Developmental Programming of Obesity and Liver Metabolism by Maternal Perinatal Nutrition Involves the Melanocortin System

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Design
2.2. Liver Histology
2.3. Plasma Analysis
2.4. mRNA Extraction and Real-Time qPCR
2.5. Statistical Analysis
3. Results
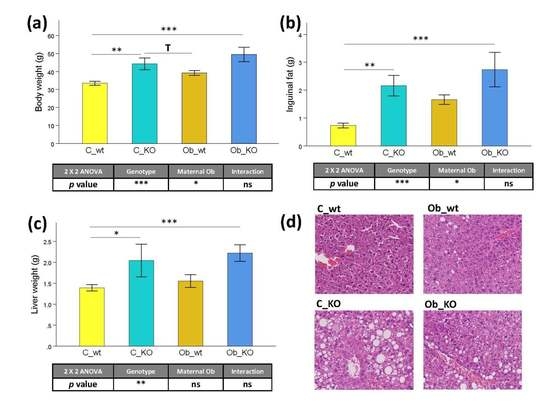
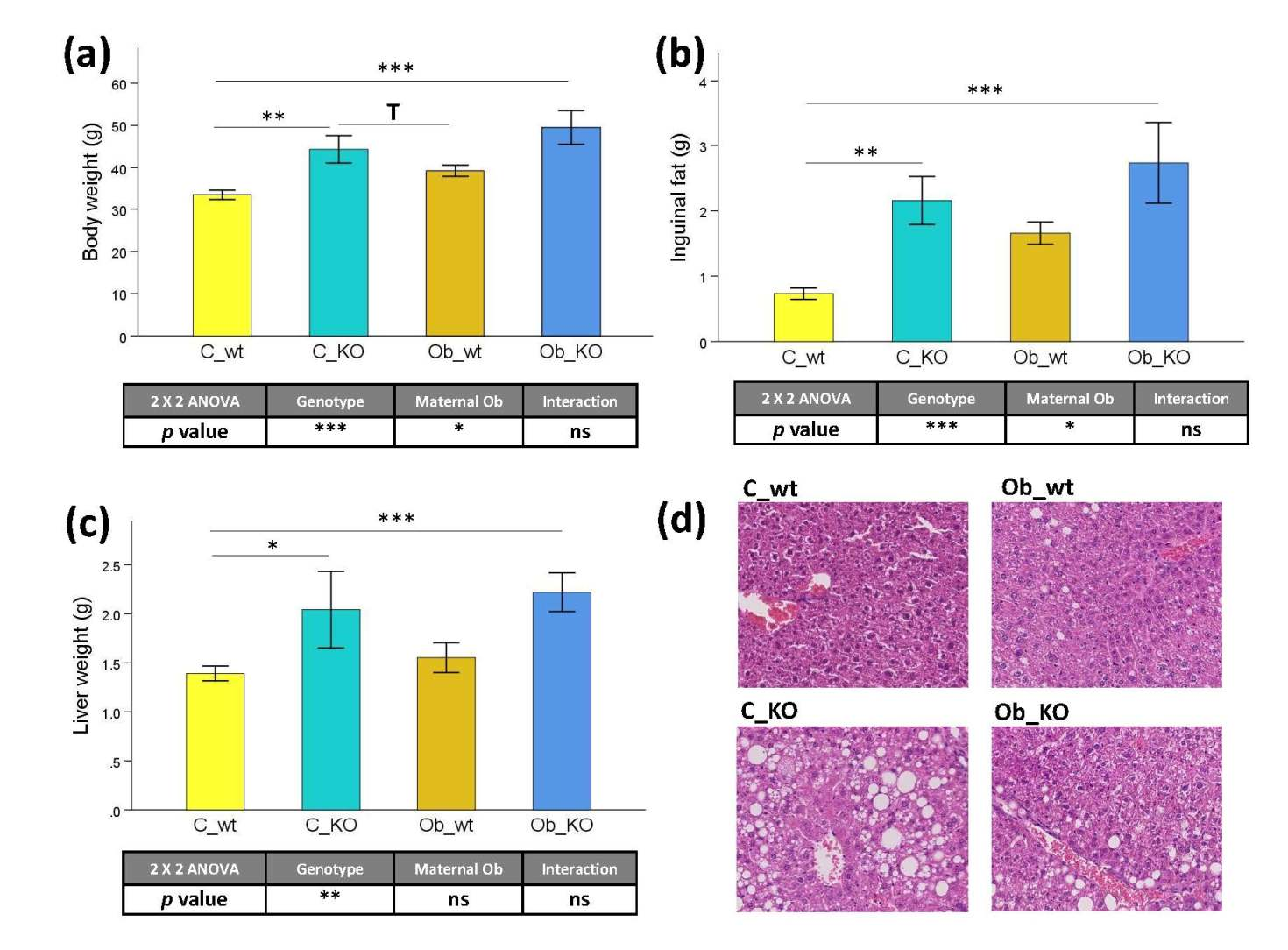
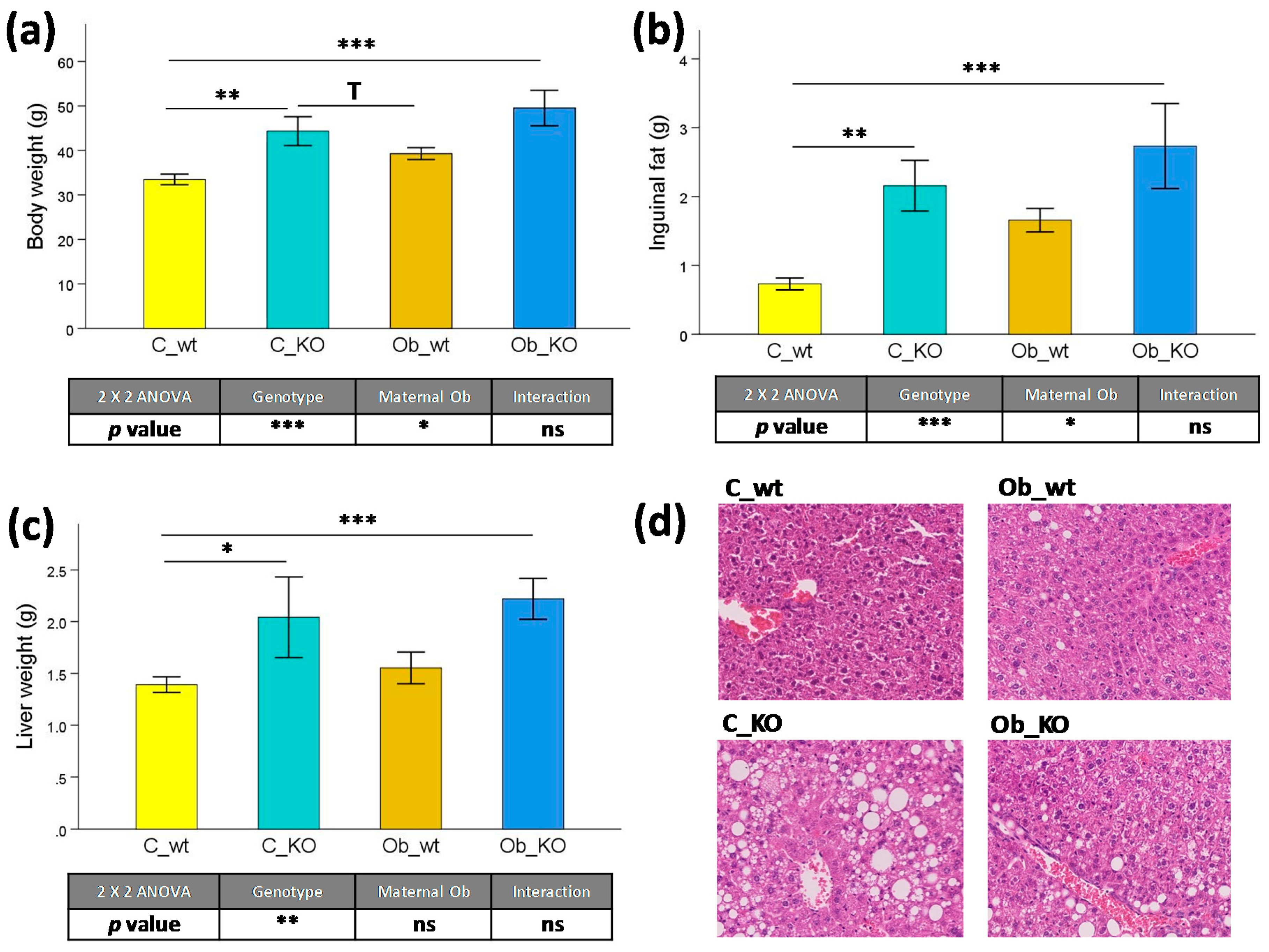
