Ascorbic Acid and the Brain: Rationale for the Use against Cognitive Decline

Abstract
:1. Introduction
2. Ascorbic Acid Biochemistry and Transport
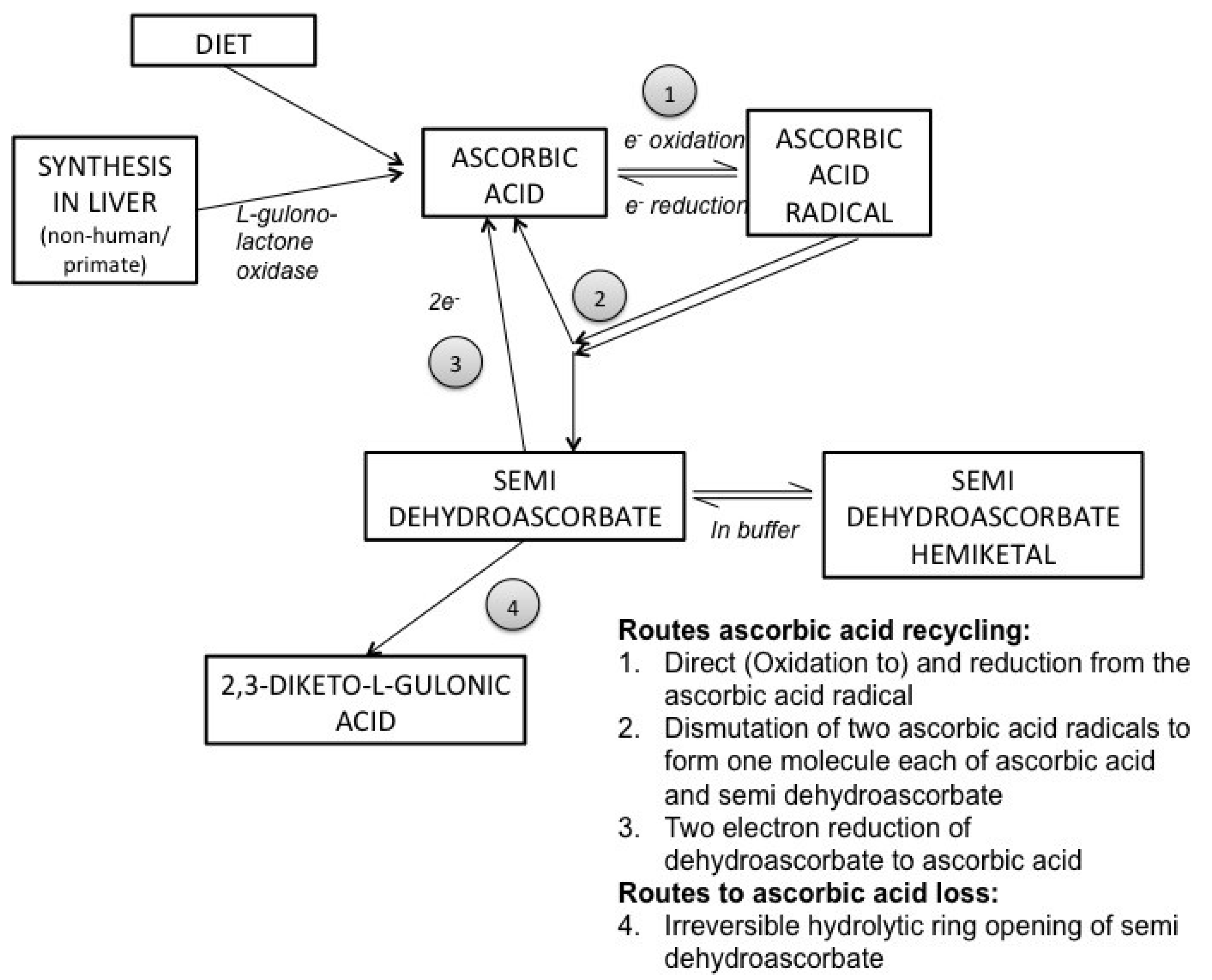

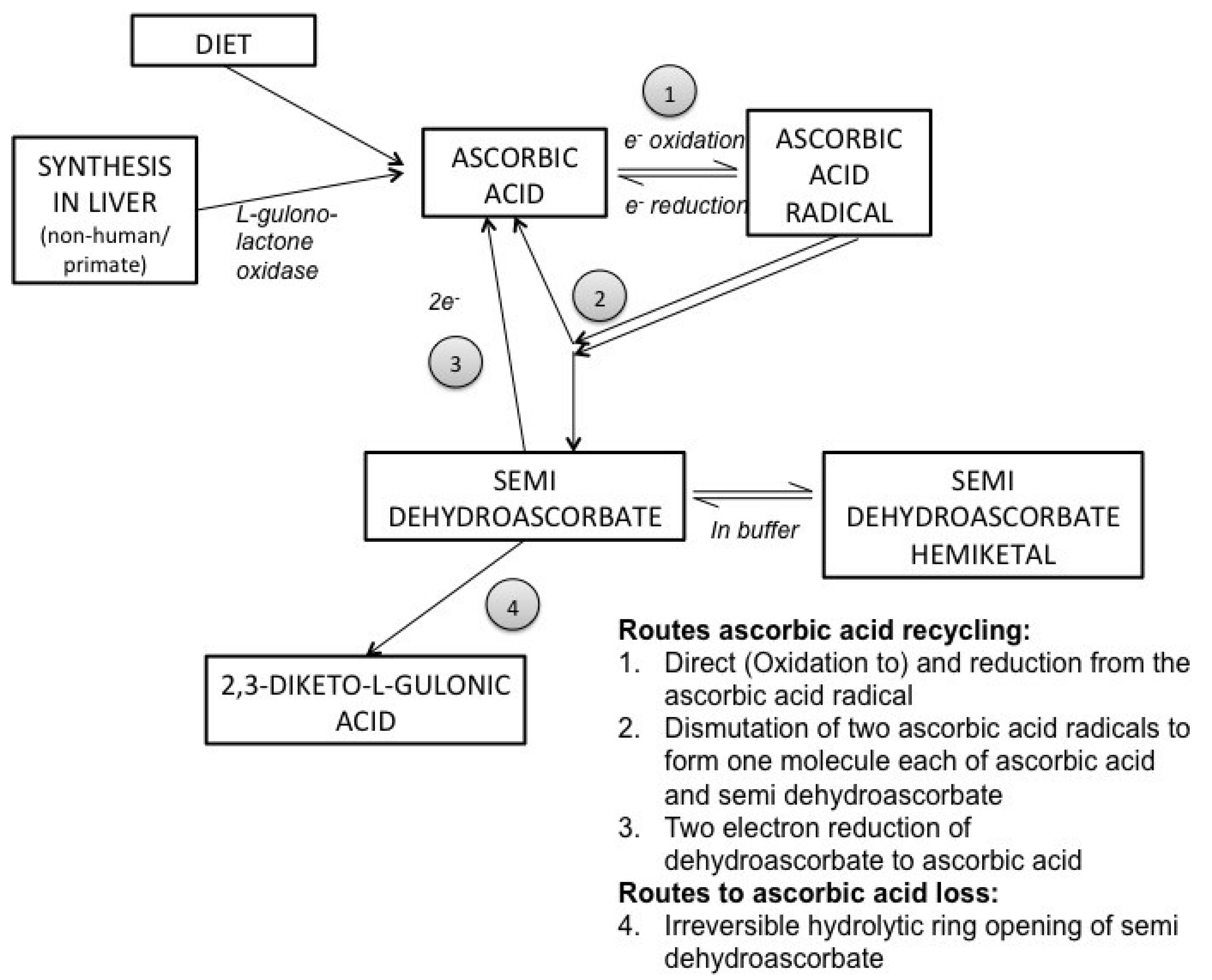
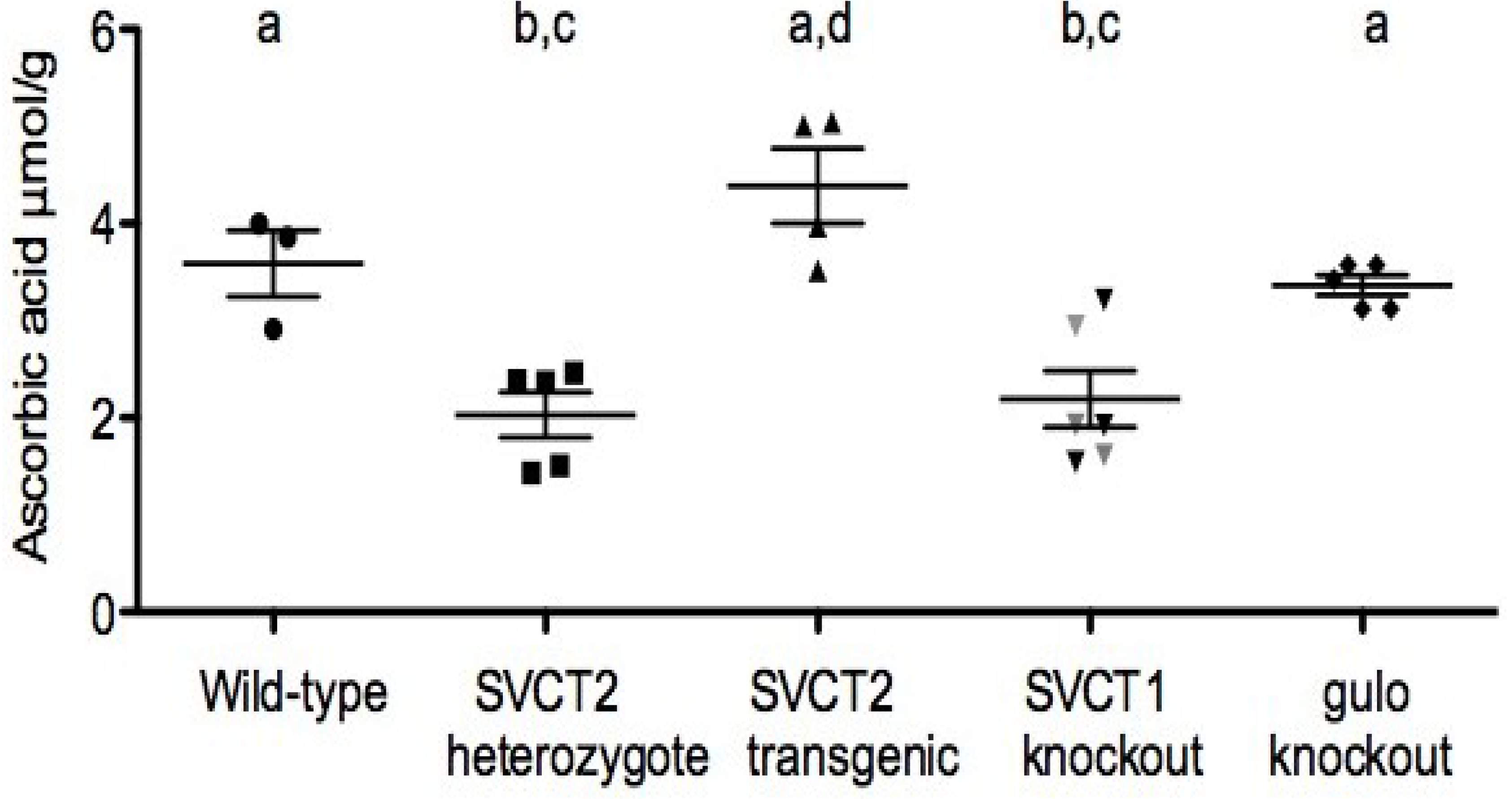
3. Ascorbic Acid Transport and Synthesis: Animal Models
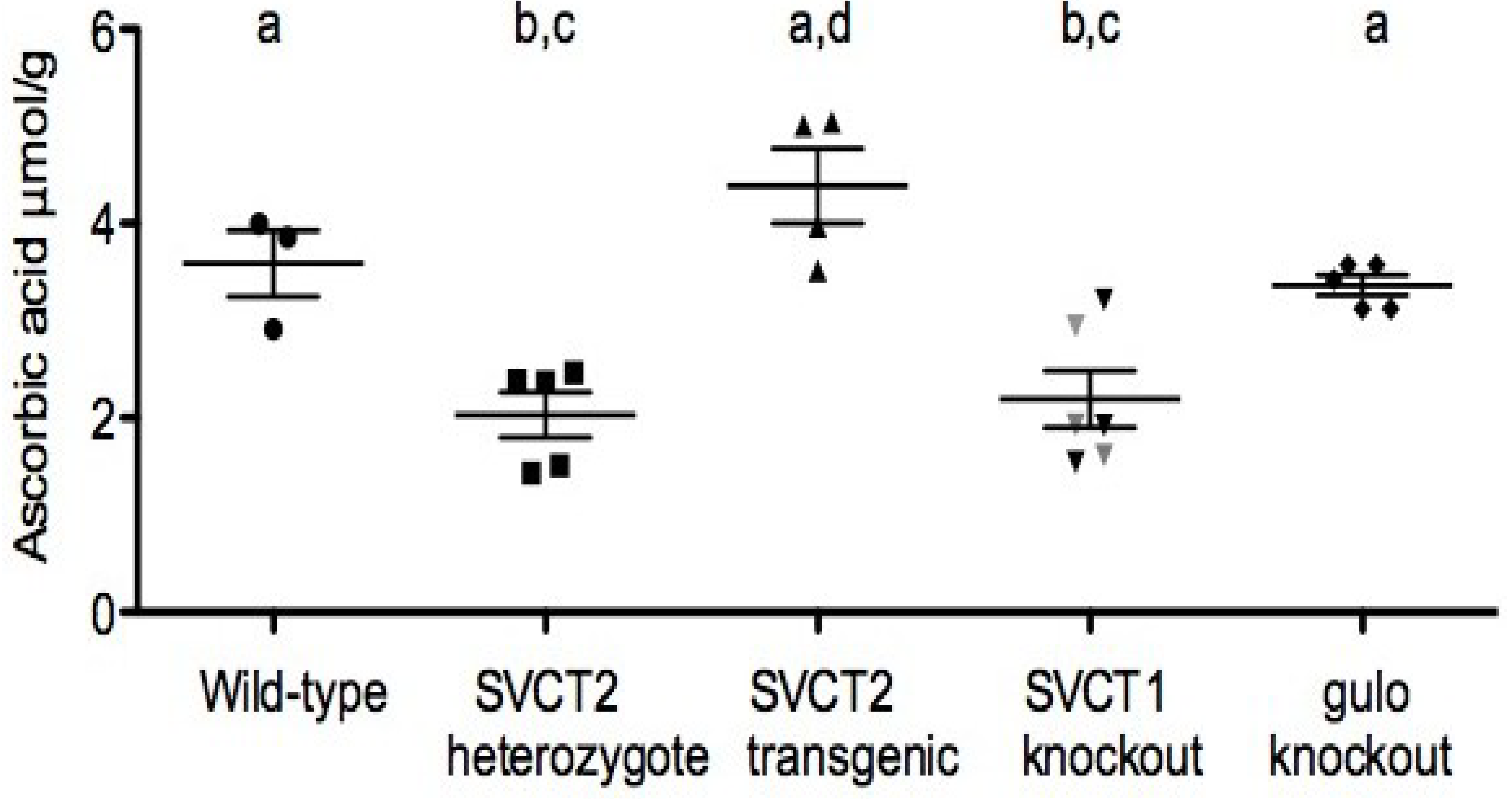
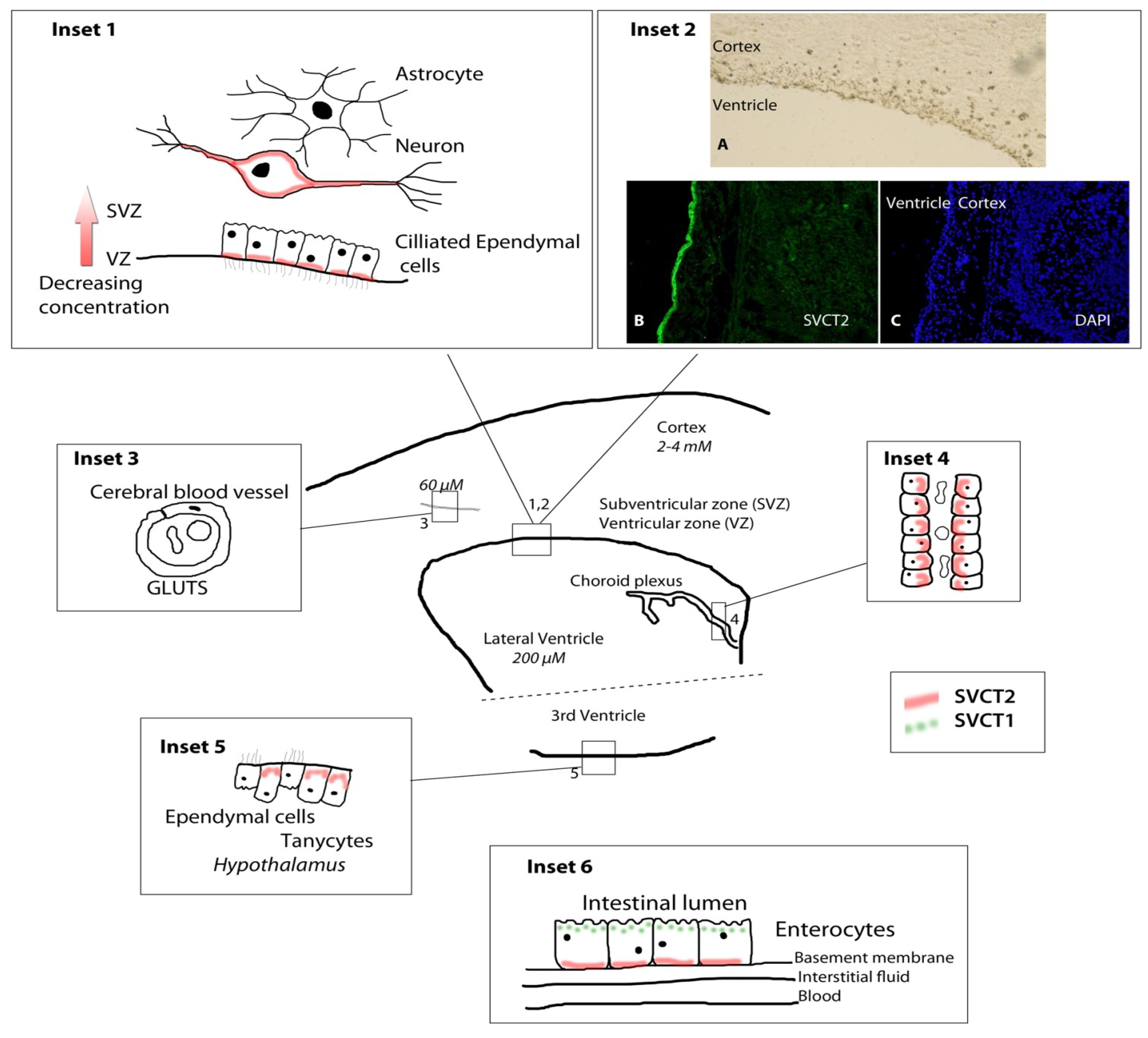
4. Relation of Mice Models to Human Studies
5. Dietary Intake of Ascorbic Acid, Cognition and Alzheimer’s Disease
6. Blood Ascorbic Acid, Cognition and Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
| Authors | Sample | Design and Methods | Conclusion | Association y/n |
|---|---|---|---|---|
| Goodwin, 1983 [123] | 260 dementia-free men/women Age ≥ 60 year Community-dwelling | Cross-sectional Plasma, 3 day diet recall Halstead-Reitan Categories Test, WMS-R delayed | Plasma AA associated with higher verbal memory; elders in the 90th percentile of plasma AA had better calculating ability and delayed recall | y |
| Gale, 1996 [124] | 921 men/women Age ≥ 65 year English-Scottish-Welsh | Cross-sectional Plasma Hodkinson test | Plasma AA ≤ 11.91 µM was associated with 1.6 higher odds of cognitive impairment (95% CI: 1.1–2.3). Less than 27 mg of AA intake per day was associated with 1.7 higher odds for impairment | y |
| Riviere, 1998 [125] | 19 ctls MMSE 24–30 20 severe AD MMSE 0–9 24 mod AD MMSE 10–23 | Cross-sectional Plasma Mini-nutritional assessment | Plasma AA was incrementally lower by degree of cognitive impairment in AD, this observation was not explained by lower intake of AA | y |
| Charlton, 2004 [126] | 93 men/women 50 ctls 43 dementia Age ≥ 65 | Cross-sectional Plasma Mini-nutritional assessment | Plasma AA was lower in subjects with dementia compared to controls, this observation was not explained by lower intake of AA | y |
| Polidori, 2002 [127] | 75 women 40 ctls (85.4 ± 4.4 year) 35 AD (85.9 ± 5.5 y) Age-matched Mean age 85 year | Cross-sectional Plasma NINCDS-ADRDA criteria | Plasma AA was lower in AD than controls (18 ± 6 µM vs. 36 ± 6, p < 0.001) AD subjects had increased plasma lipid peroxidation and less resistance to peroxyl-radical | y |
| Polidori, 2004 [128] | 141 55 ctls 63 AD 23 VD | Cross-sectional Plasma | Plasma AA was lower in AD and VD versus NIE, no differences between AD and VD | y |
| Perrig, 1997 [129] | 442 men/women (n = 312/132) Community-dwelling Age ≥ 65 (mean 75) | Prospective, cross-sectional Plasma Memory, WAIS-R vocabulary test | Plasma AA in 1971 associated with better cognitive performance in 1993; plasma AA associated with better free recall, recognition, and vocabulary, but not priming and working-memory in cross-sectional analysis | y |
7. Cerebrospinal Fluid Ascorbic Acid, Cognition, and Alzheimer’s Disease
| Authors | Sample & Design | Plasma, µM | CSF, µM | CSF:Plasma Ratio | Conclusion | |||
|---|---|---|---|---|---|---|---|---|
| Ctls | AD | Ctls | AD | Ctls | AD | |||
| Paraskevas, 1997 [131] | 32 15 ctls (58 ± 12 year) 17 AD (62 ± 7 year) Men/women Age-gender matched Cross-sectional | 43 ± 13 | 45 ± 19 | 166 ± 45 | 156 ± 38 | 3.6 ± 0.5 | 4.1 ± 1.6 | No differences between AD and controls |
| Quinn, 2003 [132] | 20 10 Ctls (MMSE 29 ± 1), 10 AD (MMSE 19 ± 7) Age-gender matched Mean age 66 y Cross-sectional | 86 ± 39 | 58 ± 42 | 238 ± 48 | 207 ± 64 | 3.1 ± 1.1 | 5.0 ± 2.6 | CSF: plasma AA ratio higher in AD (p = 0.048). Mean plasma and CSF AA were lower in AD, but not significantly. |
| Glaso, 2004 [133] | 38 18 Ctls (MMSE 27), 20 AD (MMSE 16) Women Age-matched (75–85 year) Cross- sectional | 80 ± 28 | 44 ± 25 | 167 ± 23 | 140 ± 37 | 2.1 ± 0.7 | 3.3 ± 1.4 | CSF: serum AA ratio higher in AD versus controls (p = 0.001) but both plasma (p = 0.002) and CSF (p = 0.038) AA were lower in AD. Subjects were well-nourished and without vascular disease |
| Bowman, 2009 [134] | 32 AD (MMSE 19 ± 5) Men/women Mean age 71 year Prospective | 41 ± 30 | 129 ± 52 | 4.0 ± 1.6 | Neither plasma nor CSF AA was predictive, but CSF: plasma AA ratio associated with slower cognitive decline over 1 year (age, gender, education, apoEe4, and cognitive function at baseline adjusted p = 0.025). Interaction between CSF AA Ratio and BBB integrity suggesting AA leaking from brain to periphery | |||
