer, K.; Herndon, J.E., 2nd; Kunwar, S.; McLendon, R.E.; Paolino, A.; Petry, N.A.; Provenzale, J.M.; Reardon, D.A.; Wong, T.Z.; Zalutsky, M.R.; Pastan, I.; Bigner, D.D. Intracerebral infusion of an EGFR-targeted toxin in recurrent malignant brain tumors. Neuro. Oncol. 2008, 10, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Posey, J.A.; Khazaeli, M.B.; Bookman, M.A.; Nowrouzi, A.; Grizzle, W.E.; Thornton, J.; Carey, D.E.; Lorenz, J.M.; Sing, A.P.; Siegall, C.B.; LoBuglio, A.F.; Saleh, M.N. A Phase I Trial of the Single-Chain Immunotoxin SGN-10 (BR96 sFv-PE40) in Patients with Advanced Solid Tumors. Clin. Canc. Res. 2002, 8, 3092–3099. [Google Scholar]
- Weber, F.; Asher, A.; Bucholz, R.; Berger, M.; Prados, M.; Chang, S.; Bruce, J.; Hall, W.; Rainov, N.G.; Westphal, M.; Warnick, R.E.; Rand, R.W.; Floeth, F.; Rommel, F.; Pan, H.; Hingorani, V.N.; Puri, R.K. Safety, tolerability, and tumor response of IL4-Pseudomonas exotoxin (NBI-3001) in patients with recurrent malignant glioma. J. Neurooncol. 2003, 64, 125–137. [Google Scholar]
- Weber, F.W.; Floeth, F.; Asher, A.; Bucholz, R.; Berger, M.; Prados, M.; Chang, S.; Bruce, J.; Hall, W.; Rainov, N.G.; Westphal, M.; Warnick, R.E.; Rand, R.W.; Rommell, F.; Pan, H.; Hingorani, V.N.; Puri, R.K. Local convection enhanced delivery of IL4-Pseudomonas exotoxin (NBI-3001) for treatment of patients with recurrent malignant glioma. Acta Neurochir. Suppl. 2003, 88, 93–103. [Google Scholar]
- Garland, L.; Gitlitz, B.; Ebbinghaus, S.; Pan, H.; de Haan, H.; Puri, R.K.; Von Hoff, D.; Figlin, R. Phase I trial of intravenous IL-4 pseudomonas exotoxin protein (NBI-3001) in patients with advanced solid tumors that express the IL-4 receptor. J. Immunother. 2005, 28, 376–381. [Google Scholar]
- Kreitman, R.J.; Tallman, M.S.; Coutre, S.E.; Robak, T.; Wilson, W.H.; Stetler-Stevenson, M.; Noel, P.; FitzGerald, D.J.; McDevitt, J.T.; Pastan, I. Phase I trial of recombinant immunotoxin CAT-8015 (HA22) in multiply relapsed hairy cell leukemia. J. Clin. Oncol. 2010, 28, 6523. [Google Scholar]
- Mussai, F.; Campana, D.; Bhojwani, D.; Stetler-Stevenson, M.; Steinberg, S.M.; Wayne, A.S.; Pastan, I. Cytotoxicity of the anti-CD22 immunotoxin HA22 (CAT-8015) against paediatric acute lymphoblastic leukaemia. Br. J. Haematol. 2010, 150, 352–358. [Google Scholar]
- Kunwar, S. Convection enhanced delivery of IL13-PE38QQR for treatment of recurrent malignant glioma: Presentation of interim findings from ongoing phase 1 studies. Acta Neurochir. Suppl. 2003, 88, 105–111. [Google Scholar]
- Parney, I.F.; Kunwar, S.; McDermott, M.; Berger, M.; Prados, M.; Cha, S.; Croteau, D.; Puri, R.K.; Chang, S.M. Neuroradiographic changes following convection-enhanced delivery of the recombinant cytotoxin interleukin 13-PE38QQR for recurrent malignant glioma. J. Neurosurg. 2005, 102, 267–275. [Google Scholar]
- Kunwar, S.; Prados, M.D.; Chang, S.M.; Berger, M.S.; Lang, F.F.; Piepmeier, J.M.; Sampson, J.H.; Ram, Z.; Gutin, P.H.; Gibbons, R.D.; Aldape, K.D.; Croteau, D.J.; Sherman, J.W.; Puri, R.K. Direct Intracerebral Delivery of Cintredekin Besudotox (IL13-PE38QQR) in Recurrent Malignant Glioma: A Report by the Cintredekin Besudotox Intraparenchymal Study Group. J. Clin. Oncol. 2007, 25, 837–844. [Google Scholar]
- Kunwar, S.; Chang, S.; Westphal, M.; Vogelbaum, M.; Sampson, J.; Barnett, G.; Shaffrey, M.; Ram, Z.; Piepmeier, J.; Prados, M.; Croteau, D.; Pedain, C.; Leland, P.; Husain, S.R.; Joshi, B.H.; Puri, R.K. The PRECISE Study Group. Phase III randomized trial of CED of IL13-PE38QQR vs Gliadel wafers for recurrent glioblastoma. Neuro. Oncol. 2010, 12, 871–881. [Google Scholar]
- Vitetta, E.S.; Stone, M.; Amlot, P.; Fay, J.; May, R.; Till, M.; Newman, J.; Clark, P.; Collins, R.; Cunningham, D.; Ghetie, V.; Uhr, J.W.; Thorpe, P.E. Phase I Immunotoxin Trial in Patients with B-Cell Lymphoma. Canc. Res. 1991, 51, 4052–4058. [Google Scholar]
- Amlot, P.; Stone, M.; Cunningham, D.; Fay, J.; Newman, J.; Collins, R.; May, R.; McCarthy, M.; Richardson, J.; Ghetie, V. A phase I study of an anti-CD22-deglycosylated ricin A chain immunotoxin in the treatment of B-cell lymphomas resistant to conventional therapy. Blood 1993, 82, 2624–2633. [Google Scholar]
- Sausville, E.A.; Headlee, D.; Stetler-Stevenson, M.; Jaffe, E.S.; Solomon, D.; Figg, W.D.; Herdt, J.; Kopp, W.C.; Rager, H.; Steinberg, S.M.; et al. Continuous infusion of the anti-CD22 immunotoxin IgG-RFB4-SMPT-dgA in patients with B-cell lymphoma: A phase I study. Blood 1995, 85, 3457–3465. [Google Scholar]
- Conry, R.M.; Khazaeli, M.B.; Saleh, M.N.; Ghetie, V.; Vitetta, E.S.; Liu, T.; LoBuglio, A.F. Phase I trial of an anti-CD19 deglycosylated ricin A chain immunotoxin in non-Hodgkin's lymphoma: Effect of an intensive schedule of administration. J. Immunother. Emphasis Tumor Immunol. 1995, 18, 231–241. [Google Scholar]
- Stone, M.J.; Sausville, E.A.; Fay, J.W.; Headlee, D.; Collins, R.H.; Figg, W.D.; Stetler-Stevenson, M.; Jain, V.; Jaffe, E.S.; Solomon, D.; Lush, R.M.; Senderowicz, A.; Ghetie, V.; Schindler, J.; Uhr, J.W.; Vitetta, E.S. A phase I study of bolus versus continuous infusion of the anti-CD19 immunotoxin, IgG-HD37-dgA, in patients with B-cell lymphoma. Blood 1996, 88, 1188–1197. [Google Scholar]
- Messmann, R.A.; Vitetta, E.S.; Headlee, D.; Senderowicz, A.M.; Figg, W.D.; Schindler, J.; Michiel, D.F.; Creekmore, S.; Steinberg, S.M.; Kohler, D.; Jaffe, E.S.; Stetler-Stevenson, M.; Chen, H.; Ghetie, V.; Sausville, E.A. A Phase I Study of Combination Therapy with Immunotoxins IgG-HD37-Deglycosylated Ricin A Chain (dgA) and IgG-RFB4-dgA (Combotox) in Patients with Refractory CD19(+), CD22(+) B Cell Lymphoma. Clin. Canc. Res. 2000, 6, 1302–1313. [Google Scholar]
- Herrera, L.; Bostrom, B.; Gore, L.; Sandler, E.; Lew, G.; Schlegel, P.G.; Aquino, V.; Ghetie, V.; Vitetta, E.S.; Schindler, J. A phase 1 study of Combotox in pediatric patients with refractory B-lineage acute lymphoblastic leukemia. J. Pediatr. Hematol. Oncol. 2009, 31, 936–941. [Google Scholar]
- Engert, A.; Diehl, V.; Schnell, R.; Radszuhn, A.; Hatwig, M.T.; Drillich, S.; Schon, G.; Bohlen, H.; Tesch, H.; Hansmann, M.L.; Barth, S.; Schindler, J.; Ghetie, V.; Uhr, J.; Vitetta, E. A phase-I study of an anti-CD25 ricin A-chain immunotoxin (RFT5-SMPT-dgA) in patients with refractory Hodgkin's lymphoma. Blood 1997, 89, 403–410. [Google Scholar]
- Schnell, R.; Vitetta, E.; Schindler, J.; Barth, S.; Winkler, U.; Borchmann, P.; Hansmann, M.L.; Diehl, V.; Ghetie, V.; Engert, A. Clinical trials with an anti-CD25 ricin A-chain experimental and immunotoxin (RFT5-SMPT-dgA) in Hodgkin's lymphoma. Leuk. Lymphoma 1998, 30, 525–537. [Google Scholar]
- Schnell, R.; Vitetta, E.; Schindler, J.; Borchmann, P.; Barth, S.; Ghetie, V.; Hell, K.; Drillich, S.; Diehl, V.; Engert, A. Treatment of refractory Hodgkin's lymphoma patients with an anti-CD25 ricin A-chain immunotoxin. Leukemia 2000, 14, 129–135. [Google Scholar]
- Schnell, R.; Borchmann, P.; Staak, J.O.; Schindler, J.; Ghetie, V.; Vitetta, E.S.; Engert, A. Clinical evaluation of ricin A-chain immunotoxins in patients with Hodgkin's lymphoma. Ann. Oncol. 2003, 14, 729–736. [Google Scholar]
- Martin, P.J.; Pei, J.; Gooley, T.; Anasetti, C.; Appelbaum, F.R.; Deeg, J.; Hansen, J.A.; Nash, R.A.; Petersdorf, E.W.; Storb, R.; Ghetie, V.; Schindler, J.; Vitetta, E.S. Evaluation of a CD25-specific immunotoxin for prevention of graft-versus-host disease after unrelated marrow transplantation. Biol. Blood Marrow Transplant. 2004, 10, 552–560. [Google Scholar]
- Schnell, R.; Staak, O.; Borchmann, P.; Schwartz, C.; Matthey, B.r.; Hansen, H.; Schindler, J.; Ghetie, V.; Vitetta, E.S.; Diehl, V.; Engert, A. A Phase I Study with an Anti-CD30 Ricin A-Chain Immunotoxin (Ki-4.dgA) in Patients with Refractory CD30+ Hodgkin's and Non-Hodgkin's Lymphoma. Clin. Canc. Res. 2002, 8, 1779–1786. [Google Scholar]
- Grossbard, M.L.; Gribben, J.G.; Freedman, A.S.; Lambert, J.M.; Kinsella, J.; Rabinowe, S.N.; Eliseo, L.; Taylor, J.A.; Blattler, W.A.; Epstein, C.L.; et al. Adjuvant immunotoxin therapy with anti-B4-blocked ricin after autologous bone marrow transplantation for patients with B-cell non-Hodgkin's lymphoma. Blood 1993, 81, 2263–2271. [Google Scholar]
- Grossbard, M.L.; Lambert, J.M.; Goldmacher, V.S.; Spector, N.L.; Kinsella, J.; Eliseo, L.; Coral, F.; Taylor, J.A.; Blattler, W.A.; Epstein, C.L.; et al. Anti-B4-blocked ricin: A phase I trial of 7-day continuous infusion in patients with B-cell neoplasms. J. Clin. Oncol. 1993, 11, 726–737. [Google Scholar]
- Multani, P.S.; O'Day, S.; Nadler, L.M.; Grossbard, M.L. Phase II clinical trial of bolus infusion anti-B4 blocked ricin immunoconjugate in patients with relapsed B-cell non-Hodgkin's lymphoma. Clin. Canc. Res. 1998, 4, 2599–2604. [Google Scholar]
- Grossbard, M.L.; Multani, P.S.; Freedman, A.S.; O'Day, S.; Gribben, J.G.; Rhuda, C.; Neuberg, D.; Nadler, L.M. A Phase II Study of Adjuvant Therapy with Anti-B4-blocked Ricin after Autologous Bone Marrow Transplantation for Patients with Relapsed B-Cell Non-Hodgkin's Lymphoma. Clin. Canc. Res. 1999, 5, 2392–2398. [Google Scholar]
- Zalcberg, J.R.; Pietersz, G.; Toohey, B.; Laird, J.; Huggins, R.; Zimet, A.S.; Hennessy, O.; McKenzie, A.; McKenzie, I.F.C. A phase study of the intralesional injection of ricin-monoclonal antibody conjugates in patients with hepatic metastases. Eur. J. Canc. 1994, 30, 1227–1231. [Google Scholar]
- Lynch, T.J., Jr. Immunotoxin therapy of small-cell lung cancer. N901-blocked ricin for relapsed small-cell lung cancer. Chest 1993, 103, 436S–439S. [Google Scholar] [CrossRef] [PubMed]
- Epstein, C.; Lynch, T.; Shefner, J.; Wen, P.; Maxted, D.; Braman, V.; Ariniello, P.; Coral, F.; Ritz, J. Use of the immunotoxin N901-blocked ricin in patients with small-cell lung cancer. Int. J. Canc. Suppl. 1994, 8, 57–59. [Google Scholar]
- Lynch, T., Jr; Lambert, J.; Coral, F.; Shefner, J.; Wen, P.; Blattler, W.; Collinson, A.; Ariniello, P.; Braman, G.; Cook, S.; Esseltine, D.; Elias, A.; Skarin, A.; Ritz, J. Immunotoxin therapy of small-cell lung cancer: A phase I study of N901- blocked ricin. J. Clin. Oncol. 1997, 15, 723–734. [Google Scholar] [PubMed]
- Frankel, A.E.; Laver, J.H.; Willingham, M.C.; Burns, L.J.; Kersey, J.H.; Vallera, D.A. Therapy of Patients with T-cell Lymphomas and Leukemias Using an Anti-CD7 Monoclonal Antibody-Rich a Chain Immunotoxin. Leuk. Lymphoma 1997, 26, 287–298. [Google Scholar]
- van Oosterhout, Y.V.J.M.; van Emst, L.; Schattenberg, A.V.M.B.; Tax, W.J.M.; Ruiter, D.J.; Spits, H.; Nagengast, F.M.; Masereeuw, R.; Evers, S.; de Witte, T.; Preijers, F.W.M.B. A combination of anti-CD3 and anti-CD7 ricin A-immunotoxins for the in vivo treatment of acute graft versus host disease. Blood 2000, 95, 3693–3701. [Google Scholar]
- Skyler, J.S.; Lorenz, T.J.; Schwartz, S.; Eisenbarth, G.S.; Einhorn, D.; Palmer, J.P.; Marks, J.B.; Greenbaum, C.; Saria, E.A.; Byers, V. Effects of an anti-CD5 immunoconjugate (CD5-plus) in recent onset type I diabetes mellitus: A preliminary investigation.The CD5 Diabetes Project Team. J. Diabetes Complicat. 1993, 7, 224–232. [Google Scholar]
- Strand, V.; Lipsky, P.E.; Cannon, G.W.; Calabrese, L.H.; Wiesenhutter, C.; Cohen, S.B.; Olsen, N.J.; Lee, M.L.; Lorenz, T.J.; Nelson, B. Effects of administration of an anti-CD5 plus immunoconjugate in rheumatoid arthritis.Results of two phase II studies. The CD5 Plus Rheumatoid Arthritis Investigators Group. Arthritis Rheum. 1993, 36, 620–630. [Google Scholar]
- Fishwild, D.M.; Strand, V. Administration of an anti-CD5 immunoconjugate to patients with rheumatoid arthritis: Effect on peripheral blood mononuclear cells and in vitro immune function. J. Rheumatol. 1994, 21, 596–604. [Google Scholar]
- Stafford, F.J.; Fleisher, T.A.; Lee, G.; Brown, M.; Strand, V.; Austin, H.A., 3rd; Balow, J.E.; Klippel, J.H. A pilot study of anti-CD5 ricin A chain immunoconjugate in systemic lupus erythematosus. J. Rheumatol. 1994, 21, 2068–2070. [Google Scholar] [PubMed]
- Olsen, N.J.; Brooks, R.H.; Cush, J.J.; Lipsky, P.E.; St Clair, E.W.; Matteson, E.L.; Gold, K.N.; Cannon, G.W.; Jackson, C.G.; McCune, W.J.; Fox, D.A.; Nelson, B.; Lorenz, T.; Strand, V. A double-blind, placebo-controlled study of anti-CD5 immunoconjugate in patients with rheumatoid arthritis. The Xoma RA Investigator Group. Arthritis Rheum. 1996, 39, 1102–1108. [Google Scholar]
- Kernan, N.A.; Byers, V.; Scannon, P.J.; Mischak, R.P.; Brochstein, J.; Flomenberg, N.; Dupont, B.; O'Reilly, R.J. Treatment of steroid-resistant acute graft-vs-host disease by in vivo administration of an anti-T-cell ricin A chain immunotoxin. JAMA 1988, 259, 3154–3157. [Google Scholar]
- Byers, V.; Henslee, P.; Kernan, N.; Blazar, B.; Gingrich, R.; Phillips, G.; LeMaistre, C.; Gilliland, G.; Antin, J.; Martin, P. Use of an anti-pan T-lymphocyte ricin a chain immunotoxin in steroid- resistant acute graft-versus-host disease. Blood 1990, 75, 1426–1432. [Google Scholar]
- LeMaistre, C.; Rosen, S.; Frankel, A.; Kornfeld, S.; Saria, E.; Meneghetti, C.; Drajesk, J.; Fishwild, D.; Scannon, P.; Byers, V. Phase I trial of H65-RTA immunoconjugate in patients with cutaneous T- cell lymphoma. Blood 1991, 78, 1173–1182. [Google Scholar]
- Laurent, G.; Pris, J.; Farcet, J.; Carayon, P.; Blythman, H.; Casellas, P.; Poncelet, P.; Jansen, F. Effects of therapy with T101 ricin A-chain immunotoxin in two leukemia patients. Blood 1986, 67, 1680–1687. [Google Scholar]
- Hertler, A.A.; Schlossman, D.M.; Borowitz, M.J.; Laurent, G.; Jansen, F.K.; Schmidt, C.; Frankel, A.E. A phase I study of T101-ricin A chain immunotoxin in refractory chronic lymphocytic leukemia. J. Biol. Respon. Mod. 1988, 7, 97–113. [Google Scholar]