3.1. Phenotypic and Histological Characteristics
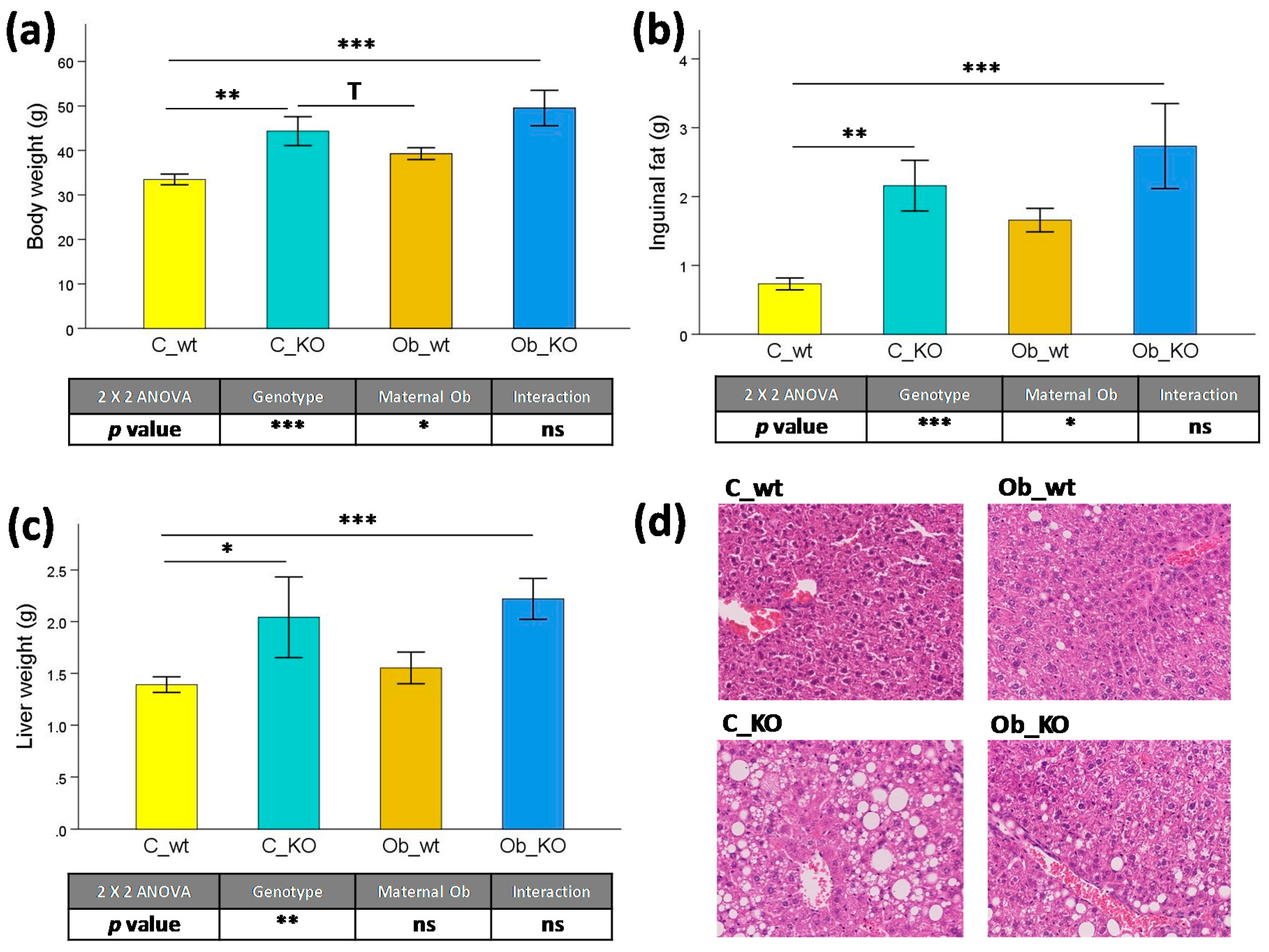
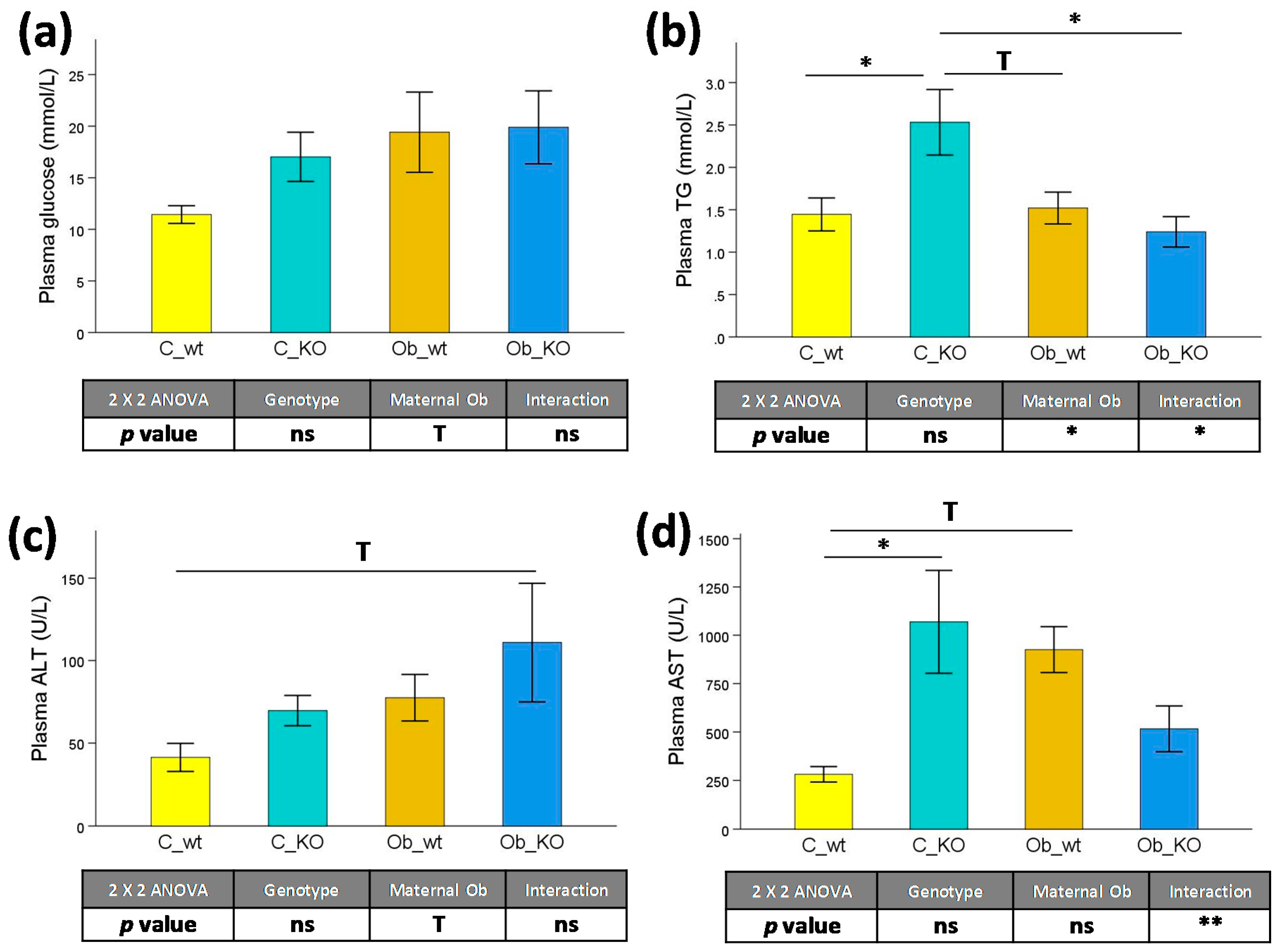
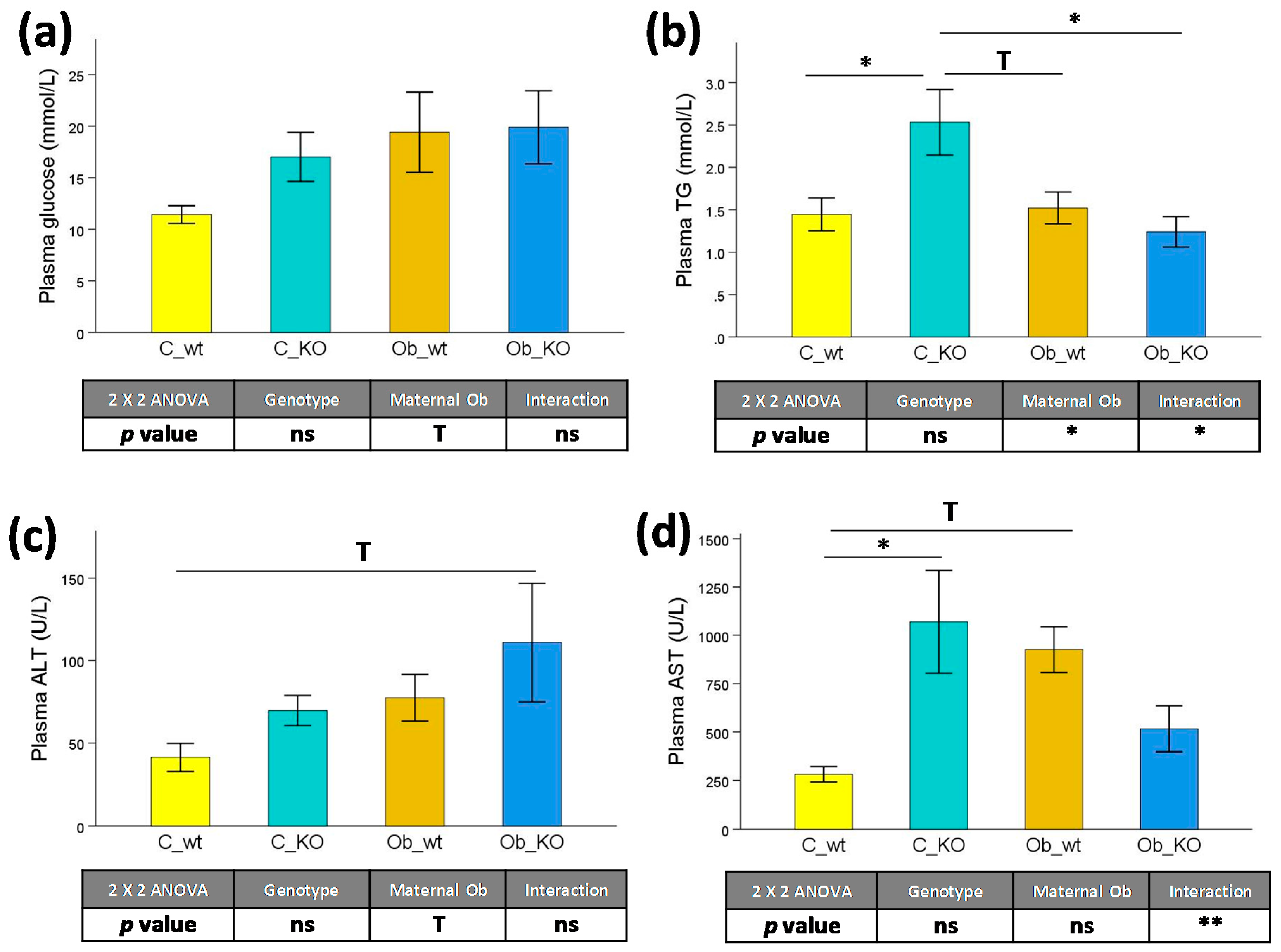
3.2. Plasma Biochemical Features
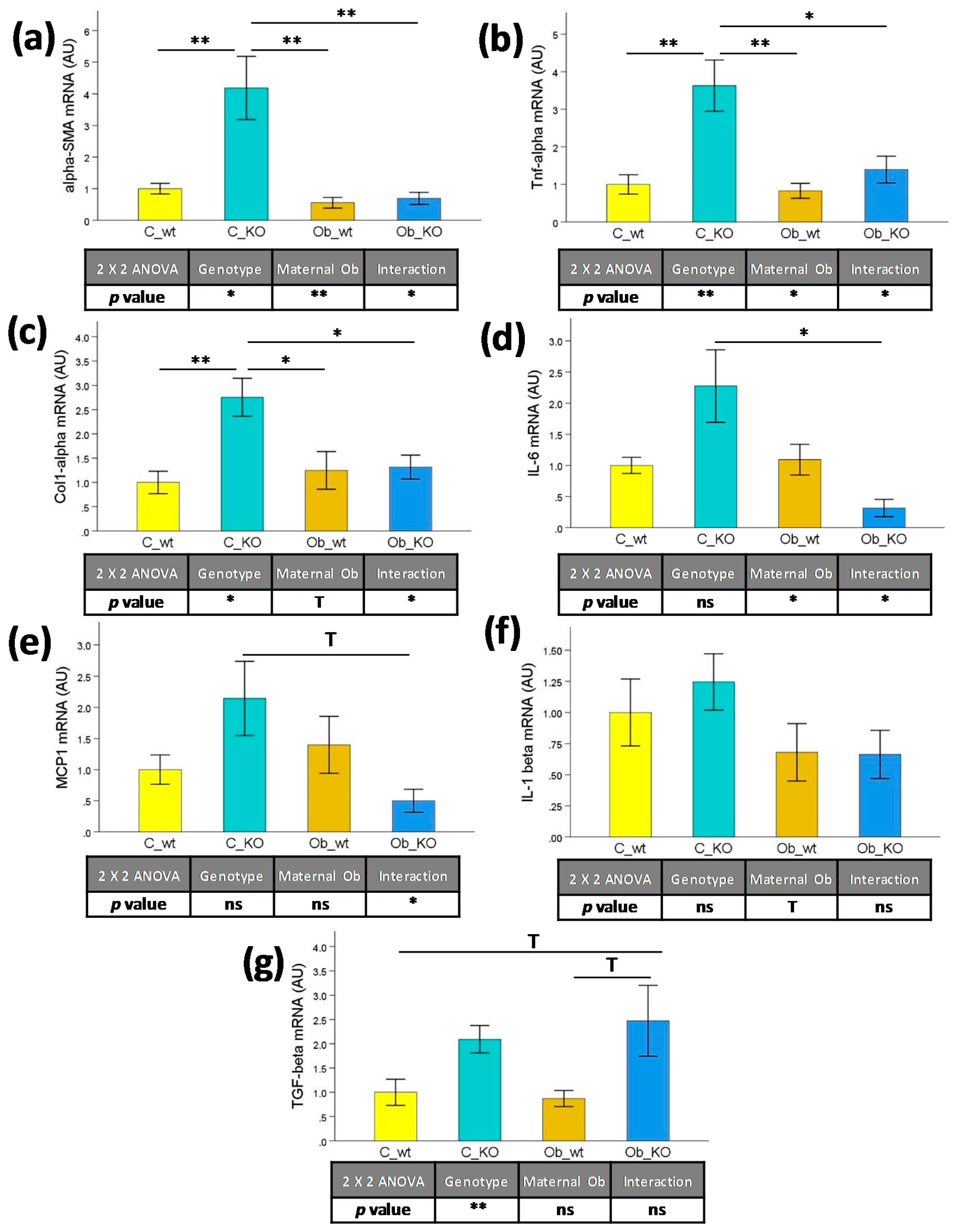
3.3. Hepatic Transcriptomic Profile
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gonzalez-Muniesa, P.; Martinez-Gonzalez, M.A.; Hu, F.B.; Despres, J.P.; Matsuzawa, Y.; Loos, R.J.F.; Moreno, L.A.; Bray, G.A.; Martinez, J.A. Obesity. Nat. Rev. Dis. Primers 2017, 3, 17034. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Obesity and Overweight. Available online: Http://www.who.int/mediacentre/factsheets/fs311/en/ (accessed on 19 July 2017).