| Arlt 2012 [135] | 23 AD men/women, already taking cholinesterase inhibitors. Randomized to AA (2 × 500 mg/day) and vitamin E (400 IU/day) (Age 67.7 ± 7.2, baseline MMSE 20.0 ± 5.3) | Baseline 201.4 ± 25.9, 1 month 229.9 ± 36.7, 12 months 233.6 ± 44.8 | Not measured | No direct effect of antioxidants on performance, however supplements did increase AA and vitamin E in CSF, with antioxidant effect. Greater oxidation of CSF lipids was associated with faster decline of cognitive ability | ||||
| No supplements (Age 73.7 ± 5.3, baseline MMSE 21.7 ± 5.5) | Not measured | Not measured | ||||||
| Galasko 2012 [136] | 16 weeks with 500 mg/day AA, plus 800 IU alpha-tocopherol and 900 mg/day alpha-lipoic acid in subjects with mild to moderate AD | Not measured | Not measured | Measures of CSF oxidative stress were decreased, but amyloid and tau markers were not. Nor were there any beneficial effects on cognitive decline. | ||||
8. Ascorbic Acid and Vascular Cognitive Aging
9. Conclusion
Acknowledgments
Conflicts of Interest
References
- Nishikimi, M.; Fukuyama, R.; Minoshima, S.; Yagi, K. Cloning and chromosomal mapping of the human nonfunctional gene for l-gulono-gamma-lactone oxidase, the enzyme for l-ascorbic acid biosynthesis missing in man. J. Biol. Chem. 1994, 269, 13685–13688. [Google Scholar]
- Frei, B.; England, L.; Ames, B.N. Ascorbate is an outstanding antioxidant in human blood plasma. Proc. Natl. Acad. Sci. USA 1989, 86, 6377–6381. [Google Scholar] [CrossRef]
- Berger, T.M.; Polidori, M.C.; Dabbagh, A.; Evans, P.J.; Halliwell, B.; Morrow, J.D.; Roberts, L.J., II; Frei, B. Antioxidant activity of vitamin C in iron-overloaded human plasma. J. Biol. Chem. 1997, 272, 15656–15660. [Google Scholar] [CrossRef]
- Traber, M.G.; Stevens, J.F. Vitamins C and E: Beneficial effects from a mechanistic perspective. Free Radic. Biol. Med. 1997, 51, 1000–1013. [Google Scholar] [CrossRef]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s Dement. 2013, 9, 63–75 e62. [Google Scholar] [CrossRef]
- Pratico, D.; Clark, C.M.; Liun, F.; Rokach, J.; Lee, V.Y.; Trojanowski, J.Q. Increase of brain oxidative stress in mild cognitive impairment: A possible predictor of Alzheimer disease. Arch. Neurol. 2002, 59, 972–976. [Google Scholar] [CrossRef]
- Rinaldi, P.; Polidori, M.C.; Metastasio, A.; Mariani, E.; Mattioli, P.; Cherubini, A.; Catani, M.; Cecchetti, R.; Senin, U.; Mecocci, P. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol. Aging 2003, 24, 915–919. [Google Scholar] [CrossRef]
- Mecocci, P.; Polidori, M.C. Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease. Biochim. Biophys. Acta 2012, 1822, 631–638. [Google Scholar]
- Mangialasche, F.; Kivipelto, M.; Mecocci, P.; Rizzuto, D.; Palmer, K.; Winblad, B.; Fratiglioni, L. High plasma levels of vitamin E forms and reduced Alzheimer’s disease risk in advanced age. J. Alzheimer’s Dis. (JAD) 2010, 20, 1029–1037. [Google Scholar]
- Raynaud-Simon, A.; Cohen-Bittan, J.; Gouronnec, A.; Pautas, E.; Senet, P.; Verny, M.; Boddaert, J. Scurvy in hospitalized elderly patients. J. Nutr. Health Aging 2010, 14, 407–410. [Google Scholar] [CrossRef]
- Harrison, F.E.; Green, R.J.; Dawes, S.M.; May, J.M. Vitamin C distribution and retention in the mouse brain. Brain Res. 2010, 1348, 181–186. [Google Scholar]
- Brubacher, D.; Moser, U.; Jordan, P. Vitamin C concentrations in plasma as a function of intake: A meta-analysis. Int. J. Vitam Nutr. Res. 2000, 70, 226–237. [Google Scholar] [CrossRef]
- Harrison, F.E.; May, J.M. Vitamin C function in the brain: vital role of the ascorbate transporter SVCT2. Free Radic Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef]
- Kaliora, A.C.; Dedoussis, G.V.; Schmidt, H. Dietary antioxidants in preventing atherogenesis. Atherosclerosis 2006, 187, 1–17. [Google Scholar] [CrossRef]
- May, J.M.; Harrison, F.E. Role of Vitamin C in the Function of the Vascular Endothelium. Antioxid. Redox Signal. 2013, 19, 2068–2083. [Google Scholar] [CrossRef]
- Proper, E.A.; Hoogland, G.; Kappen, S.M.; Jansen, G.H.; Rensen, M.G.; Schrama, L.H.; van Veelen, C.W.; van Rijen, P.C.; van Nieuwenhuizen, O.; Gispen, W.H.; et al. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain 2002, 125, 32–43. [Google Scholar] [CrossRef]
- Wilson, J.X.; Peters, C.E.; Sitar, S.M.; Daoust, P.; Gelb, A.W. Glutamate stimulates ascorbate transport by astrocytes. Brain Res. 2000, 858, 61–66. [Google Scholar] [CrossRef]
- Estrada-Sanchez, A.M.; Rebec, G.V. Corticostriatal dysfunction and glutamate transporter 1 (GLT1) in Huntington's disease: Interactions between neurons and astrocytes. Basal Ganglia 2012, 2, 57–66. [Google Scholar] [CrossRef]