- Hertler, A.A.; Schlossman, D.M.; Borowitz, M.J.; Blythman, H.E.; Casellas, P.; Frankel, A.E. An anti-CD5 immunotoxin for chronic lymphocytic leukemia: Enhancement of cytotoxicity with human serum albumin-monensin. Int. J. Canc. 1989, 43, 215–219. [Google Scholar]
- Castillo, E. MDX-RA Medarex Inc. IDrugs 1998, 1, 476–479. [Google Scholar]
- Clark, D.S.; Emery, J.M.; Munsell, M.F. Inhibition of posterior capsule opacification with an immunotoxin specific for lens epithelial cells: 24 month clinical results. J. Cataract Refract. Surg. 1998, 24, 1614–1620. [Google Scholar]
- Meacock, W.R.; Spalton, D.J.; Hollick, E.J.; Boyce, J.F.; Barman, S.; Sanguinetti, G. Double-masked prospective ocular safety study of a lens epithelial cell antibody to prevent posterior capsule opacification. J. Cataract Refract. Surg. 2000, 26, 716–721. [Google Scholar]
- Hertler, A.A.; Spitler, L.E.; Frankel, A.E. Humoral immune response to a ricin A chain immunotoxin in patients with metastatic melanoma. Canc. Drug Deliv. 1987, 4, 245–253. [Google Scholar]
- Spitler, L.E.; del Rio, M.; Khentigan, A.; Wedel, N.I.; Brophy, N.A.; Miller, L.L.; Harkonen, W.S.; Rosendorf, L.L.; Lee, H.M.; Mischak, R.P.; et al. Therapy of patients with malignant melanoma using a monoclonal antimelanoma antibody-ricin A chain immunotoxin. Canc. Res. 1987, 47, 1717–1723. [Google Scholar]
- Mischak, R.P.; Foxall, C.; Rosendorf, L.L.; Knebel, K.; Scannon, P.J.; Spitler, L.E. Human antibody responses to components of the monoclonal antimelanoma antibody ricin A chain immunotoxin XomaZyme-MEL. Mol. Biother. 1990, 2, 104–109. [Google Scholar]
- Oratz, R.; Speyer, J.L.; Wernz, J.C.; Hochster, H.; Meyers, M.; Mischak, R.; Spitler, L.E. Antimelanoma monoclonal antibody-ricin A chain immunoconjugate (XMMME-001-RTA) plus cyclophosphamide in the treatment of metastatic malignant melanoma: Results of a phase II trial. J. Biol. Response Mod. 1990, 9, 345–354. [Google Scholar]
- Gonzalez, R.; Salem, P.; Bunn, P.A., Jr.; Zukiwski, A.A.; Lamb, R.; Benjamin, R.S.; Spitler, L.; Wedel, N.; Robinson, W.A. Single-dose murine monoclonal antibody ricin A chain immunotoxin in the treatment of metastatic melanoma: A phase I trial. Mol. Biother. 1991, 3, 192–196. [Google Scholar]
- Selvaggi, K.; Saria, E.A.; Schwartz, R.; Vlock, D.R.; Ackerman, S.; Wedel, N.; Kirkwood, J.M.; Jones, H.; Ernstoff, M.S. Phase I/II study of murine monoclonal antibody-ricin A chain (XOMAZYME-Mel) immunoconjugate plus cyclosporine A in patients with metastatic melanoma. J. Immunother. Emphasis Tumor Immunol. 1993, 13, 201–207. [Google Scholar]
- Byers, V.S.; Rodvien, R.; Grant, K.; Durrant, L.G.; Hudson, K.H.; Baldwin, R.W.; Scannon, P.J. Phase I study of monoclonal antibody-ricin A chain immunotoxin XomaZyme-791 in patients with metastatic colon cancer. Canc. Res. 1989, 49, 6153–6160. [Google Scholar]
- Durrant, L.G.; Byers, V.S.; Scannon, P.J.; Rodvien, R.; Grant, K.; Robins, R.A.; Marksman, R.A.; Baldwin, R.W. Humoral immune responses to XMMCO-791-RTA immunotoxin in colorectal cancer patients. Clin. Exp. Immunol. 1989, 75, 258–264. [Google Scholar]
- LoRusso, P.M.; Lomen, P.L.; Redman, B.G.; Poplin, E.; Bander, J.J.; Valdivieso, M. Phase I study of monoclonal antibody-ricin A chain immunoconjugate Xomazyme-791 in patients with metastatic colon cancer. Am. J. Clin. Oncol. 1995, 18, 307–312. [Google Scholar]
- Laske, D.W.; Muraszko, K.M.; Oldfield, E.H.; DeVroom, H.L.; Sung, C.; Dedrick, R.L.; Simon, T.R.; Colandrea, J.; Copeland, C.; Katz, D.; Greenfield, L.; Groves, E.S.; Houston, L.L.; Youle, R.J. Intraventricular immunotoxin therapy for leptomeningeal neoplasia. Neurosurgery 1997, 41, 1039–1049, 1049–1051. [Google Scholar]
- Gould, B.J.; Borowitz, M.J.; Groves, E.S.; Carter, P.W.; Anthony, D.; Weiner, L.M.; Frankel, A.E. Phase I study of an anti-breast cancer immunotoxin by continuous infusion: Report of a targeted toxic effect not predicted by animal studies. J. Natl. Canc. Inst. 1989, 81, 775–781. [Google Scholar]
- Weiner, L.M.; O'Dwyer, J.; Kitson, J.; Comis, R.L.; Frankel, A.E.; Bauer, R.J.; Konrad, M.S.; Groves, E.S. Phase I evaluation of an anti-breast carcinoma monoclonal antibody 260F9-recombinant ricin A chain immunoconjugate. Canc. Res. 1989, 49, 4062–4067. [Google Scholar]
- Uckun, F.M. Immunotoxins for the treatment of leukaemia. Br. J. Haematol. 1993, 85, 435–438. [Google Scholar]
- Uckun, F.M.; Bellomy, K.; O'Neill, K.; Messinger, Y.; Johnson, T.; Chen, C.-L. Toxicity, Biological Activity, and Pharmacokinetics of TXU (Anti-CD7)-Pokeweed Antiviral Protein in Chimpanzees and Adult Patients Infected with Human Immunodeficiency Virus. J. Pharmacol. Exp. Therapeut. 1999, 291, 1301–1307. [Google Scholar]
- Falini, B.; Flenghi, L.; Aversa, F.; Barbabietola, G.; Martelli, M.F.; Comeli, P.; Tazzari, P.L.; Broe, M.K.; Stein, H.; Dürkop, H.; Pizzolo, G.; Bolognesi, A.; Stirpe, F.; Sabattini, E.; Pileri, S. Response of refractory Hodgkin's disease to monoclonal anti-CD30 immunotoxin. The Lancet 1992, 339, 1195–1196. [Google Scholar]
- Dean, A.; Talpaz, M.; Kantarjian, H.; Faderl, S.; Jabbour, E.; Ravandi Kashani, F.; O'Brien, S.M.; Rosenblum, M.; Cortes, J.E. Phase I clinical trial of the anti-CD33 immunotoxin HuM195/rgel in patients (pts) with advanced myeloid malignancies. J. Clin. Oncol. 2010, 28, 6549. [Google Scholar]
- Yu, L.; Gu, F.; Zhang, C.; Xie, S.; Guo, Y. Targeted diagnosis and treatment of superficial bladder cancer with monoclonal antibody BDI-1. Chin. Med. J. (Engl.) 1998, 111, 404–407. [Google Scholar] [PubMed]
- Menestrina, G.; Schiavo, G.; Montecucco, C. Molecular mechanisms of action of bacterial protein toxins. Mol. Aspect. Med. 1994, 15, 79–193. [Google Scholar]
- Falnes, P.O.; Sandvig, K. Penetration of protein toxins into cells. Curr. Opin. Cell Biol. 2000, 12, 407–413. [Google Scholar]
- Choe, S.; Bennett, M.J.; Fujii, G.; Curmi, P.M.; Kantardjieff, K.A.; Collier, R.J.; Eisenberg, D. The crystal structure of diphtheria toxin. Nature 1992, 357, 216–222. [Google Scholar]
- Tsuneoka, M.; Nakayama, K.; Hatsuzawa, K.; Komada, M.; Kitamura, N.; Mekada, E. Evidence for involvement of furin in cleavage and activation of diphtheria toxin. J. Biol. Chem. 1993, 268, 26461–26465. [Google Scholar]
- Chiron, M.F.; Fryling, C.M.; FitzGerald, D.J. Cleavage of pseudomonas exotoxin and diphtheria toxin by a furin-like enzyme prepared from beef liver. J. Biol. Chem. 1994, 269, 18167–18176. [Google Scholar]
- Donovan, J.J.; Simon, M.I.; Draper, R.K.; Montal, M. Diphtheria toxin forms transmembrane channels in planar lipid bilayers. Proc. Natl. Acad. Sci. USA 1981, 78, 172–176. [Google Scholar]
- Kagan, B.L.; Finkelstein, A.; Colombini, M. Diphtheria toxin fragment forms large pores in phospholipid bilayer membranes. Proc. Natl. Acad. Sci. USA 1981, 78, 4950–4954. [Google Scholar]
- Lemichez, E.; Bomsel, M.; Devilliers, G.; vanderSpek, J.; Murphy, J.R.; Lukianov, E.V.; Olsnes, S.; Boquet, P. Membrane translocation of diphtheria toxin fragment A exploits early to late endosome trafficking machinery. Mol. Microbiol. 1997, 23, 445–457. [Google Scholar]
- Ratts, R.; Zeng, H.; Berg, E.A.; Blue, C.; McComb, M.E.; Costello, C.E.; vanderSpek, J.C.; Murphy, J.R. The cytosolic entry of diphtheria toxin catalytic domain requires a host cell cytosolic translocation factor complex. J. Cell Biol. 2003, 160, 1139–1150. [Google Scholar]
- Trujillo, C.; Ratts, R.; Tamayo, A.; Harrison, R.; Murphy, J.R. Trojan horse or proton force: Finding the right partner(s) for toxin translocation. Neurotox. Res. 2006, 9, 63–71. [Google Scholar]
- Trujillo, C.; Taylor-Parker, J.; Harrison, R.; Murphy, J.R. Essential lysine residues within transmembrane helix 1 of diphtheria toxin facilitate COPI binding and catalytic domain entry. Mol. Microbiol. 2010, 76, 1010–1019. [Google Scholar]
- Morimoto, H.; Bonavida, B. Diphtheria toxin- and Pseudomonas A toxin-mediated apoptosis. ADP ribosylation of elongation factor-2 is required for DNA fragmentation and cell lysis and synergy with tumor necrosis factor-alpha. J. Immunol. 1992, 149, 2089–2094. [Google Scholar] [PubMed]
- Thorburn, J.; Frankel, A.E.; Thorburn, A. Apoptosis by leukemia cell-targeted diphtheria toxin occurs via receptor-independent activation of Fas-associated death domain protein. Clin. Canc. Res. 2003, 9, 861–865. [Google Scholar]
- Thorburn, A.; Thorburn, J.; Frankel, A.E. Induction of apoptosis by tumor cell-targeted toxins. Apoptosis 2004, 9, 19–25. [Google Scholar]
- Deng, Q.; Barbieri, J.T. Molecular mechanisms of the cytotoxicity of ADP-ribosylating toxins. Annu. Rev. Microbiol. 2008, 62, 271–288. [Google Scholar]
- Yamaizumi, M.; Mekada, E.; Uchida, T.; Okada, Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell 1978, 15, 245–250. [Google Scholar]
- Strauchen, J.A.; Breakstone, B.A. IL-2 receptor expression in human lymphoid lesions. Immunohistochemical study of 166 cases. Am. J. Pathol. 1987, 126, 506–512. [Google Scholar] [PubMed]
- Strauchen, J.A. Interleukin receptors in lymphoid lesions. Relevance to diagnosis, biology, and therapy. Pathol. Annu. 1989, 24, 149–165. [Google Scholar] [PubMed]
- Kodaka, T.; Uchiyama, T.; Ishikawa, T.; Kamio, M.; Onishi, R.; Itoh, K.; Hori, T.; Uchino, H.; Tsudo, M.; Araki, K. Interleukin-2 receptor beta-chain (p70-75) expressed on leukemic cells from adult T cell leukemia patients. Jpn. J. Canc. Res. 1990, 81, 902–908. [Google Scholar]
- Yagura, H.; Tamaki, T.; Furitsu, T.; Tomiyama, Y.; Nishiura, T.; Tominaga, N.; Katagiri, S.; Yonezawa, T.; Tarui, S. Demonstration of high-affinity interleukin-2 receptors on B-chronic lymphocytic leukemia cells: Functional and structural characterization. Blut 1990, 60, 181–186. [Google Scholar]
- Re, G.G.; Waters, C.; Poisson, L.; Willingham, M.C.; Sugamura, K.; Frankel, A.E. Interleukin 2 (IL-2) receptor expression and sensitivity to diphteria fusion toxin DAB389IL-2 in cultured hematopoietic cells. Canc. Res. 1996, 56, 2590–2595. [Google Scholar]
- Williams, D.P.; Snider, C.E.; Strom, T.B.; Murphy, J.R. Structure/function analysis of interleukin-2-toxin (DAB486-IL-2). Fragment B sequences required for the delivery of fragment A to the cytosol of target cells. J. Biol. Chem. 1990, 265, 11885–11889. [Google Scholar] [PubMed]
- LeMaistre, C.F.; Saleh, M.N.; Kuzel, T.M.; Foss, F.; Platanias, L.C.; Schwartz, G.; Ratain, M.; Rook, A.; Freytes, C.O.; Craig, F.; Reuben, J.; Nichols, J.C. Phase I trial of a ligand fusion-protein (DAB389IL-2) in lymphomas expressing the receptor for interleukin-2. Blood 1998, 91, 399–405. [Google Scholar]
- McGinnis, K.S.; Shapiro, M.; Junkins-Hopkins, J.M.; Smith, M.; Lessin, S.R.; Vittorio, C.C.; Rook, A.H. Denileukin diftitox for the treatment of panniculitic lymphoma. Arch. Dermatol. 2002, 138, 740–742. [Google Scholar]
- Martin, A.; Gutierrez, E.; Muglia, J.; McDonald, C.J.; Guzzo, C.; Gottlieb, A.; Pappert, A.; Garland, W.T.; Bagel, J.; Bacha, P. A multicenter dose-escalation trial with denileukin diftitox (ONTAK, DAB(389)IL-2) in patients with severe psoriasis. J. Am. Acad. Dermatol. 2001, 45, 871–881. [Google Scholar]
- Kelleher, C.A.; Wong, G.G.; Clark, S.C.; Schendel, P.F.; Minden, M.D.; McCulloch, E.A. Binding of iodinated recombinant human GM-CSF to the blast cells of acute myeloblastic leukemia. Leukemia 1988, 2, 211–215. [Google Scholar]
- Frankel, A.E.; Hall, P.D.; Burbage, C.; Vesely, J.; Willingham, M.; Bhalla, K.; Kreitman, R.J. Modulation of the apoptotic response of human myeloid leukemia cells to a diphtheria toxin granulocyte-macrophage colony-stimulating factor fusion protein. Blood 1997, 90, 3654–3661. [Google Scholar]
- Kreitman, R.J.; Pastan, I. Recombinant toxins containing human granulocyte-macrophage colony-stimulating factor and either pseudomonas exotoxin or diphtheria toxin kill gastrointestinal cancer and leukemia cells. Blood 1997, 90, 252–259. [Google Scholar]
- Recht, L.; Torres, C.O.; Smith, T.W.; Raso, V.; Griffin, T.W. Transferrin receptor in normal and neoplastic brain tissue: Implications for brain-tumor immunotherapy. J. Neurosurg. 1990, 72, 941–945. [Google Scholar]
- Greenfield, L.; Johnson, V.G.; Youle, R.J. Mutations in diphtheria toxin separate binding from entry and amplify immunotoxin selectivity. Science 1987, 238, 536–539. [Google Scholar]
- Johnson, V.G.; Wrobel, C.; Wilson, D.; Zovickian, J.; Greenfield, L.; Oldfield, E.H.; Youle, R. Improved tumor-specific immunotoxins in the treatment of CNS and leptomeningeal neoplasia. J. Neurosurg. 1989, 70, 240–248. [Google Scholar]
- Liu, P.V. Extracellular toxins of Pseudomonas aeruginosa. J. Infect. Dis. 1974, 130, S94–S99. [Google Scholar]
- Iglewski, B.H.; Kabat, D. NAD-dependent inhibition of protein synthesis by Pseudomonas aeruginosa toxin. Proc. Natl. Acad. Sci. USA 1975, 72, 2284–2288. [Google Scholar]
- Hwang, J.; Fitzgerald, D.J.; Adhya, S.; Pastan, I. Functional domains of Pseudomonas exotoxin identified by deletion analysis of the gene expressed in E coli. Cell 1987, 48, 129–136. [Google Scholar]
- Siegall, C.B.; Chaudhary, V.K.; FitzGerald, D.J.; Pastan, I. Functional analysis of domains II, Ib, and III of Pseudomonas exotoxin. J. Biol. Chem. 1989, 264, 14256–14261. [Google Scholar]
- Hessler, J.L.; Kreitman, R.J. An early step in Pseudomonas exotoxin action is removal of the terminal lysine residue, which allows binding to the KDEL receptor. Biochemistry 1997, 36, 14577–14582. [Google Scholar]
- Kounnas, M.Z.; Morris, R.E.; Thompson, M.R.; FitzGerald, D.J.; Strickland, D.K.; Saelinger, C.B. The alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein binds and internalizes Pseudomonas exotoxin A. J. Biol. Chem. 1992, 267, 12420–12423. [Google Scholar]