- Fontaine, K.R.; Redden, D.T.; Wang, C.; Westfall, A.O.; Allison, D.B. Years of life lost due to obesity. JAMA 2003, 289, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; McPherson, K.; Marsh, T.; Gortmaker, S.L.; Brown, M. Health and economic burden of the projected obesity trends in the USA and the UK. Lancet 2011, 378, 815–825. [Google Scholar] [CrossRef]
- Shalitin, S.; Battelino, T.; Moreno, L.A. Obesity, Metabolic Syndrome and Nutrition. World Rev. Nutr. Diet. 2016, 114, 21–49. [Google Scholar] [PubMed]
- Dietrich, P.; Hellerbrand, C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Temple, J.L.; Cordero, P.; Li, J.; Nguyen, V.; Oben, J.A. A Guide to Non-Alcoholic Fatty Liver Disease in Childhood and Adolescence. Int. J. Mol. Sci. 2016, 17, 947. [Google Scholar] [CrossRef] [PubMed]
- Vinciguerra, M. Protein intake, chronic liver diseases, and hepatocellular carcinoma. Hepatology 2015, 61, 730. [Google Scholar] [CrossRef] [PubMed]
- Poston, L.; Caleyachetty, R.; Cnattingius, S.; Corvalan, C.; Uauy, R.; Herring, S.; Gillman, M.W. Preconceptional and maternal obesity: Epidemiology and health consequences. Lancet Diabetes Endocrinol. 2016, 4, 1025–1036. [Google Scholar] [CrossRef]
- Martinez, J.A.; Cordero, P.; Campion, J.; Milagro, F.I. Interplay of early-life nutritional programming on obesity, inflammation and epigenetic outcomes. Proc. Nutr. Soc. 2012, 71, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Mouralidarane, A.; Soeda, J.; Sugden, D.; Bocianowska, A.; Carter, R.; Ray, S.; Saraswati, R.; Cordero, P.; Novelli, M.; Fusai, G.; et al. Maternal obesity programs offspring Non-Alcoholic Fatty Liver Disease through disruption of 24-h rhythms in mice. Int. J. Obes. 2015, 39, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Oben, J.A.; Mouralidarane, A.; Samuelsson, A.M.; Matthews, P.J.; Morgan, M.L.; McKee, C.; Soeda, J.; Fernandez-Twinn, D.S.; Martin-Gronert, M.S.; Ozanne, S.E.; et al. Maternal obesity during pregnancy and lactation programs the development of offspring Non-Alcoholic Fatty Liver Disease in mice. J. Hepatol. 2010, 52, 913–920. [Google Scholar] [CrossRef] [PubMed]
- Soeda, J.; Cordero, P.; Li, J.; Mouralidarane, A.; Asilmaz, E.; Ray, S.; Nguyen, V.; Carter, R.; Novelli, M.; Vinciguerra, M.; et al. Hepatic rhythmicity of endoplasmic reticulum stress is disrupted in perinatal and adult mice models of high-fat diet-induced obesity. Int. J. Food Sci. Nutr. 2017, 68, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Pazienza, V.; Panebianco, C.; Rappa, F.; Memoli, D.; Borghesan, M.; Cannito, S.; Oji, A.; Mazza, G.; Tamburrino, D.; Fusai, G.; et al. Histone macroH2A1.2 promotes metabolic health and leanness by inhibiting adipogenesis. Epigenet. Chromatin 2016, 9, 45. [Google Scholar] [CrossRef] [PubMed]
- Goni, L.; Milagro, F.I.; Cuervo, M.; Martinez, J.A. Single-nucleotide polymorphisms and DNA methylation markers associated with central obesity and regulation of body weight. Nutr. Rev. 2014, 72, 673–690. [Google Scholar] [CrossRef] [PubMed]
- Loos, R.J.; Lindgren, C.M.; Li, S.; Wheeler, E.; Zhao, J.H.; Prokopenko, I.; Inouye, M.; Freathy, R.M.; Attwood, A.P.; Beckmann, J.S.; et al. Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat. Genet. 2008, 40, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Xi, B.; Chandak, G.R.; Shen, Y.; Wang, Q.; Zhou, D. Association between common polymorphism near the MC4R gene and obesity risk: A systematic review and meta-analysis. PLoS ONE 2012, 7, e45731. [Google Scholar] [CrossRef] [PubMed]
- Xi, B.; Takeuchi, F.; Chandak, G.R.; Kato, N.; Pan, H.W.; Consortium, A.-T.D.; Zhou, D.H.; Pan, H.Y.; Mi, J. Common polymorphism near the MC4R gene is associated with type 2 diabetes: Data from a meta-analysis of 123,373 individuals. Diabetologia 2012, 55, 2660–2666. [Google Scholar] [CrossRef] [PubMed]