- Ballaz, S.; Morales, I.; Rodriguez, M.; Obeso, J.A. Ascorbate prevents cell death from prolonged exposure to glutamate in an in vitro model of human dopaminergic neurons. J. Neurosci. Res. 2013, 91, 1609–1617. [Google Scholar] [CrossRef]
- Dorner, J.L.; Miller, B.R.; Klein, E.L.; Murphy-Nakhnikian, A.; Andrews, R.L.; Barton, S.J.; Rebec, G.V. Corticostriatal dysfunction underlies diminished striatal ascorbate release in the R6/2 mouse model of Huntington’s disease. Brain Res. 2009, 1290, 111–120. [Google Scholar]
- Rebec, G.V.; Pierce, R.C. A vitamin as neuromodulator: Ascorbate release into the extracellular fluid of the brain regulates dopaminergic and glutamatergic transmission. Prog. Neurobiol. 1994, 43, 537–565. [Google Scholar] [CrossRef]
- Rebec, G.V.; Barton, S.J.; Marseilles, A.M.; Collins, K. Ascorbate treatment attenuates the Huntington behavioral phenotype in mice. Neuroreport 2003, 14, 1263–1265. [Google Scholar] [CrossRef]
- Spector, R.; Johanson, C.E. The nexus of vitamin homeostasis and DNA synthesis and modification in mammalian brain. Mol. Brain 2014, 7, 3. [Google Scholar] [CrossRef]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martinez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef]
- Yin, R.; Mao, S.Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 2013, 135, 10396–10403. [Google Scholar] [CrossRef]
- Minor, E.A.; Court, B.L.; Young, J.I.; Wang, G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J. Biol. Chem. 2013, 288, 13669–13674. [Google Scholar] [CrossRef]
- Rudenko, A.; Dawlaty, M.M.; Seo, J.; Cheng, A.W.; Meng, J.; Le, T.; Faull, K.F.; Jaenisch, R.; Tsai, L.H. Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron 2013, 79, 1109–1122. [Google Scholar] [CrossRef]
- Meister, A. Glutathione, ascorbate, and cellular protection. Cancer Res. 1994, 54, 1969s–1975s. [Google Scholar]
- Smuda, D.G.M. Maillard degradation pathways of vitamin C. Angew. Chem. Int. Ed. 2013, 52, 4887–4891. [Google Scholar] [CrossRef]
- Fan, X.; Reneker, L.W.; Obrenovich, M.E.; Strauch, C.; Cheng, R.; Jarvis, S.M.; Ortwerth, B.J.; Monnier, V.M. Vitamin C mediates chemical aging of lens crystallins by the Maillard reaction in a humanized mouse model. Proc. Natl. Acad. Sci. USA 2006, 103, 16912–16917. [Google Scholar] [CrossRef]
- Carr, A.; Frei, B. Does vitamin C act as a pro-oxidant under physiological conditions? FASEB J. 1999, 13, 1007–1024. [Google Scholar]
- Halliwell, B. Vitamin C: Antioxidant or pro-oxidant in vivo? Free Radic. Res. 1996, 25, 439–454. [Google Scholar] [CrossRef]
- Premkumar, K.; Bowlus, C.L. Ascorbic acid does not increase the oxidative stress induced by dietary iron in C3H mice. J. Nutr. 2004, 134, 435–438. [Google Scholar]
- Collis, C.S.; Yang, M.; Diplock, A.T.; Hallinan, T.; Rice-Evans, C.A. Effects of co-supplementation of iron with ascorbic acid on antioxidant—Pro-oxidant balance in the guinea pig. Free Radic. Res. 1997, 27, 113–121. [Google Scholar] [CrossRef]
- Gerster, H. High-dose vitamin C: A risk for persons with high iron stores? Int. J. Vitam Nutr. Res. 1999, 69, 67–82. [Google Scholar] [CrossRef]
- Loef, M.; Walach, H. Copper and iron in Alzheimer’s disease: A systematic review and its dietary implications. Br. J. Nutr. 2012, 107, 7–19. [Google Scholar] [CrossRef]
- Crespo, A.C.; Silva, B.; Marques, L.; Marcelino, E.; Maruta, C.; Costa, S.; Timoteo, A.; Vilares, A.; Couto, F.S.; Faustino, P.; et al. Genetic and biochemical markers in patients with Alzheimer’s disease support a concerted systemic iron homeostasis dysregulation. Neurobiol. Aging 2014, 35, 777–785. [Google Scholar]
- Sinha, M.; Bhowmick, P.; Banerjee, A.; Chakrabarti, S. Antioxidant role of amyloid beta protein in cell-free and biological systems: Implication for the pathogenesis of Alzheimer disease. Free Radic. Biol. Med. 2013, 56, 184–192. [Google Scholar] [CrossRef]
- Hammarstrom, L. Autoradiographic studies on the distribution of C14-labelled ascorbic acid and dehydroascorbic acid. Acta Physiol. Scand. 1966, 70, 3–83. [Google Scholar] [CrossRef]
- Spector, R.; Lorenzo, A.V. Ascorbic acid homeostasis in the central nervous system. Am. J. Physiol. 1973, 225, 757–763. [Google Scholar]
- Savini, I.; Rossi, A.; Pierro, C.; Avigliano, L.; Catani, M.V. SVCT1 and SVCT2: Key proteins for vitamin C uptake. Amino Acids 2008, 34, 347–355. [Google Scholar] [CrossRef]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature 1999, 399, 70–75. [Google Scholar] [CrossRef]