- Smith, D.C.; Spooner, R.A.; Watson, P.D.; Murray, J.L.; Hodge, T.W.; Amessou, M.; Johannes, L.; Lord, J.M.; Roberts, L.M. Internalized Pseudomonas exotoxin A can exploit multiple pathways to reach the endoplasmic reticulum. Traffic 2006, 7, 379–393. [Google Scholar]
- Fryling, C.; Ogata, M.; FitzGerald, D. Characterization of a cellular protease that cleaves Pseudomonas exotoxin. Infect. Immun. 1992, 60, 497–502. [Google Scholar]
- Ogata, M.; Fryling, C.M.; Pastan, I.; FitzGerald, D.J. Cell-mediated cleavage of Pseudomonas exotoxin between Arg279 and Gly280 generates the enzymatically active fragment which translocates to the cytosol. J. Biol. Chem. 1992, 267, 25396–25401. [Google Scholar]
- McKee, M.L.; FitzGerald, D.J. Reduction of furin-nicked Pseudomonas exotoxin A: An unfolding story. Biochemistry 1999, 38, 16507–16513. [Google Scholar]
- Lombardi, D.; Soldati, T.; Riederer, M.A.; Goda, Y.; Zerial, M.; Pfeffer, S.R. Rab9 functions in transport between late endosomes and the trans Golgi network. EMBO J. 1993, 12, 677–682. [Google Scholar]
- Chaudhary, V.K.; Jinno, Y.; FitzGerald, D.; Pastan, I. Pseudomonas exotoxin contains a specific sequence at the carboxyl terminus that is required for cytotoxicity. Proc. Natl. Acad. Sci. USA 1990, 87, 308–312. [Google Scholar]
- Kreitman, R.J.; Pastan, I. Importance of the glutamate residue of KDEL in increasing the cytotoxicity of Pseudomonas exotoxin derivatives and for increased binding to the KDEL receptor. Biochem. J. 1995, 307, 29–37. [Google Scholar]
- Jackson, M.E.; Simpson, J.C.; Girod, A.; Pepperkok, R.; Roberts, L.M.; Lord, J.M. The KDEL retrieval system is exploited by Pseudomonas exotoxin A, but not by Shiga-like toxin-1, during retrograde transport from the Golgi complex to the endoplasmic reticulum. J. Cell. Sci. 1999, 112, 467–475. [Google Scholar]
- Ogata, M.; Chaudhary, V.K.; Pastan, I.; FitzGerald, D.J. Processing of Pseudomonas exotoxin by a cellular protease results in the generation of a 37,000-Da toxin fragment that is translocated to the cytosol. J. Bio.l Chem. 1990, 265, 20678–20685. [Google Scholar]
- Theuer, C.P.; Buchner, J.; FitzGerald, D.; Pastan, I. The N-terminal region of the 37-kDa translocated fragment of Pseudomonas exotoxin A aborts translocation by promoting its own export after microsomal membrane insertion. Proc. Natl. Acad. Sci. USA 1993, 90, 7774–7778. [Google Scholar]
- Theuer, C.; Kasturi, S.; Pastan, I. Domain II of Pseudomonas exotoxin A arrests the transfer of translocating nascent chains into mammalian microsomes. Biochemistry 1994, 33, 5894–5900. [Google Scholar]
- Hazes, B.; Read, R.J. Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 1997, 36, 11051–11054. [Google Scholar]
- Koopmann, J.O.; Albring, J.; Huter, E.; Bulbuc, N.; Spee, P.; Neefjes, J.; Hammerling, G.J.; Momburg, F. Export of antigenic peptides from the endoplasmic reticulum intersects with retrograde protein translocation through the Sec61p channel. Immunity 2000, 13, 117–127. [Google Scholar]
- Spooner, R.A.; Smith, D.C.; Easton, A.J.; Roberts, L.M.; Lord, J.M. Retrograde transport pathways utilised by viruses and protein toxins. Virol. J. 2006, 3, 26. [Google Scholar]
- Iglewski, B.H.; Liu, P.V.; Kabat, D. Mechanism of action of Pseudomonas aeruginosa exotoxin Aiadenosine diphosphate-ribosylation of mammalian elongation factor 2 in vitro and in vivo. Infect. Immun. 1977, 15, 138–144. [Google Scholar]
- Carroll, S.F.; Collier, R.J. Active site of Pseudomonas aeruginosa exotoxin A. Glutamic acid 553 is photolabeled by NAD and shows functional homology with glutamic acid 148 of diphtheria toxin. J. Biol. Chem. 1987, 262, 8707–8711. [Google Scholar] [PubMed]
- Jenkins, C.E.; Swiatoniowski, A.; Issekutz, A.C.; Lin, T.J. Pseudomonas aeruginosa exotoxin A induces human mast cell apoptosis by a caspase-8 and -3-dependent mechanism. J. Biol. Chem. 2004, 279, 37201–37207. [Google Scholar]
- Wolf, P.; Elsasser-Beile, U. Pseudomonas exotoxin A: From virulence factor to anti-cancer agent. Int. J. Med. Microbiol. 2009, 299, 161–176. [Google Scholar]
- Uchiyama, T.; Broder, S.; Waldmann, T.A. A monoclonal antibody (anti-Tac) reactive with activated and functionally mature human T cells. I. Production of anti-Tac monoclonal antibody and distribution of Tac (+) cells. J. Immunol. 126, 1981, 1393–1397. [Google Scholar]
- Uchiyama, T.; Nelson, D.L.; Fleisher, T.A.; Waldmann, T.A. A monoclonal antibody (anti-Tac) reactive with activated and functionally mature human T cells. II. Expression of Tac antigen on activated cytotoxic killer T cells, suppressor cells, and on one of two types of helper T cells. J. Immunol. 1981, 126, 1398–1403. [Google Scholar] [PubMed]
- Waldmann, T.A. Anti-Tac (daclizumab, Zenapax) in the treatment of leukemia, autoimmune diseases, and in the prevention of allograft rejection: A 25-year personal odyssey. J. Clin. Immunol. 2007, 27, 1–18. [Google Scholar]
- Kreitman, R.J.; Bailon, P.; Chaudhary, V.K.; FitzGerald, D.J.; Pastan, I. Recombinant immunotoxins containing anti-Tac(Fv) and derivatives of Pseudomonas exotoxin produce complete regression in mice of an interleukin-2 receptor-expressing human carcinoma. Blood 1994, 83, 426–434. [Google Scholar]
- Kreitman, R.J.; Pastan, I. Accumulation of a recombinant immunotoxin in a tumor in vivo: Fewer than 1000 molecules per cell are sufficient for complete responses. Canc. Res. 1998, 58, 968–975. [Google Scholar]
- Robbins, D.H.; Margulies, I.; Stetler-Stevenson, M.; Kreitman, R.J. Hairy cell leukemia, a B-cell neoplasm that is particularly sensitive to the cytotoxic effect of anti-Tac(Fv)-PE38 (LMB-2). Clin Canc. Res. 2000, 6, 693–700. [Google Scholar]
- Onda, M.; Kreitman, R.J.; Vasmatzis, G.; Lee, B.; Pastan, I. Reduction of the nonspecific animal toxicity of anti-Tac(Fv)-PE38 by mutations in the framework regions of the Fv which lower the isoelectric point. J. Immunol. 1999, 163, 6072–6077. [Google Scholar]
- Kreitman, R.J. Recombinant immunotoxins for the treatment of haematological malignancies. Expert Opin. Biol. Ther. 2004, 4, 1115–1128. [Google Scholar]
- Onda, M.; Willingham, M.; Wang, Q.C.; Kreitman, R.J.; Tsutsumi, Y.; Nagata, S.; Pastan, I. Inhibition of TNF-alpha produced by Kupffer cells protects against the nonspecific liver toxicity of immunotoxin anti-Tac(Fv)-PE38, LMB-2. J. Immunol. 2000, 165, 7150–7156. [Google Scholar]
- Chakraborty, N.G.; Chattopadhyay, S.; Mehrotra, S.; Chhabra, A.; Mukherji, B. Regulatory T-cell response and tumor vaccine-induced cytotoxic T lymphocytes in human melanoma. Hum. Immunol. 2004, 65, 794–802. [Google Scholar]
- Powell, D.J., Jr.; Felipe-Silva, A.; Merino, M.J.; Ahmadzadeh, M.; Allen, T.; Levy, C.; White, D.E.; Mavroukakis, S.; Kreitman, R.J.; Rosenberg, S.A.; Pastan, I. Administration of a CD25-directed immunotoxin, LMB-2, to patients with metastatic melanoma induces a selective partial reduction in regulatory T cells in vivo. J. Immunol. 2007, 179, 4919–4928. [Google Scholar]
- Clark, E.A. CD22, a B cell-specific receptor, mediates adhesion and signal transduction. J. Immunol. 1993, 150, 4715–4718. [Google Scholar]
- Robbins, B.A.; Ellison, D.J.; Spinosa, J.C.; Carey, C.A.; Lukes, R.J.; Poppema, S.; Saven, A.; Piro, L.D. Diagnostic application of two-color flow cytometry in 161 cases of hairy cell leukemia. Blood 1993, 82, 1277–1287. [Google Scholar]
- Gudowius, S.; Recker, K.; Laws, H.J.; Dirksen, U.; Troger, A.; Wieczorek, U.; Furlan, S.; Gobel, U.; Hanenberg, H. Identification of candidate target antigens for antibody-based immunotherapy in childhood B-cell precursor ALL. Klin. Padiatr. 2006, 218, 327–333. [Google Scholar]
- Olejniczak, S.H.; Stewart, C.C.; Donohue, K.; Czuczman, M.S. A quantitative exploration of surface antigen expression in common B-cell malignancies using flow cytometry. Immunol. Invest. 2006, 35, 93–114. [Google Scholar]
- Rawstron, A.C.; de Tute, R.; Jack, A.S.; Hillmen, P. Flow cytometric protein expression profiling as a systematic approach for developing disease-specific assays: Identification of a chronic lymphocytic leukaemia-specific assay for use in rituximab-containing regimens. Leukemia 2006, 20, 2102–2110. [Google Scholar]
- Campana, D.; Janossy, G.; Bofill, M.; Trejdosiewicz, L.K.; Ma, D.; Hoffbrand, A.V.; Mason, D.Y.; Lebacq, A.M.; Forster, H.K. Human B cell development. I. Phenotypic differences of B lymphocytes in the bone marrow and peripheral lymphoid tissue. J. Immunol. 1985, 134, 1524–1530. [Google Scholar] [PubMed]
- Mansfield, E.; Amlot, P.; Pastan, I.; FitzGerald, D.J. Recombinant RFB4 immunotoxins exhibit potent cytotoxic activity for CD22-bearing cells and tumors. Blood 1997, 90, 2020–2026. [Google Scholar]
- Robak, T. Current treatment options in hairy cell leukemia and hairy cell leukemia variant. Canc. Treat. Rev. 2006, 32, 365–376. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Immunotoxins in the treatment of hematologic malignancies. Curr. Drug Targets. 2006, 7, 1301–1311. [Google Scholar]
- Kreitman, R.J. Recombinant immunotoxins for the treatment of chemoresistant hematologic malignancies. Curr. Pharm. Des. 2009, 15, 2652–2664. [Google Scholar]
- Sakamoto, J.; Furukawa, K.; Cordon-Cardo, C.; Yin, B.W.; Rettig, W.J.; Oettgen, H.F.; Old, L.J.; Lloyd, K.O. Expression of Lewisa, Lewisb, X, and Y blood group antigens in human colonic tumors and normal tissue and in human tumor-derived cell lines. Canc. Res. 1986, 46, 1553–1561. [Google Scholar]
- Miyake, M.; Taki, T.; Hitomi, S.; Hakomori, S. Correlation of expression of H/Le(y)/Le(b) antigens with survival in patients with carcinoma of the lung. N. Engl. J. Med. 1992, 327, 14–18. [Google Scholar]
- Yin, B.W.; Finstad, C.L.; Kitamura, K.; Federici, M.G.; Welshinger, M.; Kudryashov, V.; Hoskins, W.J.; Welt, S.; Lloyd, K.O. Serological and immunochemical analysis of Lewis y (Ley) blood group antigen expression in epithelial ovarian cancer. Int. J. Cancer. 1996, 65, 406–412. [Google Scholar]
- Pastan, I.; Lovelace, E.T.; Gallo, M.G.; Rutherford, A.V.; Magnani, J.L.; Willingham, M.C. Characterization of monoclonal antibodies B1 and B3 that react with mucinous adenocarcinomas. Canc. Res. 1991, 51, 3781–3787. [Google Scholar]
- Kuan, C.T.; Pai, L.H.; Pastan, I. Immunotoxins containing Pseudomonas exotoxin that target LeY damage human endothelial cells in an antibody-specific mode: Relevance to vascular leak syndrome. Clin. Canc. Res. 1995, 1, 1589–1594. [Google Scholar]
- Posey, J.A.; Khazaeli, M.B.; Bookman, M.A.; Nowrouzi, A.; Grizzle, W.E.; Thornton, J.; Carey, D.E.; Lorenz, J.M.; Sing, A.P.; Siegall, C.B.; LoBuglio, A.F.; Saleh, M.N. A phase I trial of the single-chain immunotoxin SGN-10 (BR96 sFv-PE40) in patients with advanced solid tumors. Clin. Canc. Res. 2002, 8, 3092–3099. [Google Scholar]
- Nielsen, K.; Boston, R.S. Ribosome-inactivating proteins: A plant perspective. Annu. Rev. Plant. Physiol. Plant. Mol. Biol. 2001, 52, 785–816. [Google Scholar]
- Peumans, W.J.; Hao, Q.; Van Damme, E.J. Ribosome-inactivating proteins from plants: More than RNA N-glycosidases? FASEB J 2001, 15, 1493–1506. [Google Scholar]
- Van Damme, E.J.M.; Hao, Q.; Chen, Y.; Barre, A.; Vandenbussche, F.; Desmyter, S.; Rouge, P.; Peumans, W.J. Ribosome-inactivating proteins: A family of plant proteins that do more than inactivate ribosomes. Crit. Rev. Plant Sci. 2001, 20, 395–466. [Google Scholar]
- Girbes, T.; Ferreras, J.M.; Arias, F.J.; Stirpe, F. Description, distribution, activity and phylogenetic relationship of ribosome-inactivating proteins in plants, fungi and bacteria. Mini Rev. Med. Chem. 2004, 4, 461–476. [Google Scholar]
- Hartley, M.R.; Lord, J.M. Cytotoxic ribosome-inactivating lectins from plants. Biochim. Biophys. Acta 2004, 1701, 1–14. [Google Scholar]
- Stirpe, F. Ribosome-inactivating proteins. Toxicon 2004, 44, 371–383. [Google Scholar]
- Stirpe, F.; Battelli, M.G. Ribosome-inactivating proteins: Progress and problems. Cell Mol. Life Sci. 2006, 63, 1850–1866. [Google Scholar]
- Reinbothe, S.; Reinbothe, C.; Lehmann, J.; Becker, W.; Apel, K.; Parthier, B. JIP60, a methyl jasmonate-induced ribosome-inactivating protein involved in plant stress reactions. Proc. Natl. Acad. Sci. USA 1994, 91, 7012–7016. [Google Scholar]
- Stirpe, F.; Barbieri, L.; Gorini, P.; Valbonesi, P.; Bolognesi, A.; Polito, L. Activities associated with the presence of ribosome-inactivating proteins increase in senescent and stressed leaves. FEBS Lett. 1996, 382, 309–312. [Google Scholar]
- Rippmann, J.F.; Michalowski, C.B.; Nelson, D.E.; Bohnert, H.J. Induction of a ribosome-inactivating protein upon environmental stress. Plant. Mol. Biol. 1997, 35, 701–709. [Google Scholar]
- Cola, A.D.; Poma, A.; Spanò, L. Culture senescence and abscisic acid induce saporin production in cultured roots of Saponaria officinalis. New Phytol. 1999, 141, 381–386. [Google Scholar]
- Song, S.K.; Choi, Y.; Moon, Y.H.; Kim, S.G.; Choi, Y.D.; Lee, J.S. Systemic induction of a Phytolacca insularis antiviral protein gene by mechanical wounding, jasmonic acid, and abscisic acid. Plant Mol. Biol. 2000, 43, 439–450. [Google Scholar]
- Bass, H.W.; Krawetz, J.E.; GR, O.B.; Zinselmeier, C.; Habben, J.E.; Boston, R.S. Maize ribosome-inactivating proteins (RIPs) with distinct expression patterns have similar requirements for proenzyme activation. J. Exp. Bot. 2004, 55, 2219–2233. [Google Scholar]
- Qin, W.; Ming-Xing, H.; Ying, X.; Xin-Shen, Z.; Fang, C. Expression of a ribosome inactivating protein (curcin 2) in Jatropha curcas is induced by stress. J. Biosci. 2005, 30, 351–357. [Google Scholar]
- Girbes, T.; de Torre, C.; Iglesias, R.; Miguel Ferreras, J.; Mendez, E. RIP for viruses. Nature 1996, 379, 777–778. [Google Scholar]
- Iglesias, R.; Perez, Y.; de Torre, C.; Ferreras, J.M.; Antolin, P.; Jimenez, P.; Rojo, M.A.; Mendez, E.; Girbes, T. Molecular characterization and systemic induction of single-chain ribosome-inactivating proteins (RIPs) in sugar beet (Beta vulgaris) leaves. J. Exp. Bot. 2005, 56, 1675–1684. [Google Scholar]