- Krashes, M.J.; Lowell, B.B.; Garfield, A.S. Melanocortin-4 receptor-regulated energy homeostasis. Nat. Neurosci. 2016, 19, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, A.M. New perspectives on the origin of hypertension; the role of the hypothalamic melanocortin system. Exp. Physiol. 2014, 99, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Tabachnik, T.; Kisliouk, T.; Marco, A.; Meiri, N.; Weller, A. Thyroid Hormone-Dependent Epigenetic Regulation of Melanocortin 4 Receptor Levels in Female Offspring of Obese Rats. Endocrinology 2017, 158, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Pindjakova, J.; Sartini, C.; Lo Re, O.; Rappa, F.; Coupe, B.; Lelouvier, B.; Pazienza, V.; Vinciguerra, M. Gut Dysbiosis and Adaptive Immune Response in Diet-induced Obesity vs. Systemic Inflammation. Front. Microbiol. 2017, 8, 1157. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, A.S.; Mullier, A.; Maicas, N.; Oosterhuis, N.R.; Eun Bae, S.; Novoselova, T.V.; Chan, L.F.; Pombo, J.M.; Taylor, P.D.; Joles, J.A.; et al. Central role for melanocortin-4 receptors in offspring hypertension arising from maternal obesity. Proc. Natl. Acad. Sci. USA 2016, 113, 12298–12303. [Google Scholar] [CrossRef] [PubMed]
- Rappa, F.; Greco, A.; Podrini, C.; Cappello, F.; Foti, M.; Bourgoin, L.; Peyrou, M.; Marino, A.; Scibetta, N.; Williams, R.; et al. Immunopositivity for histone macroH2A1 isoforms marks steatosis-associated hepatocellular carcinoma. PLoS ONE 2013, 8, e54458. [Google Scholar] [CrossRef]
- Balthasar, N.; Dalgaard, L.T.; Lee, C.E.; Yu, J.; Funahashi, H.; Williams, T.; Ferreira, M.; Tang, V.; McGovern, R.A.; Kenny, C.D.; et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 2005, 123, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Kooijman, S.; Boon, M.R.; Parlevliet, E.T.; Geerling, J.J.; van de Pol, V.; Romijn, J.A.; Havekes, L.M.; Meurs, I.; Rensen, P.C. Inhibition of the central melanocortin system decreases brown adipose tissue activity. J. Lipid Res. 2014, 55, 2022–2032. [Google Scholar] [CrossRef] [PubMed]
- Barb, C.R.; Hausman, G.J.; Rekaya, R.; Lents, C.A.; Lkhagvadorj, S.; Qu, L.; Cai, W.; Couture, O.P.; Anderson, L.L.; Dekkers, J.C.; et al. Gene expression in hypothalamus, liver, and adipose tissues and food intake response to melanocortin-4 receptor agonist in pigs expressing melanocortin-4 receptor mutations. Physiol. Genomics 2010, 41, 254–268. [Google Scholar] [CrossRef] [PubMed]
- Malik, I.A.; Triebel, J.; Posselt, J.; Khan, S.; Ramadori, P.; Raddatz, D.; Ramadori, G. Melanocortin receptors in rat liver cells: Change of gene expression and intracellular localization during acute-phase response. Histochem. Cell Biol. 2012, 137, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Alwahsh, S.M.; Ramadori, G.; Kollmar, O.; Slotta, J.E. Upregulation of hepatic melanocortin 4 receptor during rat liver regeneration. J. Surg. Res. 2016, 203, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.C.; Elliott, P.; Zabaneh, D.; Zhang, W.; Li, Y.; Froguel, P.; Balding, D.; Scott, J.; Kooner, J.S. Common genetic variation near MC4R is associated with waist circumference and insulin resistance. Nat. Genet. 2008, 40, 716–718. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Suganami, T.; Nakagawa, N.; Tanaka, M.; Yamamoto, Y.; Kamei, Y.; Terai, S.; Sakaida, I.; Ogawa, Y. Melanocortin 4 receptor-deficient mice as a novel Mouse model of nonalcoholic steatohepatitis. Am. J. Pathol. 2011, 179, 2454–2463. [Google Scholar] [CrossRef] [PubMed]
- Soeda, J.; Mouralidarane, A.; Cordero, P.; Li, J.; Nguyen, V.; Carter, R.; Kapur, S.R.; Pombo, J.; Poston, L.; Taylor, P.D.; et al. Maternal obesity alters endoplasmic reticulum homeostasis in offspring pancreas. J. Physiol. Biochem. 2016, 72, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Mouralidarane, A.; Soeda, J.; Visconti-Pugmire, C.; Samuelsson, A.M.; Pombo, J.; Maragkoudaki, X.; Butt, A.; Saraswati, R.; Novelli, M.; Fusai, G.; et al. Maternal obesity programs offspring nonalcoholic fatty liver disease by innate immune dysfunction in mice. Hepatology 2013, 58, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Saad, S.; Tan, Y.; Pollock, C.; Chen, H. Maternal high-fat diet induces metabolic stress response disorders in offspring hypothalamus. J. Mol. Endocrinol. 2017, 59, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Marco, A.; Kisliouk, T.; Tabachnik, T.; Meiri, N.; Weller, A. Overweight and CpG methylation of the Pomc promoter in offspring of high-fat-diet-fed dams are not “reprogrammed” by regular chow diet in rats. FASEB J. 2014, 28, 4148–4157. [Google Scholar] [CrossRef] [PubMed]