- Wang, Y.; Mackenzie, B.; Tsukaguchi, H.; Weremowicz, S.; Morton, C.C.; Hediger, M.A. Human vitamin C (l-ascorbic acid) transporter SVCT1. Biochem. Biophys. Res. Commun. 2000, 267, 488–494. [Google Scholar] [CrossRef]
- Lindblad, M.; Tveden-Nyborg, P.; Lykkesfeldt, J. Regulation of Vitamin C Homeostasis during Deficiency. Nutrients 2013, 5, 2860–2879. [Google Scholar] [CrossRef]
- Erichsen, H.C.; Eck, P.; Levine, M.; Chanock, S. Characterization of the genomic structure of the human vitamin C transporter SVCT1 (SLC23A2). J. Nutr. 2001, 131, 2623–2627. [Google Scholar]
- Erichsen, H.C.; Peters, U.; Eck, P.; Welch, R.; Schoen, R.E.; Yeager, M.; Levine, M.; Hayes, R.B.; Chanock, S. Genetic variation in sodium-dependent vitamin C transporters SLC23A1 and SLC23A2 and risk of advanced colorectal adenoma. Nutr. Cancer 2008, 60, 652–659. [Google Scholar] [CrossRef]
- Michels, A.J.; Hagen, T.M.; Frei, B. Human genetic variation influences vitamin C homeostasis by altering vitamin C transport and antioxidant enzyme function. Annu. Rev. Nutr. 2013, 33, 45–70. [Google Scholar] [CrossRef]
- Eck, P.; Erichsen, H.C.; Taylor, J.G.; Corpe, C.; Chanock, S.J.; Levine, M. Genomic and functional analysis of the sodium-dependent vitamin C transporter SLC23A1-SVCT1. Genes Nutr. 2007, 2, 143–145. [Google Scholar] [CrossRef]
- Corpe, C.P.; Tu, H.; Eck, P.; Wang, J.; Faulhaber-Walter, R.; Schnermann, J.; Margolis, S.; Padayatty, S.; Sun, H.; Wang, Y.; et al. Vitamin C transporter Slc23a1 links renal reabsorption, vitamin C tissue accumulation, and perinatal survival in mice. J. Clin. Invest. 2010, 120, 1069–1083. [Google Scholar] [CrossRef]
- Hierro, C.; Monte, M.J.; Lozano, E.; Gonzalez-Sanchez, E.; Marin, J.J.; Macias, R.I. Liver metabolic/oxidative stress induces hepatic and extrahepatic changes in the expression of the vitamin C transporters SVCT1 and SVCT2. Eur. J. Nutr. 2014, 53, 401–412. [Google Scholar] [CrossRef]
- Gess, B.; Sevimli, S.; Strecker, J.K.; Young, P.; Schabitz, W.R. Sodium-dependent vitamin C transporter 2 (SVCT2) expression and activity in brain capillary endothelial cells after transient ischemia in mice. PLoS One 2011, 6, e17139. [Google Scholar]
- May, J.M.; Qu, Z.C. Ascorbic acid prevents increased endothelial permeability caused by oxidized low density lipoprotein. Free Radic. Res. 2010, 44, 1359–1368. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.C. Ascorbic acid prevents oxidant-induced increases in endothelial permeability. Biofactors 2011, 37, 46–50. [Google Scholar] [CrossRef]
- Amano, A.; Aigaki, T.; Maruyama, N.; Ishigami, A. Ascorbic acid depletion enhances expression of the sodium-dependent vitamin C transporters, SVCT1 and SVCT2, and uptake of ascorbic acid in livers of SMP30/GNL knockout mice. Arch. Biochem. Biophys. 2010, 496, 38–44. [Google Scholar] [CrossRef]
- Meredith, M.E.; Harrison, F.E.; May, J.M. Differential regulation of the ascorbic acid transporter SVCT2 during development and in response to ascorbic acid depletion. Biochem. Biophys. Res. Commun. 2011, 414, 737–742. [Google Scholar] [CrossRef]
- Nualart, F.; Castro, T.; Low, M.; Henriquez, J.P.; Oyarce, K.; Cisternas, P.; Garcia, A.; Yanez, A.J.; Bertinat, R.; Montecinos, V.P.; et al. Dynamic expression of the sodium-vitamin C co-transporters, SVCT1 and SVCT2, during perinatal kidney development. Histochem. Cell Biol. 2013, 139, 233–247. [Google Scholar] [CrossRef]
- Jimenez-Fernandez, E.; Ponce, M.; Zuasti, E.; Fernandez-Diaz, C.; Manchado, M.; Infante, C. Molecular characterization and transcriptional regulation of the sodium-dependent vitamin C transporter genes (slc23a1 and slc23a2) in a teleost fish, the Senegalese sole (Solea senegalensis). Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2012, 161, 208–218. [Google Scholar] [CrossRef]
- Gess, B.; Lohmann, C.; Halfter, H.; Young, P. Sodium-dependent vitamin C transporter 2 (SVCT2) is necessary for the uptake of l-ascorbic acid into Schwann cells. Glia 2010, 58, 287–299. [Google Scholar]
- Maulen, N.P.; Henriquez, E.A.; Kempe, S.; Carcamo, J.G.; Schmid-Kotsas, A.; Bachem, M.; Grunert, A.; Bustamante, M.E.; Nualart, F.; Vera, J.C. Up-regulation and polarized expression of the sodium-ascorbic acid transporter SVCT1 in post-confluent differentiated CaCo-2 cells. J. Biol. Chem. 2003, 278, 9035–9041. [Google Scholar] [CrossRef]