- Xu, J.; Wang, H.; Fan, J. Expression of a ribosome-inactivating protein gene in bitter melon is induced by Sphaerotheca fuliginea and abiotic stimuli. Biotechnol. Lett. 2007, 29, 1605–1610. [Google Scholar]
- Walsh, T.A.; Morgan, A.E.; Hey, T.D. Characterization and molecular cloning of a proenzyme form of a ribosome-inactivating protein from maize. Novel mechanism of proenzyme activation by proteolytic removal of a 2.8-kilodalton internal peptide segment. J. Biol. Chem. 1991, 266, 23422–23427. [Google Scholar] [PubMed]
- Bass, H.W.; Webster, C.; GR, O.B.; Roberts, J.K.; Boston, R.S. A maize ribosome-inactivating protein is controlled by the transcriptional activator Opaque-2. Plant Cell 1992, 4, 225–234. [Google Scholar]
- Chaudhry, B.; Muller-Uri, F.; Cameron-Mills, V.; Gough, S.; Simpson, D.; Skriver, K.; Mundy, J. The barley 60 kDa jasmonate-induced protein (JIP60) is a novel ribosome-inactivating protein. Plant J. 1994, 6, 815–824. [Google Scholar]
- Nicolson, G.L.; Blaustein, J. The interaction of Ricinus communis agglutinin with normal and tumor cell surfaces. Biochim. Biophys. Acta. 1972, 266, 543–547. [Google Scholar]
- Nicolson, G.L. The interactions of lectins with animal cell surfaces. Int. Rev. Cytol. 1974, 39, 89–190. [Google Scholar]
- Nicolson, G.L. Ultrastructural analysis of toxin binding and entry into mammalian cells. Nature 1974, 251, 628–630. [Google Scholar]
- Baenziger, J.U.; Fiete, D. Structural determinants of Ricinus communis agglutinin and toxin specificity for oligosaccharides. J. Biol. Chem. 1979, 254, 9795–9799. [Google Scholar]
- Houston, L.L.; Dooley, T.P. Binding of two molecules of 4-methylumbelliferyl galactose or 4-methylumbelliferyl N-acetylgalactosamine to the B chains of ricin and Ricinus communis agglutinin and to purified ricin B chain. J. Biol. Chem. 1982, 257, 4147–4151. [Google Scholar]
- Hughes, R.C.; Mills, G. Analysis by lectin affinity chromatography of N-linked glycans of BHK cells and ricin-resistant mutants. Biochem. J. 1983, 211, 575–587. [Google Scholar]
- Skilleter, D.N.; Paine, A.J.; Stirpe, F. A comparison of the accumulation of ricin by hepatic parenchymal and non-parenchymal cells and its inhibition of protein synthesis. Biochim. Biophys. Acta 1981, 677, 495–500. [Google Scholar]
- Simmons, B.M.; Stahl, P.D.; Russell, J.H. Mannose receptor-mediated uptake of ricin toxin and ricin A chain by macrophages. Multiple intracellular pathways for a chain translocation. J. Biol. Chem. 1986, 261, 7912–7920. [Google Scholar] [PubMed]
- Magnusson, S.; Berg, T.; Turpin, E.; Frenoy, J.P. Interactions of ricin with sinusoidal endothelial rat liver cells. Different involvement of two distinct carbohydrate-specific mechanisms in surface binding and internalization. Biochem. J. 1991, 277, 855–861. [Google Scholar] [PubMed]
- Magnusson, S.; Kjeken, R.; Berg, T. Characterization of two distinct pathways of endocytosis of ricin by rat liver endothelial cells. Exp. Cell. Res. 1993, 205, 118–125. [Google Scholar]
- Riccobono, F.; Fiani, M.L. Mannose receptor dependent uptake of ricin A1 and A2 chains by macrophages. Carbohydr. Res. 1996, 282, 285–292. [Google Scholar]
- Frankel, A.E.; Fu, T.; Burbage, C.; Tagge, E.; Harris, B.; Vesely, J.; Willingham, M.C. Lectin-deficient ricin toxin intoxicates cells bearing the D-mannose receptor. Carbohydr. Res. 1997, 300, 251–258. [Google Scholar]
- Rapak, A.; Falnes, P.O.; Olsnes, S. Retrograde transport of mutant ricin to the endoplasmic reticulum with subsequent translocation to cytosol. Proc. Natl. Acad. Sci. USA 1997, 94, 3783–3788. [Google Scholar]
- Sandvig, K.; van Deurs, B. Endocytosis and intracellular transport of ricin: Recent discoveries. FEBS Lett. 1999, 452, 67–70. [Google Scholar]
- Simpson, J.C.; Roberts, L.M.; Romisch, K.; Davey, J.; Wolf, D.H.; Lord, J.M. Ricin A chain utilises the endoplasmic reticulum-associated protein degradation pathway to enter the cytosol of yeast. FEBS Lett. 1999, 459, 80–84. [Google Scholar]
- Wesche, J.; Rapak, A.; Olsnes, S. Dependence of ricin toxicity on translocation of the toxin A-chain from the endoplasmic reticulum to the cytosol. J. Biol. Chem. 1999, 274, 34443–34449. [Google Scholar]
- Deeks, E.D.; Cook, J.P.; Day, P.J.; Smith, D.C.; Roberts, L.M.; Lord, J.M. The low lysine content of ricin A chain reduces the risk of proteolytic degradation after translocation from the endoplasmic reticulum to the cytosol. Biochemistry 2002, 41, 3405–3413. [Google Scholar]
- Wesche, J. Retrograde transport of ricin. Int. J. Med. Microbiol. 2002, 291, 517–521. [Google Scholar]
- Lord, J.M.; Deeks, E.; Marsden, C.J.; Moore, K.; Pateman, C.; Smith, D.C.; Spooner, R.A.; Watson, P.; Roberts, L.M. Retrograde transport of toxins across the endoplasmic reticulum membrane. Biochem. Soc. Trans. 2003, 31, 1260–1262. [Google Scholar]
- Roberts, L.M.; Lord, J.M. Ribosome-inactivating proteins: Entry into mammalian cells and intracellular routing. Mini Rev. Med. Chem. 2004, 4, 505–512. [Google Scholar]
- Spooner, R.A.; Watson, P.D.; Marsden, C.J.; Smith, D.C.; Moore, K.A.; Cook, J.P.; Lord, J.M.; Roberts, L.M. Protein disulphide-isomerase reduces ricin to its A and B chains in the endoplasmic reticulum. Biochem. J. 2004, 383, 285–293. [Google Scholar]
- Vago, R.; Marsden, C.J.; Lord, J.M.; Ippoliti, R.; Flavell, D.J.; Flavell, S.U.; Ceriotti, A.; Fabbrini, M.S. Saporin and ricin A chain follow different intracellular routes to enter the cytosol of intoxicated cells. FEBS J. 2005, 272, 4983–4995. [Google Scholar]
- Cavallaro, U.; Nykjaer, A.; Nielsen, M.; Soria, M.R. Alpha 2-macroglobulin receptor mediates binding and cytotoxicity of plant ribosome-inactivating proteins. Eur. J. Biochem. 1995, 232, 165–171. [Google Scholar]
- Cavallaro, U.; Soria, M.R. Targeting plant toxins to the urokinase and alpha 2-macroglobulin receptors. Semin. Canc. Biol. 1995, 6, 269–278. [Google Scholar]
- Chan, W.L.; Shaw, P.C.; Tam, S.C.; Jacobsen, C.; Gliemann, J.; Nielsen, M.S. Trichosanthin interacts with and enters cells via LDL receptor family members. Biochem. Biophys. Res. Commun. 2000, 270, 453–457. [Google Scholar]
- Stirpe, F.; Olsnes, S.; Pihl, A. Gelonin, a new inhibitor of protein synthesis, nontoxic to intact cells. Isolation, characterization, and preparation of cytotoxic complexes with concanavalin A. J. Biol. Chem. 1980, 255, 6947–6953. [Google Scholar] [PubMed]
- Bolognesi, A.; Tazzari, P.L.; Tassi, C.; Gromo, G.; Gobbi, M.; Stirpe, F. A comparison of anti-lymphocyte immunotoxins containing different ribosome-inactivating proteins and antibodies. Clin. Exp. Immunol. 1992, 89, 341–346. [Google Scholar]
- Bolognesi, A.; Polito, L. Immunotoxins and other conjugates: Pre-clinical studies. Mini Rev. Med. Chem. 2004, 4, 563–583. [Google Scholar]
- Fracasso, G.; Bellisola, G.; Castelletti, D.; Tridente, G.; Colombatti, M. Immunotoxins and other conjugates: Preparation and general characteristics. Mini Rev. Med. Chem. 2004, 4, 545–562. [Google Scholar]
- Geden, S.E.; Gardner, R.A.; Fabbrini, M.S.; Ohashi, M.; Phanstiel Iv, O.; Teter, K. Lipopolyamine treatment increases the efficacy of intoxication with saporin and an anticancer saporin conjugate. FEBS J. 2007, 274, 4825–4836. [Google Scholar]
- Endo, Y.; Tsurugi, K. The RNA N-glycosidase activity of ricin A-chain. The characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J. Biol. Chem. 1988, 263, 8735–8739. [Google Scholar] [PubMed]
- Endo, Y.; Tsurugi, K.; Lambert, J.M. The site of action of six different ribosome-inactivating proteins from plants on eukaryotic ribosomes: The RNA N-glycosidase activity of the proteins. Biochem. Biophys. Res. Commun. 1988, 150, 1032–1036. [Google Scholar]
- Stirpe, F.; Bailey, S.; Miller, S.P.; Bodley, J.W. Modification of ribosomal RNA by ribosome-inactivating proteins from plants. Nucl. Acid. Res. 1988, 16, 1349–1357. [Google Scholar]
- Montanaro, L.; Sperti, S.; Mattioli, A.; Testoni, G.; Stirpe, F. Inhibition by ricin of protein synthesis in vitro. Inhibition of the binding of elongation factor 2 and of adenosine diphosphate-ribosylated elongation factor 2 to ribosomes. Biochem. J. 1975, 146, 127–131. [Google Scholar] [PubMed]
- Nilsson, L.; Asano, K.; Svensson, B.; Poulsen, F.M.; Nygard, O. Reduced turnover of the elongation factor EF-1 X ribosome complex after treatment with the protein synthesis inhibitor II from barley seeds. Biochim. Biophys. Acta 1986, 868, 62–70. [Google Scholar]
- Griffiths, G.D.; Leek, M.D.; Gee, D.J. The toxic plant proteins ricin and abrin induce apoptotic changes in mammalian lymphoid tissues and intestine. J. Pathol. 1987, 151, 221–229. [Google Scholar]
- Brigotti, M.; Rambelli, F.; Zamboni, M.; Montanaro, L.; Sperti, S. Effect of alpha-sarcin and ribosome-inactivating proteins on the interaction of elongation factors with ribosomes. Biochem. J. 1989, 257, 723–727. [Google Scholar]
- Osborn, R.W.; Hartley, M.R. Dual effects of the ricin A chain on protein synthesis in rabbit reticulocyte lysate. Inhibition of initiation and translocation. Eur. J. Biochem. 1990, 193, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, G.; Perfetti, V.; Tonon, L.; Novella, A.; Lucotti, C.; Danova, M.; Glennie, M.J.; Merlini, G.; Cazzola, M. Saporin, a ribosome-inactivating protein used to prepare immunotoxins, induces cell death via apoptosis. Br. J. Haematol. 1996, 93, 789–794. [Google Scholar]
- Bolognesi, A.; Tazzari, P.L.; Olivieri, F.; Polito, L.; Falini, B.; Stirpe, F. Induction of apoptosis by ribosome-inactivating proteins and related immunotoxins. Int. J. Canc. 1996, 68, 349–355. [Google Scholar]
- Bantel, H.; Engels, I.H.; Voelter, W.; Schulze-Osthoff, K.; Wesselborg, S. Mistletoe lectin activates caspase-8/FLICE independently of death receptor signaling and enhances anticancer drug-induced apoptosis. Canc. Res. 1999, 59, 2083–2090. [Google Scholar]
- Narayanan, S.; Surolia, A.; Karande, A.A. Ribosome-inactivating protein and apoptosis: Abrin causes cell death via mitochondrial pathway in Jurkat cells. Biochem. J. 2004, 377, 233–240. [Google Scholar]
- Mi, S.L.; An, C.C.; Wang, Y.; Chen, J.Y.; Che, N.Y.; Gao, Y.; Chen, Z.L. Trichomislin, a novel ribosome-inactivating protein, induces apoptosis that involves mitochondria and caspase-3. Arch. Biochem. Biophys. 2005, 434, 258–265. [Google Scholar]
- Rao, P.V.; Jayaraj, R.; Bhaskar, A.S.; Kumar, O.; Bhattacharya, R.; Saxena, P.; Dash, P.K.; Vijayaraghavan, R. Mechanism of ricin-induced apoptosis in human cervical cancer cells. Biochem. Pharmacol. 2005, 69, 855–865. [Google Scholar]
- Parikh, B.A.; Tumer, N.E. Antiviral activity of ribosome inactivating proteins in medicine. Mini Rev. Med. Chem. 2004, 4, 523–543. [Google Scholar]
- Barbieri, L.; Valbonesi, P.; Bonora, E.; Gorini, P.; Bolognesi, A.; Stirpe, F. Polynucleotide: Adenosine glycosidase activity of ribosome-inactivating proteins: Effect on DNA, RNA and poly(A). Nucl. Acid. Res. 1997, 25, 518–522. [Google Scholar]
- Barbieri, L.; Bolognesi, A.; Valbonesi, P.; Polito, L.; Olivieri, F.; Stirpe, F. Polynucleotide: Adenosine glycosidase activity of immunotoxins containing ribosome-inactivating proteins. J. Drug Target 2000, 8, 281–288. [Google Scholar]
- Barbieri, L.; Valbonesi, P.; Govoni, M.; Pession, A.; Stirpe, F. Polynucleotide: Adenosine glycosidase activity of saporin-L1: Effect on various forms of mammalian DNA. Biochim. Biophys. Acta 2000, 1480, 258–266. [Google Scholar]
- Barbieri, L.; Valbonesi, P.; Righi, F.; Zuccheri, G.; Monti, F.; Gorini, P.; Samori, B.; Stirpe, F. Polynucleotide: Adenosine glycosidase is the sole activity of ribosome-inactivating proteins on DNA. J. Biochem. 2000, 128, 883–889. [Google Scholar]
- Nicolas, E.; Beggs, J.M.; Taraschi, T.F. Gelonin is an unusual DNA glycosylase that removes adenine from single-stranded DNA, normal base pairs and mismatches. J. Biol. Chem. 2000, 275, 31399–31406. [Google Scholar]
- Li, M.X.; Yeung, H.W.; Pan, L.P.; Chan, S.I. Trichosanthin, a potent HIV-1 inhibitor, can cleave supercoiled DNA in vitro. Nucl. Acid. Res. 1991, 19, 6309–6312. [Google Scholar]
- Ling, J.; Li, X.; Wu, X.; Liu, W. Topological requirements for recognition and cleavage of DNA by ribosome-inactivating proteins. Biol. Chem. Hoppe. Seyler 1995, 376, 637–641. [Google Scholar]
- Roncuzzi, L.; Gasperi-Campani, A. DNA-nuclease activity of the single-chain ribosome-inactivating proteins dianthin 30, saporin 6 and gelonin. FEBS Lett. 1996, 392, 16–20. [Google Scholar]
- Nicolas, E.; Beggs, J.M.; Haltiwanger, B.M.; Taraschi, T.F. Direct evidence for the deoxyribonuclease activity of the plant ribosome inactivating protein gelonin. FEBS Lett. 1997, 406, 162–164. [Google Scholar]
- Wang, P.; Tumer, N.E. Pokeweed antiviral protein cleaves double-stranded supercoiled DNA using the same active site required to depurinate rRNA. Nucl. Acid. Res. 1999, 27, 1900–1905. [Google Scholar]
- Bagga, S.; Seth, D.; Batra, J.K. The cytotoxic activity of ribosome-inactivating protein saporin-6 is attributed to its rRNA N-glycosidase and internucleosomal DNA fragmentation activities. J. Biol. Chem. 2003, 278, 4813–4820. [Google Scholar]
- Gasperi-Campani, A.; Brognara, I.; Baiocchi, D.; Roncuzzi, L. Mitochondrial DNA D-loop as a new target of Saporin 6 nuclease activity. Toxicon 2005, 45, 475–480. [Google Scholar]
- Choudhary, N.L.; Yadav, O.P.; Lodha, M.L. Ribonuclease, deoxyribonuclease, and antiviral activity of Escherichia coli-expressed Bougainvillea xbuttiana antiviral protein 1. Biochemistry (Mosc) 2008, 73, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Mock, J.W.; Ng, T.B.; Wong, R.N.; Yao, Q.Z.; Yeung, H.W.; Fong, W.P. Demonstration of ribonuclease activity in the plant ribosome-inactivating proteins alpha- and beta-momorcharins. Life Sci. 1996, 59, 1853–1859. [Google Scholar]