- Marco, A.; Kisliouk, T.; Tabachnik, T.; Weller, A.; Meiri, N. DNA CpG Methylation (5-Methylcytosine) and Its Derivative (5-Hydroxymethylcytosine) Alter Histone Posttranslational Modifications at the Pomc Promoter, Affecting the Impact of Perinatal Diet on Leanness and Obesity of the Offspring. Diabetes 2016, 65, 2258–2267. [Google Scholar] [CrossRef] [PubMed]
- Oben, J.A.; Patel, T.; Mouralidarane, A.; Samuelsson, A.M.; Matthews, P.; Pombo, J.; Morgan, M.; McKee, C.; Soeda, J.; Novelli, M.; et al. Maternal obesity programmes offspring development of non-alcoholic fatty pancreas disease. Biochem. Biophys. Res. Commun. 2010, 394, 24–28. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dietary Composition (g/Kg) | Control | Obesogenic | Condensed Milk |
|---|---|---|---|
| Protein | 144 | 230 | 80 |
| Amino Acids | |||
| Glutamic Acid | 31.7 | 45.5 | 16.6 |
| Proline | 12 | 24.8 | 7.7 |
| Leucine | 9.8 | 20.5 | 7.8 |
| Aspartic Acid | 6.7 | 15.4 | 6 |
| Serine | 5.6 | 12.9 | 4.3 |
| Valine | 6.9 | 14.5 | 5.3 |
| Lysine | 6.6 | 18.9 | 6.3 |
| Glycine | 11.1 | 4.1 | 1.7 |
| Arginine | 9.1 | 8.1 | 2.9 |
| Others | 44.5 | 65.3 | 20.5 |
| Carbohydrates | |||
| Polysaccharides | 500 | 283 | 0 |
| Simple sugars | 40 | 105 | 550 |
| Cellulose | 43.2 | 61.7 | |
| Hemicellulose | 101.7 | ||
| Lipid | 27 | 226 | 90 |
| Saturated Fatty Acids | 5.1 | 76.2 | 59.4 |
| Monounsaturated Fatty Acids | 8.8 | 85.2 | 24.3 |
| Polyunsaturated Fatty Acids | 8.8 | 39.1 | 3.4 |
| Mineral content | 35 | ||
| Vitamin content | 4.1 | ||
| AIN-93G mineral mix | 1.68 | ||
| AIN-93M mineral mix | 43 | ||
| Vitamin mix | 12 | ||
| Energy (kcal/g) | 3.52 | 4.54 | 3.22 |
| Gene | Primer Sequence |
|---|---|
| 18S | sense: AGTCCCTGCCCTTTGTACACA |
| antisense: CGATCCGAGGGCCTCACTA | |
| Gapdh | sense: CGTCCCGTAGACAAAATGGT |
| antisense: TCAATGAAGGGGTCGTTGAT | |
| α-SMA | sense: CTCTTGCTCTGGGCTTCATC |
| antisense: GGCTGTTTTCCCATCCATC | |
| TNF-α | sense: CCACCACGCTCTTCTGTCTA |
| antisense: AGGGTCTGGGCCATAGAACT | |
| Col-1α | sense: GTCCCCGAGGCAGAGATG |
| antisense: GTCCAGGGCCAGATGAAACT | |
| IL6 | sense: TCAATTCCAGAAACCGCTATG |
| antisense: GTCTCCTCTCCGGACTTGTG | |
| MCP1 | sense: CCCACTCACCTGCTGCTACT |
| antisense: TCTGGACCCATTCCTTCTTG | |
| IL-1β | sense: CAACCAACAAGTGATATTCTCCATG |
| antisense: GATCCACACTCTCCAGCTGCA | |
| TGF-β | sense: AAAATCAAGTGTGGAGCAAC |
| antisense: CCACGTGGAGTTTGTTATCT |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordero, P.; Li, J.; Nguyen, V.; Pombo, J.; Maicas, N.; Novelli, M.; Taylor, P.D.; Samuelsson, A.-M.; Vinciguerra, M.; Oben, J.A. Developmental Programming of Obesity and Liver Metabolism by Maternal Perinatal Nutrition Involves the Melanocortin System. Nutrients 2017, 9, 1041. https://doi.org/10.3390/nu9091041
Cordero P, Li J, Nguyen V, Pombo J, Maicas N, Novelli M, Taylor PD, Samuelsson A-M, Vinciguerra M, Oben JA. Developmental Programming of Obesity and Liver Metabolism by Maternal Perinatal Nutrition Involves the Melanocortin System. Nutrients. 2017; 9(9):1041. https://doi.org/10.3390/nu9091041
Chicago/Turabian StyleCordero, Paul, Jiawei Li, Vi Nguyen, Joaquim Pombo, Nuria Maicas, Marco Novelli, Paul D. Taylor, Anne-Maj Samuelsson, Manlio Vinciguerra, and Jude A. Oben. 2017. "Developmental Programming of Obesity and Liver Metabolism by Maternal Perinatal Nutrition Involves the Melanocortin System" Nutrients 9, no. 9: 1041. https://doi.org/10.3390/nu9091041