- Boyer, J.C.; Campbell, C.E.; Sigurdson, W.J.; Kuo, S.M. Polarized localization of vitamin C transporters, SVCT1 and SVCT2, in epithelial cells. Biochem. Biophys. Res. Commun. 2005, 334, 150–156. [Google Scholar] [CrossRef]
- MacDonald, L.; Thumser, A.E.; Sharp, P. Decreased expression of the vitamin C transporter SVCT1 by ascorbic acid in a human intestinal epithelial cell line. Br. J. Nutr. 2002, 87, 97–100. [Google Scholar] [CrossRef]
- Varma, S.; Sobey, K.; Campbell, C.E.; Kuo, S.M. Hierarchal contribution of N- and C-terminal sequences to the differential localization of homologous sodium-dependent vitamin C transporters, SVCT1 and SVCT2, in epithelial cells. Biochemistry 2009, 48, 2969–2980. [Google Scholar] [CrossRef]
- May, J.M.; Li, L.; Qu, Z.C. Oxidized LDL up-regulates the ascorbic acid transporter SVCT2 in endothelial cells. Mol. Cell. Biochem. 2010, 343, 217–222. [Google Scholar] [CrossRef]
- Qiao, H.; May, J.M. Development of ascorbate transporters in brain cortical capillary endothelial cells in culture. Brain Res. 2008, 1208, 79–86. [Google Scholar]
- Chothe, P.P.; Chutkan, N.; Sangani, R.; Wenger, K.H.; Prasad, P.D.; Thangaraju, M.; Hamrick, M.W.; Isales, C.M.; Ganapathy, V.; Fulzele, S. Sodium-coupled vitamin C transporter (SVCT2): expression, function, and regulation in intervertebral disc cells. Spine J. 2013, 13, 549–557. [Google Scholar] [CrossRef]
- Berger, U.V.; Hediger, M.A. The vitamin C transporter SVCT2 is expressed by astrocytes in culture but not in situ. Neuroreport 2000, 11, 1395–1399. [Google Scholar] [CrossRef]
- Korcok, J.; Yan, R.; Siushansian, R.; Dixon, S.J.; Wilson, J.X. Sodium-ascorbate cotransport controls intracellular ascorbate concentration in primary astrocyte cultures expressing the SVCT2 transporter. Brain Res. 2000, 881, 144–151. [Google Scholar] [CrossRef]
- Oke, A.F.; May, L.; Adams, R.N. Ascorbic acid distribution patterns in human brain. A comparison with nonhuman mammalian species. Ann. N. Y. Acad. Sci. 1987, 498, 1–12. [Google Scholar] [CrossRef]
- Mefford, I.N.; Oke, A.F.; Adams, R.N. Regional distribution of ascorbate in human brain. Brain Res. 1981, 212, 223–226. [Google Scholar] [CrossRef]
- Rice, M.E.; Russo-Menna, I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1998, 82, 1213–1223. [Google Scholar] [CrossRef]
- Caprile, T.; Salazar, K.; Astuya, A.; Cisternas, P.; Silva-Alvarez, C.; Montecinos, H.; Millan, C.; de Los Angeles Garcia, M.; Nualart, F. The Na+-dependent L-ascorbic acid transporter SVCT2 expressed in brainstem cells, neurons, and neuroblastoma cells is inhibited by flavonoids. J. Neurochem. 2009, 108, 563–577. [Google Scholar] [CrossRef]
- Qiu, S.; Li, L.; Weeber, E.J.; May, J.M. Ascorbate transport by primary cultured neurons and its role in neuronal function and protection against excitotoxicity. J. Neurosci. Res. 2007, 85, 1046–1056. [Google Scholar] [CrossRef]
- Mun, G.H.; Kim, M.J.; Lee, J.H.; Kim, H.J.; Chung, Y.H.; Chung, Y.B.; Kang, J.S.; Hwang, Y.I.; Oh, S.H.; Kim, J.G.; et al. Immunohistochemical study of the distribution of sodium-dependent vitamin C transporters in adult rat brain. J. Neurosci. Res. 2006, 83, 919–928. [Google Scholar] [CrossRef]
- Garcia Mde, L.; Salazar, K.; Millan, C.; Rodriguez, F.; Montecinos, H.; Caprile, T.; Silva, C.; Cortes, C.; Reinicke, K.; Vera, J.C.; et al. Sodium vitamin C cotransporter SVCT2 is expressed in hypothalamic glial cells. Glia 2005, 50, 32–47. [Google Scholar] [CrossRef]
- Chinoy, N.J.; Sanjeevan, A.G. On the specificity of alcoholic acidic silver nitrate reagent for the histochemical localization of ascorbic acid. A reappraisal. Histochemistry 1978, 56, 275–282. [Google Scholar]
- Berger, U.V.; Lu, X.C.; Liu, W.; Tang, Z.; Slusher, B.S.; Hediger, M.A. Effect of middle cerebral artery occlusion on mRNA expression for the sodium-coupled vitamin C transporter SVCT2 in rat brain. J. Neurochem. 2003, 86, 896–906. [Google Scholar] [CrossRef]
- McHenry, E.W.; Reedman, E.J.; Sheppard, M. The physiological properties of ascorbic acid: An effect upon the weights of guinea-pigs. Biochem. J. 1938, 32, 1302–1304. [Google Scholar]
- Svirbely, J.L.; Szent-Gyorgyi, A. The chemical nature of vitamin C. Biochem. J. 1932, 26, 865–870. [Google Scholar]
- Szent-Gyorgyi, A.H.W.N. “Hexuronic Acid” (Ascorbic Acid) as the Antiscorbutic Factor. Nature 1933, 131, 24. [Google Scholar] [CrossRef]