- Fong, W.P.; Mock, W.Y.; Ng, T.B. Intrinsic ribonuclease activities in ribonuclease and ribosome-inactivating proteins from the seeds of bitter gourd. Int. J. Biochem. Cell Biol. 2000, 32, 571–577. [Google Scholar]
- Herceg, Z.; Wang, Z.Q. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Mutat. Res. 2001, 477, 97–110. [Google Scholar]
- Barbieri, L.; Brigotti, M.; Perocco, P.; Carnicelli, D.; Ciani, M.; Mercatali, L.; Stirpe, F. Ribosome-inactivating proteins depurinate poly(ADP-ribosyl)ated poly(ADP-ribose) polymerase and have transforming activity for 3T3 fibroblasts. FEBS Lett. 2003, 538, 178–182. [Google Scholar]
- Hudak, K.A.; Wang, P.; Tumer, N.E. A novel mechanism for inhibition of translation by pokeweed antiviral protein: Depurination of the capped RNA template. RNA 2000, 6, 369–380. [Google Scholar]
- Vivanco, J.M.; Tumer, N.E. Translation Inhibition of Capped and Uncapped Viral RNAs Mediated by Ribosome-Inactivating Proteins. Phytopathology 2003, 93, 588–595. [Google Scholar]
- Li, X.D.; Chen, W.F.; Liu, W.Y.; Wang, G.H. Large-scale preparation of two new ribosome-inactivating proteins—Cinnamomin and camphorin from the seeds of Cinnamomum camphora. Protein Expr. Purif. 1997, 10, 27–31. [Google Scholar]
- Sharma, N.; Park, S.W.; Vepachedu, R.; Barbieri, L.; Ciani, M.; Stirpe, F.; Savary, B.J.; Vivanco, J.M. Isolation and characterization of an RIP (ribosome-inactivating protein)-like protein from tobacco with dual enzymatic activity. Plant Physiol. 2004, 134, 171–181. [Google Scholar]
- Barbieri, L.; Polito, L.; Bolognesi, A.; Ciani, M.; Pelosi, E.; Farini, V.; Jha, A.K.; Sharma, N.; Vivanco, J.M.; Chambery, A.; Parente, A.; Stirpe, F. Ribosome-inactivating proteins in edible plants and purification and characterization of a new ribosome-inactivating protein from Cucurbita moschata. Biochim. Biophys. Acta 2006, 1760, 783–792. [Google Scholar]
- Helmy, M.; Lombard, S.; Pieroni, G. Ricin RCA60: Evidence of its phospholipase activity. Biochem. Biophy.s Res. Commun. 1999, 258, 252–255. [Google Scholar]
- Nicolas, E.; Goodyer, I.D.; Taraschi, T.F. An additional mechanism of ribosome-inactivating protein cytotoxicity: Degradation of extrachromosomal DNA. Biochem. J. 1997, 327, 413–417. [Google Scholar]
- Brigotti, M.; Alfieri, R.; Sestili, P.; Bonelli, M.; Petronini, P.G.; Guidarelli, A.; Barbieri, L.; Stirpe, F.; Sperti, S. Damage to nuclear DNA induced by Shiga toxin 1 and ricin in human endothelial cells. FASEB J. 2002, 16, 365–372. [Google Scholar]
- Sestili, P.; Alfieri, R.; Carnicelli, D.; Martinelli, C.; Barbieri, L.; Stirpe, F.; Bonelli, M.; Petronini, P.G.; Brigotti, M. Shiga toxin 1 and ricin inhibit the repair of H2O2-induced DNA single strand breaks in cultured mammalian cells. DNA Repair (Amst) 2005, 4, 271–277. [Google Scholar]
- Brigotti, M.; Carnicelli, D.; Ravanelli, E.; Vara, A.G.; Martinelli, C.; Alfieri, R.R.; Petronini, P.G.; Sestili, P. Molecular damage and induction of proinflammatory cytokines in human endothelial cells exposed to Shiga toxin 1, Shiga toxin 2, and alpha-sarcin. Infect. Immun. 2007, 75, 2201–2207. [Google Scholar]
- Li, X.P.; Baricevic, M.; Saidasan, H.; Tumer, N.E. Ribosome depurination is not sufficient for ricin-mediated cell death in Saccharomyces cerevisiae. Infect. Immun. 2007, 75, 417–428. [Google Scholar]
- Sikriwal, D.; Ghosh, P.; Batra, J.K. Ribosome inactivating protein saporin induces apoptosis through mitochondrial cascade, independent of translation inhibition. Int. J. Biochem. Cell Biol. 2008, 40, 2880–2888. [Google Scholar]
- Polito, L.; Bortolotti, M.; Farini, V.; Battelli, M.G.; Barbieri, L.; Bolognesi, A. Saporin induces multiple death pathways in lymphoma cells with different intensity and timing as compared to ricin. Int. J. Biochem. Cell Biol. 2009, 41, 1055–1061. [Google Scholar]
- Stein, H.; Mason, D.Y.; Gerdes, J.; O'Connor, N.; Wainscoat, J.; Pallesen, G.; Gatter, K.; Falini, B.; Delsol, G.; Lemke, H.; et al. The expression of the Hodgkin's disease associated antigen Ki-1 in reactive and neoplastic lymphoid tissue: Evidence that Reed-Sternberg cells and histiocytic malignancies are derived from activated lymphoid cells. Blood 1985, 66, 848–858. [Google Scholar]
- Froese, P.; Lemke, H.; Gerdes, J.; Havsteen, B.; Schwarting, R.; Hansen, H.; Stein, H. Biochemical characterization and biosynthesis of the Ki-1 antigen in Hodgkin-derived and virus-transformed human B and T lymphoid cell lines. J. Immunol. 1987, 139, 2081–2087. [Google Scholar]
- Agnarsson, B.A.; Kadin, M.E. The immunophenotype of Reed-Sternberg cells. A study of 50 cases of Hodgkin's disease using fixed frozen tissues. Cancer 1989, 63, 2083–2087. [Google Scholar] [CrossRef] [PubMed]
- Foxwell, B.M.; Donovan, T.A.; Thorpe, P.E.; Wilson, G. The removal of carbohydrates from ricin with endoglycosidases H, F and D and alpha-mannosidase. Biochim. Biophys. Acta 1985, 840, 193–203. [Google Scholar]
- Blakey, D.C.; Thorpe, P.E. Effect of chemical deglycosylation on the in vivo fate of ricin A-chain. Cancer Drug Deliv. 1986, 3, 189–196. [Google Scholar]
- Bourrie, B.J.; Casellas, P.; Blythman, H.E.; Jansen, F.K. Study of the plasma clearance of antibody--ricin-A-chain immunotoxins. Evidence for specific recognition sites on the A chain that mediate rapid clearance of the immunotoxin. Eur. J. Biochem. 1986, 155, 1–10. [Google Scholar]
- Skilleter, D.N.; Foxwell, B.M. Selective uptake of ricin A-chain by hepatic non-parenchymal cells in vitro. Importance of mannose oligosaccharides in the toxin. FEBS Lett. 1986, 196, 344–348. [Google Scholar]
- Blakey, D.C.; Watson, G.J.; Knowles, P.P.; Thorpe, P.E. Effect of chemical deglycosylation of ricin A chain on the in vivo fate and cytotoxic activity of an immunotoxin composed of ricin A chain and anti-Thy 1.1 antibody. Canc. Res. 1987, 47, 947–952. [Google Scholar]
- Fulton, R.J.; Tucker, T.F.; Vitetta, E.S.; Uhr, J.W. Pharmacokinetics of tumor-reactive immunotoxins in tumor-bearing mice: Effect of antibody valency and deglycosylation of the ricin A chain on clearance and tumor localization. Canc. Res. 1988, 48, 2618–2625. [Google Scholar]
- Trown, P.W.; Reardan, D.T.; Carroll, S.F.; Stoudemire, J.B.; Kawahata, R.T. Improved pharmacokinetics and tumor localization of immunotoxins constructed with the Mr 30,000 form of ricin A chain. Canc. Res. 1991, 51, 4219–4225. [Google Scholar]
- Mason, D.Y.; Stein, H.; Gerdes, J.; Pulford, K.A.; Ralfkiaer, E.; Falini, B.; Erber, W.N.; Micklem, K.; Gatter, K.C. Value of monoclonal anti-CD22 (p135) antibodies for the detection of normal and neoplastic B lymphoid cells. Blood 1987, 69, 836–840. [Google Scholar]
- Tedder, T.F.; Tuscano, J.; Sato, S.; Kehrl, J.H. CD22, a B lymphocyte-specific adhesion molecule that regulates antigen receptor signaling. Annu. Rev. Immunol. 1997, 15, 481–504. [Google Scholar]
- Fujimoto, M.; Poe, J.C.; Inaoki, M.; Tedder, T.F. CD19 regulates B lymphocyte responses to transmembrane signals. Semin. Immunol. 1998, 10, 267–277. [Google Scholar]
- Poe, J.C.; Hasegawa, M.; Tedder, T.F. CD19, CD21, and CD22: Multifaceted response regulators of B lymphocyte signal transduction. Int. Rev. Immunol. 2001, 20, 739–762. [Google Scholar]
- Shen, G.L.; Li, J.L.; Ghetie, M.A.; Ghetie, V.; May, R.D.; Till, M.; Brown, A.N.; Relf, M.; Knowles, P.; Uhr, J.W.; et al. Evaluation of four CD22 antibodies as ricin A chain-containing immunotoxins for the in vivo therapy of human B-cell leukemias and lymphomas. Int. J. Canc. 1988, 42, 792–797. [Google Scholar]
- Pezzutto, A.; Dorken, B.; Rabinovitch, P.S.; Ledbetter, J.A.; Moldenhauer, G.; Clark, E.A. CD19 monoclonal antibody HD37 inhibits anti-immunoglobulin-induced B cell activation and proliferation. J. Immunol. 1987, 138, 2793–2799. [Google Scholar]
- Amlot, P.L.; Stone, M.J.; Cunningham, D.; Fay, J.; Newman, J.; Collins, R.; May, R.; McCarthy, M.; Richardson, J.; Ghetie, V.; et al. A phase I study of an anti-CD22-deglycosylated ricin A chain immunotoxin in the treatment of B-cell lymphomas resistant to conventional therapy. Blood 1993, 82, 2624–2633. [Google Scholar]
- Fathman, C.G.; Costa, G.L.; Seroogy, C.M. Gene therapy for autoimmune disease. Clin. Immunol. 2000, 95, S39–S43. [Google Scholar]
- Kawashiri, M.-A.; Rader, D.J. Gene therapy for lipid disorders. Curr. Control Trials Cardiovasc. Med. 2000, 1, 120–127. [Google Scholar]
- Kouraklis, G. Gene therapy for cancer: From the laboratory to the patient. Dig. Dis. Sci. 2000, 45, 1045–1052. [Google Scholar]
- Lyngstadaas, A. Status and potential of gene therapy in clinical medicine. Assessment of an emerging health technology through systematic survey of clinical gene therapy protocols and published results. Int. J. Technol. Assess. Health. Care 2002, 18, 645–674. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, P.D.; Zielske, S.P.; Roth, J.C.; Ballas, C.B.; Bowman, J.E.; Gerson, S.L. Cancer gene therapy: Scientific basis. Annu. Rev. Med. 2002, 53, 437–452. [Google Scholar]
- Dishart, K.L.; Work, L.M.; Denby, L.; Baker, A.H. Gene Therapy for Cardiovascular Disease. J. Biomed. Biotechnol. 2003, 2003, 138–148. [Google Scholar] [Green Version]
- Selkirk, S.M. Gene therapy in clinical medicine. Postgrad. Med. J. 2004, 80, 560–570. [Google Scholar]
- Cathomen, T.; Weitzman, M.D. Gene repair: Pointing the finger at genetic disease. Gene Ther. 2005, 12, 1415–1416. [Google Scholar]
- Cross, D.; Burmester, J.K. Gene therapy for cancer treatment: Past, present and future. Clin. Med. Res. 2006, 4, 218–227. [Google Scholar]
- Chuang, Y.H.; Yang, Y.H.; Wu, S.J.; Chiang, B.L. Gene therapy for allergic diseases. Curr. Gene Ther. 2009, 9, 185–191. [Google Scholar]
- Cao, S.; Cripps, A.; Wei, M.Q. New strategies for cancer gene therapy: Progress and opportunities. Clin. Exp. Pharm. Physiol. 2010, 37, 108–114. [Google Scholar]
- Itaka, K.; Kataoka, K. Recent development of nonviral gene delivery systems with virus-like structures and mechanisms. Eur. J. Pharm. Biopharm. 2009, 71, 475–483. [Google Scholar]
- Pathak, A.; Patnaik, S.; Gupta, K.C. Recent trends in non-viral vector-mediated gene delivery. Biotechnol. J. 2009, 4, 1559–1572. [Google Scholar]
- Heilbronn, R.; Weger, S. Viral vectors for gene transfer: Current status of gene therapeutics. Handbook Exp. Pharm. 2010, 197, 143–170. [Google Scholar]
- Moolten, F.L. Drug sensitivity ("suicide") genes for selective cancer chemotherapy. Canc. Gene Ther. 1994, 1, 279–287. [Google Scholar]
- Spencer, D.M. Developments in suicide genes for preclinical and clinical applications. Curr. Opin. Mol. Ther. 2000, 2, 433–440. [Google Scholar]
- Ozawa, T.; Hu, J.L.; Hu, L.J.; Kong, E.L.; Bollen, A.W.; Lamborn, K.R.; Deen, D.F. Functionality of hypoxia-induced BAX expression in a human glioblastoma xenograft model. Canc. Gene Ther. 2005, 12, 449–455. [Google Scholar]
- Yang, W.S.; Park, S.O.; Yoon, A.R.; Yoo, J.Y.; Kim, M.K.; Yun, C.O.; Kim, C.W. Suicide cancer gene therapy using pore-forming toxin, streptolysin O. Mol. Canc. Ther. 2006, 5, 1610–1619. [Google Scholar]
- Altaner, C. Prodrug cancer gene therapy. Canc. Lett 2008, 270, 191–201. [Google Scholar]
- Maxwell, I.H.; Maxwell, F.O.; Glode, L.M. Regulated Expression of a Diphtheria Toxin A-Chain Gene Transfected into Human Cells: Possible Strategy for Inducing Cancer Cell Suicide. Canc. Res. 1986, 46, 4660–4664. [Google Scholar]
- Walther, W.; Stein, U. Cell type specific and inducible promoters for vectors in gene therapy as an approach for cell targeting. J. Mol. Med. 1996, 74, 379–392. [Google Scholar]
- Peng, K.W. Strategies for targeting therapeutic gene delivery. Mol. Med. Today 1999, 5, 448–453. [Google Scholar]
- Nettelbeck, D.M.; Jerome, V.; Muller, R. Gene therapy: Designer promoters for tumour targeting. Trends Genet. 2000, 16, 174–181. [Google Scholar]
- Haviv, Y.S.; Blackwell, J. Transcriptional regulation in cancer gene therapy. Isr. Med. Assoc. J. 2001, 3, 517–522. [Google Scholar]
- Robson, T.; Hirst, D.G. Transcriptional Targeting in Cancer Gene Therapy. J. Biomed. Biotechnol. 2003, 2003, 110–137. [Google Scholar] [Green Version]
- Dorer, D.E.; Nettelbeck, D.M. Targeting cancer by transcriptional control in cancer gene therapy and viral oncolysis. Adv.Drug. Deliv. Rev. 2009, 61, 554–571. [Google Scholar]
- Li, Y.; McCadden, J.; Ferrer, F.; Kruszewski, M.; Carducci, M.; Simons, J.; Rodriguez, R. Prostate-specific expression of the diphtheria toxin A chain (DT-A): Studies of inducibility and specificity of expression of prostate-specific antigen promoter-driven DT-A adenoviral-mediated gene transfer. Canc. Res. 2002, 62, 2576–2582. [Google Scholar]
- Peng, W.; Chen, J.; Huang, Y.H.; Sawicki, J.A. Tightly-regulated suicide gene expression kills PSA-expressing prostate tumor cells. Gene Ther. 2005, 12, 1573–1580. [Google Scholar]
- Peng, W.; Anderson, D.G.; Bao, Y.; Padera, R.F., Jr.; Langer, R.; Sawicki, J.A. Nanoparticulate delivery of suicide DNA to murine prostate and prostate tumors. Prostate 2007, 67, 855–862. [Google Scholar]
- Lidor, Y.J.; Lee, W.E.; Nilson, J.H.; Maxwell, I.H.; Su, L.J.; Brand, E.; Glode, L.M. In vitro expression of the diphtheria toxin A-chain gene under the control of human chorionic gonadotropin gene promoters as a means of directing toxicity to ovarian cancer cell lines. Am. J. Obstet. Gynecol. 1997, 177, 579–585. [Google Scholar]
- Huang, Y.H.; Zugates, G.T.; Peng, W.; Holtz, D.; Dunton, C.; Green, J.J.; Hossain, N.; Chernick, M.R.; Padera, R.F., Jr.; Langer, R.; Anderson, D.G.; Sawicki, J.A. Nanoparticle-delivered suicide gene therapy effectively reduces ovarian tumor burden in mice. Canc. Res. 2009, 69, 6184–6191. [Google Scholar]