- Anderson WES, A.H. The effect of acute scurvy on the subsequent nutrition and growth of guinea pigs. J. Biol. Chem. 1924, 61, 181–191. [Google Scholar]
- Burk, R.F.; Christensen, J.M.; Maguire, M.J.; Austin, L.M.; Whetsell, W.O., Jr.; May, J.M.; Hill, K.E.; Ebner, F.F. A combined deficiency of vitamins E and C causes severe central nervous system damage in guinea pigs. J. Nutr. 2006, 136, 1576–1581. [Google Scholar]
- Hill, K.E.; Motley, A.K.; May, J.M.; Burk, R.F. Combined selenium and vitamin C deficiency causes cell death in guinea pig skeletal muscle. Nutr. Res. 2009, 29, 213–219. [Google Scholar] [CrossRef]
- Frikke-Schmidt, H.; Lykkesfeldt, J. Role of marginal vitamin C deficiency in atherogenesis: In vivo models and clinical studies. Basic Clin. Pharmacol. Toxicol. 2009, 104, 419–433. [Google Scholar] [CrossRef]
- Maeda, N.; Hagihara, H.; Nakata, Y.; Hiller, S.; Wilder, J.; Reddick, R. Aortic wall damage in mice unable to synthesize ascorbic acid. Proc. Natl. Acad. Sci. USA 2000, 97, 841–846. [Google Scholar]
- Harrison, F.E.; Meredith, M.E.; Dawes, S.M.; Saskowski, J.L.; May, J.M. Low ascorbic acid and increased oxidative stress in gulo(−/−) mice during development. Brain Res. 2010, 1349, 143–152. [Google Scholar]
- Harrison, F.E.; Yu, S.S.; Van Den Bossche, K.L.; Li, L.; May, J.M.; McDonald, M.P. Elevated oxidative stress and sensorimotor deficits but normal cognition in mice that cannot synthesize ascorbic acid. J. Neurochem. 2008, 106, 1198–1208. [Google Scholar] [CrossRef]
- Chen, Y.; Curran, C.P.; Nebert, D.W.; Patel, K.V.; Williams, M.T.; Vorhees, C.V. Effect of vitamin C deficiency during postnatal development on adult behavior: functional phenotype of Gulo(−/−) knockout mice. Genes Brain Behav. 2012, 11, 269–277. [Google Scholar] [CrossRef]
- Harrison, F.E.; May, J.M.; McDonald, M.P. Vitamin C deficiency increases basal exploratory activity but decreases scopolamine-induced activity in APP/PSEN1 transgenic mice. Pharmacol. Biochem. Behav. 2010, 94, 543–552. [Google Scholar] [CrossRef]
- Duggan, G.E.; Joan Miller, B.; Jirik, F.R.; Vogel, H.J. Metabolic profiling of vitamin C deficiency in Gulo−/− mice using proton NMR spectroscopy. J. Biomol. NMR 2011, 49, 165–173. [Google Scholar] [CrossRef]
- Vissers, M.C.; Wilkie, R.P. Ascorbate deficiency results in impaired neutrophil apoptosis and clearance and is associated with up-regulation of hypoxia-inducible factor 1alpha. J. Leukoc. Biol. 2007, 81, 1236–1244. [Google Scholar] [CrossRef]
- Ishigami, A.; Fujita, T.; Handa, S.; Shirasawa, T.; Koseki, H.; Kitamura, T.; Enomoto, N.; Sato, N.; Shimosawa, T.; Maruyama, N. Senescence marker protein-30 knockout mouse liver is highly susceptible to tumor necrosis factor-alpha- and Fas-mediated apoptosis. Am. J. Pathol. 2002, 161, 1273–1281. [Google Scholar] [CrossRef]
- Kondo, Y.; Inai, Y.; Sato, Y.; Handa, S.; Kubo, S.; Shimokado, K.; Goto, S.; Nishikimi, M.; Maruyama, N.; Ishigami, A. Senescence marker protein 30 functions as gluconolactonase in l-ascorbic acid biosynthesis, and its knockout mice are prone to scurvy. Proc. Natl. Acad. Sci. USA 2006, 103, 5723–5728. [Google Scholar] [CrossRef]
- Kondo, Y.; Sasaki, T.; Sato, Y.; Amano, A.; Aizawa, S.; Iwama, M.; Handa, S.; Shimada, N.; Fukuda, M.; Akita, M.; et al. Vitamin C depletion increases superoxide generation in brains of SMP30/GNL knockout mice. Biochem. Biophys. Res. Commun. 2008, 377, 291–296. [Google Scholar] [CrossRef]
- Son, T.G.; Zou, Y.; Jung, K.J.; Yu, B.P.; Ishigami, A.; Maruyama, N.; Lee, J. SMP30 deficiency causes increased oxidative stress in brain. Mech. Ageing Dev. 2006, 127, 451–457. [Google Scholar] [CrossRef]
- Beamer, W.G.; Rosen, C.J.; Bronson, R.T.; Gu, W.; Donahue, L.R.; Baylink, D.J.; Richardson, C.C.; Crawford, G.C.; Barker, J.E. Spontaneous fracture (sfx): A mouse genetic model of defective peripubertal bone formation. Bone 2000, 27, 619–626. [Google Scholar] [CrossRef]
- Mohan, S.; Kapoor, A.; Singgih, A.; Zhang, Z.; Taylor, T.; Yu, H.; Chadwick, R.B.; Chung, Y.S.; Donahue, L.R.; Rosen, C.; et al. Spontaneous fractures in the mouse mutant sfx are caused by deletion of the gulonolactone oxidase gene, causing vitamin C deficiency. J. Bone Miner. Res. 2005, 20, 1597–1610. [Google Scholar] [CrossRef]
- Jiao, Y.; Zhang, J.; Yan, J.; Stuart, J.; Gibson, G.; Lu, L.; Willaims, R.; Wang, Y.J.; Gu, W. Differential gene expression between wild-type and Gulo-deficient mice supplied with vitamin C. Genet. Mol. Biol. 2011, 34, 386–395. [Google Scholar] [CrossRef]
- Ward, M.S.; Lamb, J.; May, J.M.; Harrison, F.E. Behavioral and monoamine changes following severe vitamin C deficiency. J. Neurochem. 2013, 124, 363–375. [Google Scholar] [CrossRef]
- Amano, A.; Tsunoda, M.; Aigaki, T.; Maruyama, N.; Ishigami, A. Effect of ascorbic acid deficiency on catecholamine synthesis in adrenal glands of SMP30/GNL knockout mice. Eur. J. Nutr. 2014, 53, 177–185. [Google Scholar] [CrossRef]
- Sotiriou, S.; Gispert, S.; Cheng, J.; Wang, Y.; Chen, A.; Hoogstraten-Miller, S.; Miller, G.F.; Kwon, O.; Levine, M.; Guttentag, S.H.; et al. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat. Med. 2002, 8, 514–517. [Google Scholar] [CrossRef]
- Harrison, F.E.; Dawes, S.M.; Meredith, M.E.; Babaev, V.R.; Li, L.; May, J.M. Low vitamin C and increased oxidative stress and cell death in mice that lack the sodium-dependent vitamin C transporter SVCT2. Free Radic. Biol. Med. 2010, 49, 821–829. [Google Scholar] [CrossRef]
- Bornstein, S.R.; Yoshida-Hiroi, M.; Sotiriou, S.; Levine, M.; Hartwig, H.G.; Nussbaum, R.L.; Eisenhofer, G. Impaired adrenal catecholamine system function in mice with deficiency of the ascorbic acid transporter (SVCT2). FASEB J. 2003, 17, 1928–1930. [Google Scholar]
- Meredith, M.E.; May, J.M. Regulation of embryonic neurotransmitter and tyrosine hydroxylase protein levels by ascorbic acid. Brain Res. 2013, 1539, 7–14. [Google Scholar]
- Harrison, F.E.; Best, J.L.; Meredith, M.E.; Gamlin, C.R.; Borza, D.B.; May, J.M. Increased expression of SVCT2 in a new mouse model raises ascorbic acid in tissues and protects against paraquat-induced oxidative damage in lung. PLoS One 2012, 7, e35623. [Google Scholar]
- Babaev, V.R.; Whitesell, R.R.; Li, L.; Linton, M.F.; Fazio, S.; May, J.M. Selective macrophage ascorbate deficiency suppresses early atherosclerosis. Free Radic. Biol. Med. 2011, 50, 27–36. [Google Scholar] [CrossRef]
- Babaev, V.R.; Li, L.; Shah, S.; Fazio, S.; Linton, M.F.; May, J.M. Combined vitamin C and vitamin E deficiency worsens early atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1751–1757. [Google Scholar] [CrossRef]
- Kook, S.Y.; Lee, K.M.; Kim, Y.; Cha, M.Y.; Kang, S.; Baik, S.H.; Lee, H.; Park, R.; Mook-Jung, I. High-dose of vitamin C supplementation reduces amyloid plaque burden and ameliorates pathological changes in the brain of 5XFAD mice. Cell. Death Dis. 2014, 5, e1083. [Google Scholar] [CrossRef]
- Pierce, M.R.; Diasio, D.L.; Rodrigues, L.M.; Harrison, F.E.; May, J.M. Combined vitamin C and E deficiency induces motor defects in gulo(−/−)/SVCT2(+/−) mice. Nutr. Neurosci. 2013, 16, 160–173. [Google Scholar] [CrossRef]
- Morris, M.C.; Beckett, L.A.; Scherr, P.A.; Hebert, L.E.; Bennett, D.A.; Field, T.S.; Evans, D.A. Vitamin E and vitamin C supplement use and risk of incident Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1998, 12, 121–126. [Google Scholar] [CrossRef]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Tangney, C.C.; Bennett, D.A.; Aggarwal, N.; Wilson, R.S.; Scherr, P.A. Dietary intake of antioxidant nutrients and the risk of incident Alzheimer disease in a biracial community study. JAMA 2002, 287, 3230–3237. [Google Scholar] [CrossRef]
- Carr, A.C.; Bozonet, S.M.; Pullar, J.M.; Simcock, J.W.; Vissers, M.C. A Randomized Steady-State Bioavailability Study of Synthetic versus Natural (Kiwifruit-Derived) Vitamin C. Nutrients 2013, 5, 3684–3695. [Google Scholar] [CrossRef]
- Gray, S.L.; Anderson, M.L.; Crane, P.K.; Breitner, J.C.; McCormick, W.; Bowen, J.D.; Teri, L.; Larson, E. Antioxidant vitamin supplement use and risk of dementia or Alzheimer’s disease in older adults. J. Am. Geriatr. Soc. 2008, 56, 291–295. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Mayeux, R. Antioxidant vitamin intake and risk of Alzheimer disease. Arch. Neurol. 2003, 60, 203–208. [Google Scholar] [CrossRef]
- Masaki, K.H.; Losonczy, K.G.; Izmirlian, G.; Foley, D.J.; Ross, G.W.; Petrovitch, H.; Havlik, R.; White, L.R. Association of vitamin E and C supplement use with cognitive function and dementia in elderly men. Neurology 2000, 54, 1265–1272. [Google Scholar] [CrossRef]
- Zandi, P.P.; Anthony, J.C.; Khachaturian, A.S.; Stone, S.V.; Gustafson, D.; Tschanz, J.T.; Norton, M.C.; Welsh-Bohmer, K.A.; Breitner, J.C. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: the Cache County Study. Arch. Neurol. 2004, 61, 82–88. [Google Scholar] [CrossRef]