- Sidi, A.A.; Ohana, P.; Benjamin, S.; Shalev, M.; Ransom, J.H.; Lamm, D.; Hochberg, A.; Leibovitch, I. Phase I/II marker lesion study of intravesical BC-819 DNA plasmid in H19 over expressing superficial bladder cancer refractory to bacillus Calmette-Guerin. J. Urol. 2008, 180, 2379–2383. [Google Scholar]
- Mizrahi, A.; Czerniak, A.; Levy, T.; Amiur, S.; Gallula, J.; Matouk, I.; Abu-lail, R.; Sorin, V.; Birman, T.; de Groot, N.; Hochberg, A.; Ohana, P. Development of targeted therapy for ovarian cancer mediated by a plasmid expressing diphtheria toxin under the control of H19 regulatory sequences. J. Transl. Med. 2009, 7, 69. [Google Scholar]
- Mizrahi, A.; Czerniak, A.; Ohana, P.; Amiur, S.; Gallula, J.; Matouk, I.; Abu-Lail, R.; Birman, T.; Hochberg, A.; Levy, T. Treatment of ovarian cancer ascites by intra-peritoneal injection of diphtheria toxin A chain-H19 vector: A case report. J. Med. Case. Rep. 2010, 4, 228. [Google Scholar]
- Abdul-Ghani, R.; Ohana, P.; Matouk, I.; Ayesh, S.; Ayesh, B.; Laster, M.; Bibi, O.; Giladi, H.; Molnar-Kimber, K.; Sughayer, M.A.; de Groot, N.; Hochberg, A. Use of Transcriptional Regulatory Sequences of Telomerase (hTER and hTERT) for Selective Killing of Cancer Cells. Mol. Ther. 2000, 2, 539–544. [Google Scholar]
- Harrison, G.S.; Long, C.J.; Curiel, T.J.; Maxwell, F.; Maxwell, I.H. Inhibition of human immunodeficiency virus-1 production resulting from transduction with a retrovirus containing an HIV-regulated diphtheria toxin A chain gene. Hum. Gene. Ther. 1992, 3, 461–469. [Google Scholar]
- Dinges, M.M.; Cook, D.R.; King, J.; Curiel, T.J.; Zhang, X.Q.; Harrison, G.S. HIV-regulated diphtheria toxin A chain gene confers long-term protection against HIV type 1 infection in the human promonocytic cell line U937. Hum. Gene. Ther. 1995, 6, 1437–1445. [Google Scholar]
- Konopka, K.; Harrison, G.S.; Felgner, P.L.; Duzgunes, N. Cationic liposome-mediated expression of HIV-regulated luciferase and diphtheria toxin a genes in HeLa cells infected with or expressing HIV. Biochim. Biophys. Acta 1997, 1356, 185–197. [Google Scholar]
- Wang, Z.; Tang, Z.; Zheng, Y.; Yu, D.; Spear, M.; Iyer, S.R.; Bishop, B.; Wu, Y. Development of a nonintegrating Rev-dependent lentiviral vector carrying diphtheria toxin A chain and human TRAF6 to target HIV reservoirs. Gene Ther. 2010, 17, 1063–1076. [Google Scholar]
- Massuda, E.S.; Dunphy, E.J.; Redman, R.A.; Schreiber, J.J.; Nauta, L.E.; Barr, F.G.; Maxwell, I.H.; Cripe, T.P. Regulated expression of the diphtheria toxin A chain by a tumor-specific chimeric transcription factor results in selective toxicity for alveolar rhabdomyosarcoma cells. Proc. Natl. Acad. Sci. USA 1997, 94, 14701–14706. [Google Scholar]
- Maxwell, I.H.; Kaletta, C.; Naujoks, K.; Maxwell, F. Targeting diphtheria toxin A-chain transcription to activated endothelial cells using an E-selectin promoter. Angiogenesis 2003, 6, 31–38. [Google Scholar]
- Lee, E.J.; Jameson, J.L. Cell-specific Cre-mediated activation of the diphtheria toxin gene in pituitary tumor cells: Potential for cytotoxic gene therapy. Hum. Gene Ther. 2002, 13, 533–542. [Google Scholar]
- Wang, C.Y.; Li, F.; Yang, Y.; Guo, H.Y.; Wu, C.X.; Wang, S. Recombinant baculovirus containing the diphtheria toxin A gene for malignant glioma therapy. Canc. Res. 2006, 66, 5798–5806. [Google Scholar]
- Cao, G.W.; Qi, Z.T.; Pan, X.; Zhang, X.Q.; Miao, X.H.; Feng, Y.; Lu, X.H.; Kuriyama, S.; Du, P. Gene therapy for human colorectal carcinoma using human CEA promoter contro led bacterial ADP-ribosylating toxin genes human CEA: PEA & DTA gene transfer. World J. Gastroenterol. 1998, 4, 388–391. [Google Scholar]
- Hine, C.M.; Seluanov, A.; Gorbunova, V. Use of the Rad51 promoter for targeted anti-cancer therapy. Proc.Natl. Acad. Sci. USA 2008, 105, 20810–20815. [Google Scholar]
- Murayama, Y.; Tadakuma, T.; Kunitomi, M.; Kumai, K.; Tsutsui, K.; Yasuda, T.; Kitajima, M. Cell-specific expression of the diphtheria toxin A-chain coding sequence under the control of the upstream region of the human alpha-fetoprotein gene. J. Surg. Oncol. 1999, 70, 145–149. [Google Scholar]
- Kunitomi, M.; Takayama, E.; Suzuki, S.; Yasuda, T.; Tsutsui, K.; Nagaike, K.; Hiroi, S.; Tadakuma, T. Selective inhibition of hepatoma cells using diphtheria toxin A under the control of the promoter/enhancer region of the human alpha-fetoprotein gene. Jpn. J. Canc. Res. 2000, 91, 343–350. [Google Scholar]
- Maxwell, I.H.; Glode, L.M.; Maxwell, F. Expression of the diphtheria toxin A-chain coding sequence under the control of promoters and enhancers from immunoglobulin genes as a means of directing toxicity to B-lymphoid cells. Canc. Res. 1991, 51, 4299–4304. [Google Scholar]
- Rothfels, H.; Paschen, A.; Schadendorf, D. Evaluation of combined gene regulatory elements for transcriptional targeting of suicide gene expression to malignant melanoma. Exp. Dermatol. 2003, 12, 799–810. [Google Scholar]
- Martin, V.; Cortes, M.L.; de Felipe, P.; Farsetti, A.; Calcaterra, N.B.; Izquierdo, M. Cancer gene therapy by thyroid hormone-mediated expression of toxin genes. Canc. Res. 2000, 60, 3218–3224. [Google Scholar]
- Showalter, S.L.; Huang, Y.H.; Witkiewicz, A.; Costantino, C.L.; Yeo, C.J.; Green, J.J.; Langer, R.; Anderson, D.G.; Sawicki, J.A.; Brody, J.R. Nanoparticulate delivery of diphtheria toxin DNA effectively kills Mesothelin expressing pancreatic cancer cells. Canc. Biol. Ther. 2008, 7, 1584–1590. [Google Scholar]
- Fogar, P.; Navaglia, F.; Basso, D.; Zambon, C.F.; Moserle, L.; Indraccolo, S.; Stranges, A.; Greco, E.; Fadi, E.; Padoan, A.; Pantano, G.; Sanzari, M.C.; Pedrazzoli, S.; Montecucco, C.; Plebani, M. Heat-induced transcription of diphtheria toxin A or its variants, CRM176 and CRM197: Implications for pancreatic cancer gene therapy. Canc. Gene Ther. 2010, 17, 58–68. [Google Scholar]
- Lee, J.T.; Watarai, S.; Kakidani, H.; Onuma, M.; Zhao, D.D.; Yasuda, T. Evaluation of cationic liposomes for delivery of diphtheria toxin A-chain gene to cells infected with bovine leukemia virus. J. Vet. Med. Sci. 1997, 59, 169–174. [Google Scholar]
- Tana; Watarai, S.; Lee, J.T.; Onuma, M.; Ochiai, K.; Kakidani, H.; Yasuda, T. In vivo antitumor effect of cationic liposomes containing diphtheria toxin A-chain gene on cells infected with bovine leukemia virus. J. Vet. Med. Sci. 1997, 59, 617–619. [Google Scholar]
- Diaw, L.; Woodson, K.; Gillespie, J.W. Prostate cancer epigenetics: A review on gene regulation. Gene Regul. Syst. Bio. 2007, 1, 313–325. [Google Scholar]
- Lilja, H. A kallikrein-like serine protease in prostatic fluid cleaves the predominant seminal vesicle protein. J. Clin. Invest. 1985, 76, 1899–1903. [Google Scholar]
- Lilja, H. Structure, function, and regulation of the enzyme activity of prostate-specific antigen. World J. Urol. 1993, 11, 188–191. [Google Scholar]
- Aumuller, G.; Seitz, J.; Lilja, H.; Abrahamsson, P.A.; von der Kammer, H.; Scheit, K.H. Species- and organ-specificity of secretory proteins derived from human prostate and seminal vesicles. Prostate 1990, 17, 31–40. [Google Scholar]
- Wu, L.; Matherly, J.; Smallwood, A.; Adams, J.Y.; Billick, E.; Belldegrun, A.; Carey, M. Chimeric PSA enhancers exhibit augmented activity in prostate cancer gene therapy vectors. Gene Ther. 2001, 8, 1416–1426. [Google Scholar]
- Mabjeesh, N.J.; Zhong, H.; Simons, J.W. Gene therapy of prostate cancer: Current and future directions. Endocr. Relat. Canc. 2002, 9, 115–139. [Google Scholar]
- Makarov, D.V.; Loeb, S.; Getzenberg, R.H.; Partin, A.W. Biomarkers for prostate cancer. Annu. Rev. Med. 2009, 60, 139–151. [Google Scholar]
- Zheng, J.Y.; Chen, D.; Chan, J.; Yu, D.; Ko, E.; Pang, S. Regression of prostate cancer xenografts by a lentiviral vector specifically expressing diphtheria toxin A. Canc. Gene Ther. 2003, 10, 764–770. [Google Scholar]
- Anderson, D.G.; Peng, W.; Akinc, A.; Hossain, N.; Kohn, A.; Padera, R.; Langer, R.; Sawicki, J.A. A polymer library approach to suicide gene therapy for cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 16028–16033. [Google Scholar]
- Picard, D. Regulation of protein function through expression of chimaeric proteins. Curr. Opin. Biotechnol. 1994, 5, 511–515. [Google Scholar]
- Logie, C.; Stewart, A.F. Ligand-regulated site-specific recombination. Proc. Natl. Acad. Sci USA 1995, 92, 5940–5944. [Google Scholar]
- Peng, W.; Verbitsky, A.; Bao, Y.; Sawicki, J. Regulated expression of diphtheria toxin in prostate cancer cells. Mol. Ther. 2002, 6, 537–545. [Google Scholar]
- Cho, K.R.; Shih Ie, M. Ovarian cancer. Annu Rev Pathol 2009, 4, 287–313. [Google Scholar]
- Tsampalas, M.; Gridelet, V.; Berndt, S.; Foidart, J.M.; Geenen, V.; Perrier d'Hauterive, S. Human chorionic gonadotropin: A hormone with immunological and angiogenic properties. J. Reprod. Immunol. 2010, 85, 93–98. [Google Scholar]
- Eagle, K.; Ledermann, J.A. Tumor Markers in Ovarian Malignancies. The Oncologist 1997, 2, 324–329. [Google Scholar]
- Demirtas, E.; Krishnamurthy, S.; Tulandi, T. Elevated serum beta-human chorionic gonadotropin In nonpregnant conditions. Obstet. Gynecol. Surv. 2007, 62, 675–679. [Google Scholar]
- Scholler, N.; Fu, N.; Yang, Y.; Ye, Z.; Goodman, G.E.; Hellstrom, K.E.; Hellstrom, I. Soluble member(s) of the mesothelin/megakaryocyte potentiating factor family are detectable in sera from patients with ovarian carcinoma. Proc. Natl. Acad. Sci. USA 1999, 96, 11531–11536. [Google Scholar]
- Schummer, M.; Ng, W.V.; Bumgarner, R.E.; Nelson, P.S.; Schummer, B.; Bednarski, D.W.; Hassell, L.; Baldwin, R.L.; Karlan, B.Y.; Hood, L. Comparative hybridization of an array of 21 500 ovarian cDNAs for the discovery of genes overexpressed in ovarian carcinomas. Gene 1999, 238, 375–385. [Google Scholar]
- Wang, K.; Gan, L.; Jeffery, E.; Gayle, M.; Gown, A.M.; Skelly, M.; Nelson, P.S.; Ng, W.V.; Schummer, M.; Hood, L.; Mulligan, J. Monitoring gene expression profile changes in ovarian carcinomas using cDNA microarray. Gene 1999, 229, 101–108. [Google Scholar]
- Hough, C.D.; Sherman-Baust, C.A.; Pizer, E.S.; Montz, F.J.; Im, D.D.; Rosenshein, N.B.; Cho, K.R.; Riggins, G.J.; Morin, P.J. Large-scale serial analysis of gene expression reveals genes differentially expressed in ovarian cancer. Canc. Res. 2000, 60, 6281–6287. [Google Scholar]
- Frierson, H.F., Jr.; Moskaluk, C.A.; Powell, S.M.; Zhang, H.; Cerilli, L.A.; Stoler, M.H.; Cathro, H.; Hampton, G.M. Large-scale molecular and tissue microarray analysis of mesothelin expression in common human carcinomas. Hum. Pathol. 2003, 34, 605–609. [Google Scholar]
- Ordonez, N.G. Application of mesothelin immunostaining in tumor diagnosis. Am. J. Surg. Pathol. 2003, 27, 1418–1428. [Google Scholar]
- Li, J.; Dowdy, S.; Tipton, T.; Podratz, K.; Lu, W.G.; Xie, X.; Jiang, S.W. HE4 as a biomarker for ovarian and endometrial cancer management. Expert Rev. Mol. Diagn. 2009, 9, 555–566. [Google Scholar]
- Tanos, V.; Prus, D.; Ayesh, S.; Weinstein, D.; Tykocinski, M.L.; De-Groot, N.; Hochberg, A.; Ariel, I. Expression of the imprinted H19 oncofetal RNA in epithelial ovarian cancer. Eur. J. Obstet. Gynecol. Reprod. Biol. 1999, 85, 7–11. [Google Scholar]
- Steenman, M.J.; Rainier, S.; Dobry, C.J.; Grundy, P.; Horon, I.L.; Feinberg, A.P. Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms' tumour. Nat. Genet. 1994, 7, 433–439. [Google Scholar]
- Ilana, A.; Orit, L.; Tamar, S.; Galina, P.; Mally, S.; Nathan, D.-G.; Abraham, H. The imprinted H19 gene as a tumor marker in bladder carcinoma. Urology 1995, 45, 335–338. [Google Scholar]
- Kondo, M.; Suzuki, H.; Ueda, R.; Osada, H.; Takagi, K.; Takahashi, T. Frequent loss of imprinting of the H19 gene is often associated with its overexpression in human lung cancers. Oncogene 1995, 10, 1193–1198. [Google Scholar]
- Douc-Rasy, S.; Barrois, M.; Fogel, S.; Ahomadegbe, J.C.; Stehelin, D.; Coll, J.; Riou, G. High incidence of loss of heterozygosity and abnormal imprinting of H19 and IGF2 genes in invasive cervical carcinomas. Uncoupling of H19 and IGF2 expression and biallelic hypomethylation of H19. Oncogene 1996, 12, 423–430. [Google Scholar] [PubMed]
- Hibi, K.; Nakamura, H.; Hirai, A.; Fujikake, Y.; Kasai, Y.; Akiyama, S.; Ito, K.; Takagi, H. Loss of H19 imprinting in esophageal cancer. Canc. Res. 1996, 56, 480–482. [Google Scholar]
- Ariel, I.; Ayesh, S.; Perlman, E.J.; Pizov, G.; Tanos, V.; Schneider, T.; Erdmann, V.A.; Podeh, D.; Komitowski, D.; Quasem, A.S.; de Groot, N.; Hochberg, A. The product of the imprinted H19 gene is an oncofetal RNA. Mol. Pathol. 1997, 50, 34–44. [Google Scholar]
- Ariel, I.; Miao, H.Q.; Ji, X.R.; Schneider, T.; Roll, D.; de Groot, N.; Hochberg, A.; Ayesh, S. Imprinted H19 oncofetal RNA is a candidate tumour marker for hepatocellular carcinoma. Mol. Pathol. 1998, 51, 21–25. [Google Scholar]
- Jirtle, R.L. Genomic imprinting and cancer. Exp. Cell. Res. 1999, 248, 18–24. [Google Scholar]
- Erdmann, V.A.; Barciszewska, M.Z.; Szymanski, M.; Hochberg, A.; Groot, N.d.; Barciszewski, J. The non-coding RNAs as riboregulators. Nucl. Acid. Res. 2001, 29, 189–193. [Google Scholar]
- Pow-Sang, J.M.; Seigne, J.D. Contemporary management of superficial bladder cancer. Canc. Control 2000, 7, 335–339. [Google Scholar]
- Jacobs, B.L.; Lee, C.T.; Montie, J.E. Bladder cancer in 2010: How far have we come? CA Canc. J. Clin. 2010, 60, 244–272. [Google Scholar]
- Shay, J.W.; Zou, Y.; Hiyama, E.; Wright, W.E. Telomerase and cancer. Hum. Mol. Genet. 2001, 10, 677–685. [Google Scholar]
- Blackburn, E.H. Switching and Signaling at the Telomere. Cell 2001, 106, 661–673. [Google Scholar]
- Mark, J.C.; Martin, F.; Dymitr, K.; Alexander, S.; Ekkehard, S.; Ilana, A.; Mark, L.T.; Stela, M.; Joseph, I.; Nathan De, G.; Abraham, H. Developmentally Imprinted Genes as Markers for Bladder Tumor Progression. J. Urol. 1996, 155, 2120–2127. [Google Scholar]
- Ohana, P.; Bibi, O.; Matouk, I.; Levy, C.; Birman, T.; Ariel, I.; Schneider, T.; Ayesh, S.; Giladi, H.; Laster, M.; Groot, N.d.; Hochberg, A. Use of H19 regulatory sequences for targeted gene therapy in cancer. Int. J. Canc. 2002, 98, 645–650. [Google Scholar]
- Ohana, P.; Gofrit, O.; Ayesh, S.; Al-Sharef, W.; Mizrahi, A.; Birman, T.; Schneider, T.; Matouk, I.; Groot, N.d.; Tavdy, E.; Sidi, A.A.; Hochberg, A. Regulatory sequences of the H19 gene in DNA based therapy of bladder cancer. Gene Ther. Mol. Biol. 2004, 8, 181–192. [Google Scholar]
- Freed, E.O. HIV-1 replication. Somat. Cell. Mol. Genet. 2001, 26, 13–33. [Google Scholar]
- Liang, C.; Wainberg, M.A. The role of Tat in HIV-1 replication: An activator and/or a suppressor? AIDS Rev. 2002, 4, 41–49. [Google Scholar]
- Wu, Y.; Marsh, J.W. Gene transcription in HIV infection. Microb. Infect. 2003, 5, 1023–1027. [Google Scholar]
- Pugliese, A.; Vidotto, V.; Beltramo, T.; Petrini, S.; Torre, D. A review of HIV-1 Tat protein biological effects. Cell Biochem. Funct. 2005, 23, 223–227. [Google Scholar]
- Suhasini, M.; Reddy, T.R. Cellular proteins and HIV-1 Rev function. Curr. HIV Res. 2009, 7, 91–100. [Google Scholar]
- Fanales-Belasio, E.; Raimondo, M.; Suligoi, B.; Butto, S. HIV virology and pathogenetic mechanisms of infection: A brief overview. Ann. Ist. Super. Sanita 2010, 46, 5–14. [Google Scholar]
- Harrison, G.S.; Maxwell, F.; Long, C.J.; Rosen, C.A.; Glode, L.M.; Maxwell, I.H. Activation of a diphtheria toxin A gene by expression of human immunodeficiency virus-1 Tat and Rev proteins in transfected cells. Hum. Gene. Ther. 1991, 2, 53–60. [Google Scholar]
- Harrison, G.S.; Long, C.J.; Maxwell, F.; Glode, L.M.; Maxwell, I.H. Inhibition of HIV production in cells containing an integrated, HIV-regulated diphtheria toxin A chain gene. AIDS Res. Hum. Retroviruses 1992, 8, 39–45. [Google Scholar]
- Curiel, T.J.; Cook, D.R.; Wang, Y.; Hahn, B.H.; Ghosh, S.K.; Harrison, G.S. Long-term inhibition of clinical and laboratory human immunodeficiency virus strains in human T-cell lines containing an HIV-regulated diphtheria toxin A chain gene. Hum. Gene Ther. 1993, 4, 741–747. [Google Scholar]
- Kedzierska, K.; Crowe, S.M. The role of monocytes and macrophages in the pathogenesis of HIV-1 infection. Curr. Med. Chem. 2002, 9, 1893–1903. [Google Scholar]
- Williams, D.P.; Wen, Z.; Watson, R.S.; Boyd, J.; Strom, T.B.; Murphy, J.R. Cellular processing of the interleukin-2 fusion toxin DAB486-IL-2 and efficient delivery of diphtheria fragment A to the cytosol of target cells requires Arg194. J. Biol. Chem. 1990, 265, 20673–20677. [Google Scholar]
- Klimpel, K.R.; Molloy, S.S.; Thomas, G.; Leppla, S.H. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc. Natl. Acad.Sci. USA 1992, 89, 10277–10281. [Google Scholar]
- Gordon, V.M.; Leppla, S.H. Proteolytic activation of bacterial toxins: Role of bacterial and host cell proteases. Infect. Immun. 1994, 62, 333–340. [Google Scholar]
- Beauregard, K.E.; Collier, R.J.; Swanson, J.A. Proteolytic activation of receptor-bound anthrax protective antigen on macrophages promotes its internalization. Cell. Microbiol. 2000, 2, 251–258. [Google Scholar]
- Liu, S.; Netzel-Arnett, S.; Birkedal-Hansen, H.; Leppla, S.H. Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Canc. Res. 2000, 60, 6061–6067. [Google Scholar]
- Liu, S.; Wang, H.; Currie, B.M.; Molinolo, A.; Leung, H.J.; Moayeri, M.; Basile, J.R.; Alfano, R.W.; Gutkind, J.S.; Frankel, A.E.; Bugge, T.H.; Leppla, S.H. Matrix metalloproteinase-activated anthrax lethal toxin demonstrates high potency in targeting tumor vasculature. J. Biol. Chem. 2008, 283, 529–540. [Google Scholar]
- Alfano, R.W.; Leppla, S.H.; Liu, S.; Bugge, T.H.; Meininger, C.J.; Lairmore, T.C.; Mulne, A.F.; Davis, S.H.; Duesbery, N.S.; Frankel, A.E. Matrix metalloproteinase-activated anthrax lethal toxin inhibits endothelial invasion and neovasculature formation during in vitro morphogenesis. Mol. Canc. Res. 2009, 7, 452–461. [Google Scholar]
- Alfano, R.W.; Leppla, S.H.; Liu, S.; Bugge, T.H.; Ortiz, J.M.; Lairmore, T.C.; Duesbery, N.S.; Mitchell, I.C.; Nwariaku, F.; Frankel, A.E. Inhibition of tumor angiogenesis by the matrix metalloproteinase-activated anthrax lethal toxin in an orthotopic model of anaplastic thyroid carcinoma. Mol. Canc. Ther. 2010, 9, 190–201. [Google Scholar]
- Liu, S.; Bugge, T.H.; Leppla, S.H. Targeting of tumor cells by cell surface urokinase plasminogen activator-dependent anthrax toxin. J. Biol. Chem. 2001, 276, 17976–17984. [Google Scholar]
- Liu, S.; Aaronson, H.; Mitola, D.J.; Leppla, S.H.; Bugge, T.H. Potent antitumor activity of a urokinase-activated engineered anthrax toxin. Proc. Natl. Acad. Sci. USA 2003, 100, 657–662. [Google Scholar]
- Abi-Habib, R.J.; Singh, R.; Liu, S.; Bugge, T.H.; Leppla, S.H.; Frankel, A.E. A urokinase-activated recombinant anthrax toxin is selectively cytotoxic to many human tumor cell types. Mol. Canc. Ther. 2006, 5, 2556–2562. [Google Scholar]
- Rono, B.; Romer, J.; Liu, S.; Bugge, T.H.; Leppla, S.H.; Kristjansen, P.E. Antitumor efficacy of a urokinase activation-dependent anthrax toxin. Mol. Canc. Ther. 2006, 5, 89–96. [Google Scholar]
- Su, Y.; Ortiz, J.; Liu, S.; Bugge, T.H.; Singh, R.; Leppla, S.H.; Frankel, A.E. Systematic urokinase-activated anthrax toxin therapy produces regressions of subcutaneous human non-small cell lung tumor in athymic nude mice. Canc. Res. 2007, 67, 3329–3336. [Google Scholar]
- Liu, S.; Redeye, V.; Kuremsky, J.G.; Kuhnen, M.; Molinolo, A.; Bugge, T.H.; Leppla, S.H. Intermolecular complementation achieves high-specificity tumor targeting by anthrax toxin. Nat. Biotechnol. 2005, 23, 725–730. [Google Scholar]
- Tcherniuk, S.O.; Chroboczek, J.; Balakirev, M.Y. Construction of tumor-specific toxins using ubiquitin fusion technique. Mol. Ther. 2005, 11, 196–204. [Google Scholar]
- Abi-Habib, R.J.; Liu, S.; Bugge, T.H.; Leppla, S.H.; Frankel, A.E. A urokinase-activated recombinant diphtheria toxin targeting the granulocyte-macrophage colony-stimulating factor receptor is selectively cytotoxic to human acute myeloid leukemia blasts. Blood 2004, 104, 2143–2148. [Google Scholar]
- Falnes, P.O.; Welker, R.; Krausslich, H.G.; Olsnes, S. Toxins that are activated by HIV type-1 protease through removal of a signal for degradation by the N-end-rule pathway. Biochem. J. 1999, 343, 199–207. [Google Scholar]
- Law, S.K. ; Wang, R.R.; Mak, A.N.; Wong, K.B.; Zheng, Y.T.; Shaw, P.C. A switch-on mechanism to activate maize ribosome-inactivating protein for targeting HIV-infected cells. Nucl. Acid. Res. 2010. [Google Scholar]
- Westermarck, J.; Kahari, V.-M. Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J. 1999, 13, 781–792. [Google Scholar]
- Stamenkovic, I. Matrix metalloproteinases in tumor invasion and metastasis. Semin. Canc. Biol. 2000, 10, 415–433. [Google Scholar]
- Konjević, G.; Stanković, S. Matrix metalloproteinases in the process of invasion and metastasis of breast cancer. Arch. Oncol. 2006, 14, 136–140. [Google Scholar]
- Fingleton, B. Matrix metalloproteinases as valid clinical targets. Curr. Pharm. Des. 2007, 13, 333–346. [Google Scholar]
- Roy, R.; Yang, J.; Moses, M.A. Matrix Metalloproteinases As Novel Biomarkers and Potential Therapeutic Targets in Human Cancer. J. Clin. Oncol. 2009, 27, 5287–5297. [Google Scholar]
- Ascenzi, P.; Visca, P.; Ippolito, G.; Spallarossa, A.; Bolognesi, M.; Montecucco, C. Anthrax toxin: A tripartite lethal combination. FEBS Lett. 2002, 531, 384–388. [Google Scholar]
- Collier, R.J.; Young, J.A. Anthrax toxin. Annu. Rev. Cell Dev. Biol. 2003, 19, 45–70. [Google Scholar]
- Young, J.A.; Collier, R.J. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Annu. Rev. Biochem. 2007, 76, 243–265. [Google Scholar]
- Bradley, K.A.; Mogridge, J.; Mourez, M.; Collier, R.J.; Young, J.A. Identification of the cellular receptor for anthrax toxin. Nature 2001, 414, 225–229. [Google Scholar]
- Scobie, H.M.; Rainey, G.J.; Bradley, K.A.; Young, J.A. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 5170–5174. [Google Scholar]
- Milne, J.C.; Furlong, D.; Hanna, P.C.; Wall, J.S.; Collier, R.J. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J. Biol. Chem. 1994, 269, 20607–20612. [Google Scholar]
- Abrami, L.; Liu, S.; Cosson, P.; Leppla, S.H.; van der Goot, F.G. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J. Cell. Biol. 2003, 160, 321–328. [Google Scholar]
- Abrami, L.; Bischofberger, M.; Kunz, B.; Groux, R.; Van der Goot, F. Endocytosis of the Anthrax Toxin Is Mediated by Clathrin, Actin and Unconventional Adaptors. PLoS Pathog. 2010, 6, e1000792. [Google Scholar]
- Abrami, L.; Lindsay, M.; Parton, R.G.; Leppla, S.H.; van der Goot, F.G. Membrane insertion of anthrax protective antigen and cytoplasmic delivery of lethal factor occur at different stages of the endocytic pathway. J. Cell. Biol. 2004, 166, 645–651. [Google Scholar] [Green Version]
- Blaustein, R.O.; Koehler, T.M.; Collier, R.J.; Finkelstein, A. Anthrax toxin: Channel-forming activity of protective antigen in planar phospholipid bilayers. Proc. Natl. Acad. Sci. USA 1989, 86, 2209–2213. [Google Scholar]
- Milne, J.C.; Collier, R.J. pH-dependent permeabilization of the plasma membrane of mammalian cells by anthrax protective antigen. Mol. Microbiol. 1993, 10, 647–653. [Google Scholar]
- Finkelstein, A. The channel formed in planar lipid bilayers by the protective antigen component of anthrax toxin. Toxicology 1994, 87, 29–41. [Google Scholar]
- Krantz, B.A.; Melnyk, R.A.; Zhang, S.; Juris, S.J.; Lacy, D.B.; Wu, Z.; Finkelstein, A.; Collier, R.J. A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science 2005, 309, 777–781. [Google Scholar]
- Krantz, B.A.; Finkelstein, A.; Collier, R.J. Protein Translocation through the Anthrax Toxin Transmembrane Pore is Driven by a Proton Gradient. J. Mol. Biol. 2006, 355, 968–979. [Google Scholar]
- Tamayo, A.G.; Bharti, A.; Trujillo, C.; Harrison, R.; Murphy, J.R. COPI coatomer complex proteins facilitate the translocation of anthrax lethal factor across vesicular membranes in vitro. Proc. Natl. Acad.Sci. USA 2008, 105, 5254–5259. [Google Scholar]
- Collier, R.J. Membrane translocation by anthrax toxin. Mol. Aspects Med. 2009, 30, 413–422. [Google Scholar]
- Finkelstein, A. Proton-coupled protein transport through the anthrax toxin channel. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2009, 364, 209–215. [Google Scholar]
- Van der Goot, G.; Young, J.A. Receptors of anthrax toxin and cell entry. Mol. Aspects Med. 2009, 30, 406–412. [Google Scholar]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; Vande Woude, G.F. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar]
- Duesbery, N.S.; Vande Woude, G.F. Anthrax lethal factor causes proteolytic inactivation of mitogen-activated protein kinase kinase. J. Appl. Microbiol. 1999, 87, 289–293. [Google Scholar]
- Pellizzari, R.; Guidi-Rontani, C.; Vitale, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFNgamma-induced release of NO and TNFalpha. FEBS Lett. 1999, 462, 199–204. [Google Scholar]
- Vitale, G.; Pellizzari, R.; Recchi, C.; Napolitani, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves the N-terminus of MAPKKS and induces tyrosine/threonine phosphorylation of MAPKS in cultured macrophages. J. Appl. Microbiol. 1999, 87, 288. [Google Scholar]
- Vitale, G.; Bernardi, L.; Napolitani, G.; Mock, M.; Montecucco, C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000, 352, 739–745. [Google Scholar]
- Park, J.M.; Greten, F.R.; Li, Z.W.; Karin, M. Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science 2002, 297, 2048–2051. [Google Scholar]
- Agrawal, A.; Lingappa, J.; Leppla, S.H.; Agrawal, S.; Jabbar, A.; Quinn, C.; Pulendran, B. Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature 2003, 424, 329–334. [Google Scholar]
- Chopra, A.P.; Boone, S.A.; Liang, X.; Duesbery, N.S. Anthrax lethal factor proteolysis and inactivation of MAPK kinase. J. Biol. Chem. 2003, 278, 9402–9406. [Google Scholar]
- Tonello, F.; Montecucco, C. The anthrax lethal factor and its MAPK kinase-specific metalloprotease activity. Mol. Aspects Med. 2009, 30, 431–438. [Google Scholar]
- Leppla, S.H. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar]
- Leppla, S.H. Bacillus anthracis calmodulin-dependent adenylate cyclase: Chemical and enzymatic properties and interactions with eucaryotic cells. Adv. Cyclic Nucl. Protein Phosph. Res. 1984, 17, 189–198. [Google Scholar]
- Drum, C.L.; Yan, S.Z.; Sarac, R.; Mabuchi, Y.; Beckingham, K.; Bohm, A.; Grabarek, Z.; Tang, W.J. An extended conformation of calmodulin induces interactions between the structural domains of adenylyl cyclase from Bacillus anthracis to promote catalysis. J. Biol. Chem. 2000, 275, 36334–36340. [Google Scholar]
- Drum, C.L.; Yan, S.Z.; Bard, J.; Shen, Y.Q.; Lu, D.; Soelaiman, S.; Grabarek, Z.; Bohm, A.; Tang, W.J. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 2002, 415, 396–402. [Google Scholar]
- Ulmer, T.S.; Soelaiman, S.; Li, S.; Klee, C.B.; Tang, W.J.; Bax, A. Calcium dependence of the interaction between calmodulin and anthrax edema factor. J. Biol. Chem. 2003, 278, 29261–29266. [Google Scholar]
- Shen, Y.; Zhukovskaya, N.L.; Guo, Q.; Florian, J.; Tang, W.J. Calcium-independent calmodulin binding and two-metal-ion catalytic mechanism of anthrax edema factor. EMBO J. 2005, 24, 929–941. [Google Scholar]
- Di Nezza, L.A.; Misajon, A.; Zhang, J.; Jobling, T.; Quinn, M.A.; Ostor, A.G.; Nie, G.; Lopata, A.; Salamonsen, L.A. Presence of active gelatinases in endometrial carcinoma and correlation of matrix metalloproteinase expression with increasing tumor grade and invasion. Cancer 2002, 94, 1466–1475. [Google Scholar]
- Sato, T.; Sakai, T.; Noguchi, Y.; Takita, M.; Hirakawa, S.; Ito, A. Tumor-stromal cell contact promotes invasion of human uterine cervical carcinoma cells by augmenting the expression and activation of stromal matrix metalloproteinases. Gynecol. Oncol. 2004, 92, 47–56. [Google Scholar]
- Berube, M.; Deschambeault, A.; Boucher, M.; Germain, L.; Petitclerc, E.; Guerin, S.L. MMP-2 expression in uveal melanoma: Differential activation status dictated by the cellular environment. Mol. Vis. 2005, 11, 1101–1111. [Google Scholar]
- Roomi, M.W.; Monterrey, J.C.; Kalinovsky, T.; Rath, M.; Niedzwiecki, A. Patterns of MMP-2 and MMP-9 expression in human cancer cell lines. Oncol. Rep. 2009, 21, 1323–1333. [Google Scholar]
- Arora, N.; Klimpel, K.R.; Singh, Y.; Leppla, S.H. Fusions of anthrax toxin lethal factor to the ADP-ribosylation domain of Pseudomonas exotoxin A are potent cytotoxins which are translocated to the cytosol of mammalian cells. J. Biol. Chem. 1992, 267, 15542–15548. [Google Scholar]
- Arora, N.; Leppla, S.H. Residues 1-254 of anthrax toxin lethal factor are sufficient to cause cellular uptake of fused polypeptides. J. Biol. Chem. 1993, 268, 3334–3341. [Google Scholar]
- Milne, J.C.; Blanke, S.R.; Hanna, P.C.; Collier, R.J. Protective antigen-binding domain of anthrax lethal factor mediates translocation of a heterologous protein fused to its amino- or carboxy-terminus. Mol. Microbiol. 1995, 15, 661–666. [Google Scholar]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; Davis, N.; Dicks, E.; Ewing, R.; Floyd, Y.; Gray, K.; Hall, S.; Hawes, R.; Hughes, J.; Kosmidou, V.; Menzies, A.; Mould, C.; Parker, A.; Stevens, C.; Watt, S.; Hooper, S.; Wilson, R.; Jayatilake, H.; Gusterson, B.A.; Cooper, C.; Shipley, J.; Hargrave, D.; Pritchard-Jones, K.; Maitland, N.; Chenevix-Trench, G.; Riggins, G.J.; Bigner, D.D.; Palmieri, G.; Cossu, A.; Flanagan, A.; Nicholson, A.; Ho, J.W.C.; Leung, S.Y.; Yuen, S.T.; Weber, B.L.; Seigler, H.F.; Darrow, T.L.; Paterson, H.; Marais, R.; Marshall, C.J.; Wooster, R.; Stratton, M.R.; Futreal, P.A. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar]
- Koo, H.M.; VanBrocklin, M.; McWilliams, M.J.; Leppla, S.H.; Duesbery, N.S.; Woude, G.F. Apoptosis and melanogenesis in human melanoma cells induced by anthrax lethal factor inactivation of mitogen-activated protein kinase kinase. Proc. Natl. Acad. Sci. USA 2002, 99, 3052–3057. [Google Scholar]
- Sebolt-Leopold, J.S.; Herrera, R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat. Rev. Canc. 2004, 4, 937–947. [Google Scholar]
- Abi-Habib, R.J.; Urieto, J.O.; Liu, S.; Leppla, S.H.; Duesbery, N.S.; Frankel, A.E. BRAF status and mitogen-activated protein/extracellular signal-regulated kinase kinase 1/2 activity indicate sensitivity of melanoma cells to anthrax lethal toxin. Mol. Canc. Ther. 2005, 4, 1303–1310. [Google Scholar]
- Abi-Habib, R.J.; Singh, R.; Leppla, S.H.; Greene, J.J.; Ding, Y.; Berghuis, B.; Duesbery, N.S.; Frankel, A.E. Systemic anthrax lethal toxin therapy produces regressions of subcutaneous human melanoma tumors in athymic nude mice. Clin. Canc. Res. 2006, 12, 7437–7443. [Google Scholar]
- Moayeri, M.; Haines, D.; Young, H.A.; Leppla, S.H. Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J. Clin. Invest. 2003, 112, 670–682. [Google Scholar]
- Turpeenniemi-Hujanen, T. Gelatinases (MMP-2 and -9) and their natural inhibitors as prognostic indicators in solid cancers. Biochimie 2010, 87, 287–297. [Google Scholar]
- Kurizaki, T.; Toi, M.; Tominaga, T. Relationship between matrix metalloproteinase expression and tumor angiogenesis in human breast carcinoma. Oncol. Rep. 1998, 5, 673–677. [Google Scholar]
- Rundhaug, J.E. Matrix Metalloproteinases, Angiogenesis, and Cance. Clin. Canc. Res. 2003, 9, 551–554. [Google Scholar]
- Rundhaug, J.E. Matrix metalloproteinases and angiogenesis. J. Cell. Mol. Med. 2005, 9, 267–285. [Google Scholar]
- Alfano, R.W.; Leppla, S.H.; Liu, S.; Bugge, T.H.; Duesbery, N.S.; Frankel, A.E. Potent inhibition of tumor angiogenesis by the matrix metalloproteinase-activated anthrax lethal toxin: Implications for broad anti-tumor efficacy. Cell Cycle 2008, 7, 745–749. [Google Scholar]
- Syrovets, T.; Simmet, T. Novel aspects and new roles for the serine protease plasmin. Cell. Mol. Life Sci. 2004, 61, 873–885. [Google Scholar]
- Petersen, L.C. Kinetics of reciprocal pro-urokinase/plasminogen activation—Stimulation by a template formed by the urokinase receptor bound to poly(D-lysine). Eur. J. Biochem. 1997, 245, 316–323. [Google Scholar]
- Duffy, M.J.; Duggan, C. The urokinase plasminogen activator system: A rich source of tumour markers for the individualised management of patients with cancer. Clin. Biochem. 2004, 37, 541–548. [Google Scholar]
- Kilpatrick, L.M.; Harris, R.L.; Owen, K.A.; Bass, R.; Ghorayeb, C.; Bar-Or, A.; Ellis, V. Initiation of plasminogen activation on the surface of monocytes expressing the type II transmembrane serine protease matriptase. Blood 2006, 108, 2616–2623. [Google Scholar]
- Moran, P.; Li, W.; Fan, B.; Vij, R.; Eigenbrot, C.; Kirchhofer, D. Pro-urokinase-type plasminogen activator is a substrate for hepsin. J. Biol. Chem. 2006, 281, 30439–30446. [Google Scholar]
- McMahon, B.; Kwaan, H.C. The plasminogen activator system and cancer. Pathophysiol. Haemost. Thromb. 2008, 36, 184–194. [Google Scholar]
- Andronicos, N.M.; Chen, E.I.; Baik, N.; Bai, H.; Parmer, C.M.; Kiosses, W.B.; Kamps, M.P.; Yates, J.R., III; Parmer, R.J.; Miles, L.A. Proteomics-based discovery of a novel, structurally unique, and developmentally regulated plasminogen receptor, Plg-RKT, a major regulator of cell surface plasminogen activation. Blood 2010, 115, 1319–1330. [Google Scholar]
- Strickland, D.K. A new plasminogen receptor. Blood 2010, 115, 1315–1316. [Google Scholar]
- Andreasen, P.A.; Egelund, R.; Petersen, H.H. The plasminogen activation system in tumor growth, invasion, and metastasis. Cell. Mol. Life. Sci. 2000, 57, 25–40. [Google Scholar]
- Dass, K.; Ahmad, A.; Azmi, A.S.; Sarkar, S.H.; Sarkar, F.H. Evolving role of uPA/uPAR system in human cancers. Canc. Treat. Rev. 2008, 34, 122–136. [Google Scholar]
- Cunningham, K.; Lacy, D.B.; Mogridge, J.; Collier, R.J. Mapping the lethal factor and edema factor binding sites on oligomeric anthrax protective antigen. Proc. Nat.l Acad. Sci. USA 2002, 99, 7049–7053. [Google Scholar]
- Mogridge, J.; Cunningham, K.; Lacy, D.B.; Mourez, M.; Collier, R.J. The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proc. Natl. Acad. Sci. USA 2002, 99, 7045–7048. [Google Scholar]
- Patick, A.K.; Potts, K.E. Protease inhibitors as antiviral agents. Clin. Microbiol. Rev. 1998, 11, 614–627. [Google Scholar]
- Tong, L. Viral proteases. Chem. Rev. 2002, 102, 4609–4626. [Google Scholar]
- Hsu, J.T.; Wang, H.C.; Chen, G.W.; Shih, S.R. Antiviral drug discovery targeting to viral proteases. Curr. pharm. Des. 2006, 12, 1301–1314. [Google Scholar]
- Varshavsky, A. The N-end rule. Cold Spring Harb. Symp. Quant. Biol. 1995, 60, 461–478. [Google Scholar]
- Varshavsky, A. The N-end rule: Functions, mysteries, uses. Proc. Natl. Acad. Sci. USA 1996, 93, 12142–12149. [Google Scholar]
- Varshavsky, A. The N-end rule pathway of protein degradation. Genes Cells 1997, 2, 13–28. [Google Scholar]
- Varshavsky, A. The N-end rule and regulation of apoptosis. Nat. Cell Biol. 2003, 5, 373–376. [Google Scholar]
- Mogk, A.; Schmidt, R.; Bukau, B. The N-end rule pathway for regulated proteolysis: Prokaryotic and eukaryotic strategies. Trends Cell Biol. 2007, 17, 165–172. [Google Scholar]
- Ravid, T.; Hochstrasser, M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell. Biol. 2008, 9, 679–689. [Google Scholar]
- Falnes, P.O.; Olsnes, S. Modulation of the intracellular stability and toxicity of diphtheria toxin through degradation by the N-end rule pathway. EMBO J. 1998, 17, 615–625. [Google Scholar]
- Wesche, J.; Elliott, J.L.; Falnes, P.O.; Olsnes, S.; Collier, R.J. Characterization of Membrane Translocation by Anthrax Protective Antigen. Biochemistry 1998, 37, 15737–15746. [Google Scholar]
- Kaplan, A.H.; Swanstrom, R. Human immunodeficiency virus type 1 Gag proteins are processed in two cellular compartments. Proc. Natl.Acad. Sci. USA 1991, 88, 4528–4532. [Google Scholar]
- Riviere, Y.; Blank, V.; Kourilsky, P.; Israel, A. Processing of the precursor of NF-kappa B by the HIV-1 protease during acute infection. Nature 1991, 350, 625–626. [Google Scholar]
- Adams, L.D.; Tomasselli, A.G.; Robbins, P.; Moss, B.; Heinrikson, R.L. HIV-1 protease cleaves actin during acute infection of human T-lymphocytes. AIDS Res. Hum. Retroviruses 1992, 8, 291–295. [Google Scholar]
- Konvalinka, J.; Litterst, M.A.; Welker, R.; Kottler, H.; Rippmann, F.; Heuser, A.M.; Krausslich, H.G. An active-site mutation in the human immunodeficiency virus type 1 proteinase (PR) causes reduced PR activity and loss of PR-mediated cytotoxicity without apparent effect on virus maturation and infectivity. J. Virol. 1995, 69, 7180–7186. [Google Scholar]
- Nie, Z.; Bren, G.D.; Vlahakis, S.R.; Schimnich, A.A.; Brenchley, J.M.; Trushin, S.A.; Warren, S.; Schnepple, D.J.; Kovacs, C.M.; Loutfy, M.R.; Douek, D.C.; Badley, A.D. Human immunodeficiency virus type 1 protease cleaves procaspase 8 in vivo. J. Virol. 2007, 81, 6947–6956. [Google Scholar]
- Gump, J.M.; Dowdy, S.F. TAT transduction: The molecular mechanism and therapeutic prospects. Trends Mol. Med. 2007, 13, 443–448. [Google Scholar]
- Frankel, A.E.; Tagge, E.P.; Willingham, M.C. Clinical trials of targeted toxins. Semin. Canc. Biol. 1995, 6, 307–317. [Google Scholar]
- Baluna, R.; Vitetta, E.S. Vascular leak syndrome: A side effect of immunotherapy. Immunopharmacology 1997, 37, 117–132. [Google Scholar]
- Baluna, R.; Rizo, J.; Gordon, B.E.; Ghetie, V.; Vitetta, E.S. Evidence for a structural motif in toxins and interleukin-2 that may be responsible for binding to endothelial cells and initiating vascular leak syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 3957–3962. [Google Scholar]
- Kreitman, R.J. Taming ricin toxin. Nat. Biotech. 2003, 21, 372–374. [Google Scholar]
- Smallshaw, J.E.; Ghetie, V.; Rizo, J.; Fulmer, J.R.; Trahan, L.L.; Ghetie, M.-A.; Vitetta, E.S. Genetic engineering of an immunotoxin to eliminate pulmonary vascular leak in mice. Nat. Biotech. 2003, 21, 387–391. [Google Scholar]
- Zhang, Q.; Chen, G.; Liu, X.; Qian, Q. Monoclonal antibodies as therapeutic agents in oncology and antibody gene therapy. Cell Res. 2007, 17, 89–99. [Google Scholar]
- da Silva, F.A.; Corte-Real, S.; Goncalves, J. Recombinant Antibodies as Therapeutic Agents: Pathways for Modeling New Biodrugs. BioDrugs 2008, 22, 301–314. [Google Scholar]
- Tsutsumi, Y.; Onda, M.; Nagata, S.; Lee, B.; Kreitman, R.J.; Pastan, I. Site-specific chemical modification with polyethylene glycol of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) improves antitumor activity and reduces animal toxicity and immunogenicity. Proc. Natl. Acad. Sci. USA 2000, 97, 8548–8553. [Google Scholar]
- Hu, R.-G.; Zhai, Q.-W.; He, W.-J.; Mei, L.; Liu, W.-Y. Bioactivities of ricin retained and its immunoreactivity to anti-ricin polyclonal antibodies alleviated through pegylation. Int. J. Biochem. Cell Biol. 2002, 34, 396–402. [Google Scholar]
- Filpula, D.; Yang, K.; Basu, A.; Hassan, R.; Xiang, L.; Zhang, Z.; Wang, M.; Wang, Q.-c.; Ho, M.; Beers, R.; Zhao, H.; Peng, P.; Zhou, J.; Li, X.; Petti, G.; Janjua, A.; Liu, J.; Wu, D.; Yu, D.; Zhang, Z.; Longley, C.; FitzGerald, D.; Kreitman, R.J.; Pastan, I. Releasable PEGylation of Mesothelin Targeted Immunotoxin SS1P Achieves Single Dosage Complete Regression of a Human Carcinoma in Mice. Bioconjug. Chem. 2007, 18, 773–784. [Google Scholar]
- Pai, L.H.; FitzGerald, D.J.; Tepper, M.; Schacter, B.; Spitalny, G.; Pastan, I. Inhibition of Antibody Response to Pseudomonas Exotoxin and an Immunotoxin Containing Pseudomonas Exotoxin by 15-Deoxyspergualin in Mice. Canc. Res. 1990, 50, 7750–7753. [Google Scholar]
- Siegall, C.B.; Haggerty, H.G.; Warner, G.L.; Chace, D.; Mixan, B.; Linsley, P.S.; Davidson, T. Prevention of immunotoxin-induced immunogenicity by coadministration with CTLA4Ig enhances antitumor efficacy. J. Immunol. 1997, 159, 5168–5173. [Google Scholar]
- Gelber, E.E.; Vitetta, E.S. Effect of immunosuppressive agents on the immunogenicity and efficacy of an immunotoxin in mice. Clin. Canc. Res. 1998, 4, 1297–1304. [Google Scholar]
- Onda, M.; Nagata, S.; FitzGerald, D.J.; Beers, R.; Fisher, R.J.; Vincent, J.J.; Lee, B.; Nakamura, M.; Hwang, J.; Kreitman, R.J.; Hassan, R.; Pastan, I. Characterization of the B cell epitopes associated with a truncated form of Pseudomonas exotoxin (PE38) used to make immunotoxins for the treatment of cancer patients. J. Immunol. 2006, 177, 8822–8834. [Google Scholar]
- Onda, M.; Beers, R.; Xiang, L.; Nagata, S.; Wang, Q.C.; Pastan, I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc. Natl. Acad. Sci. USA 2008, 105, 11311–11316. [Google Scholar]
- Stish, B.J.; Oh, S.; Chen, H.; Dudek, A.Z.; Kratzke, R.A.; Vallera, D.A. Design and modification of EGF4KDEL 7Mut, a novel bispecific ligand-directed toxin, with decreased immunogenicity and potent anti-mesothelioma activity. Br. J. Canc. 2009, 101, 1114–1123. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shapira, A.; Benhar, I. Toxin-Based Therapeutic Approaches. Toxins 2010, 2, 2519-2583. https://doi.org/10.3390/toxins2112519
Shapira A, Benhar I. Toxin-Based Therapeutic Approaches. Toxins. 2010; 2(11):2519-2583. https://doi.org/10.3390/toxins2112519
Chicago/Turabian StyleShapira, Assaf, and Itai Benhar. 2010. "Toxin-Based Therapeutic Approaches" Toxins 2, no. 11: 2519-2583. https://doi.org/10.3390/toxins2112519