- Grodstein, F.; Chen, J.; Willett, W.C. High-dose antioxidant supplements and cognitive function in community-dwelling elderly women. Am. J. Clin. Nutr. 2003, 77, 975–984. [Google Scholar]
- Devore, E.E.; Kang, J.H.; Stampfer, M.J.; Grodstein, F. The association of antioxidants and cognition in the Nurses' Health Study. Am. J. Epidemiol. 2013, 177, 33–41. [Google Scholar] [CrossRef]
- Engelhart, M.J.; Geerlings, M.I.; Ruitenberg, A.; van Swieten, J.C.; Hofman, A.; Witteman, J.C.; Breteler, M.M. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA 2002, 287, 3223–3229. [Google Scholar] [CrossRef]
- Bowman, G.L.; Shannon, J.; Ho, E.; Traber, M.G.; Frei, B.; Oken, B.S.; Kaye, J.A.; Quinn, J.F. Reliability and validity of food frequency questionnaire and nutrient biomarkers in elders with and without mild cognitive impairment. Alzheimer Dis. Assoc. Disord. 2011, 25, 49–57. [Google Scholar] [CrossRef]
- Rodrigue, K.M.; Kennedy, K.M.; Devous, M.D., Sr.; Rieck, J.R.; Hebrank, A.C.; Diaz-Arrastia, R.; Mathews, D.; Park, D.C. beta-Amyloid burden in healthy aging: Regional distribution and cognitive consequences. Neurology 2012, 78, 387–395. [Google Scholar] [CrossRef]
- Bowman, G.L.; Silbert, L.C.; Howieson, D.; Dodge, H.H.; Traber, M.G.; Frei, B.; Kaye, J.A.; Shannon, J.; Quinn, J.F. Nutrient biomarker patterns, cognitive function, and MRI measures of brain aging. Neurology 2012, 78, 241–249. [Google Scholar] [CrossRef]
- Bowman, G.L. Ascorbic acid, cognitive function, and Alzheimer’s disease: A current review and future direction. Biofactors 2012, 38, 114–122. [Google Scholar] [CrossRef]
- Goodwin, J.S.; Goodwin, J.M.; Garry, P.J. Association between nutritional status and cognitive functioning in a healthy elderly population. JAMA 1983, 249, 2917–2921. [Google Scholar] [CrossRef]
- Gale, C.R.; Martyn, C.N.; Cooper, C. Cognitive impairment and mortality in a cohort of elderly people. BMJ 1996, 312, 608–611. [Google Scholar] [CrossRef]
- Riviere, S.; Birlouez-Aragon, I.; Nourhashemi, F.; Vellas, B. Low plasma vitamin C in Alzheimer patients despite an adequate diet. Int. J. Geriatr. Psychiatry 1998, 13, 749–754. [Google Scholar] [CrossRef]
- Charlton, K.E.; Rabinowitz, T.L.; Geffen, L.N.; Dhansay, M.A. Lowered plasma vitamin C, but not vitamin E, concentrations in dementia patients. J. Nutr. Health Aging 2004, 8, 99–107. [Google Scholar]
- Polidori, M.C.; Mecocci, P. Plasma susceptibility to free radical-induced antioxidant consumption and lipid peroxidation is increased in very old subjects with Alzheimer disease. J. Alzheimer’s Dis. (JAD) 2002, 4, 517–522. [Google Scholar]
- Polidori, M.C.; Mattioli, P.; Aldred, S.; Cecchetti, R.; Stahl, W.; Griffiths, H.; Senin, U.; Sies, H.; Mecocci, P. Plasma antioxidant status, immunoglobulin g oxidation and lipid peroxidation in demented patients: relevance to Alzheimer disease and vascular dementia. Dement. Geriatr. Cogn. Disord. 2004, 18, 265–270. [Google Scholar] [CrossRef]
- Perrig, W.J.; Perrig, P.; Stahelin, H.B. The relation between antioxidants and memory performance in the old and very old. J. Am. Geriatr. Soc. 1997, 45, 718–724. [Google Scholar]
- Spector, R. Nutrient transport systems in brain: 40 years of progress. J. Neurochem. 2009, 111, 315–320. [Google Scholar] [CrossRef]
- Paraskevas, G.P.; Kapaki, E.; Libitaki, G.; Zournas, C.; Segditsa, I.; Papageorgiou, C. Ascorbate in healthy subjects, amyotrophic lateral sclerosis and Alzheimer’s disease. Acta Neurol. Scand. 1997, 96, 88–90. [Google Scholar]
- Quinn, J.; Suh, J.; Moore, M.M.; Kaye, J.; Frei, B. Antioxidants in Alzheimer's disease-vitamin C delivery to a demanding brain. J. Alzheimers Dis. 2003, 5, 309–313. [Google Scholar]
- Glaso, M.; Nordbo, G.; Diep, L.; Bohmer, T. Reduced concentrations of several vitamins in normal weight patients with late-onset dementia of the Alzheimer type without vascular disease. J. Nutr. Health Aging 2004, 8, 407–413. [Google Scholar]
- Bowman, G.L.; Dodge, H.; Frei, B.; Calabrese, C.; Oken, B.S.; Kaye, J.A.; Quinn, J.F. Ascorbic acid and rates of cognitive decline in Alzheimer's disease. J. Alzheimers Dis. 2009, 16, 93–98. [Google Scholar]
- Arlt, S.; Muller-Thomsen, T.; Beisiegel, U.K.; Ontush, A. Effect of One-Year Vitamin C- and E-Supplementation on Cerebrospinal Fluid Oxidation Parameters and Clinical Course in Alzheimer’s Disease. Neurochem. Res. 2012, 37, 2706–2714. [Google Scholar] [CrossRef]
- Galasko, D.R.; Peskind, E.; Clark, C.M.; Quinn, J.F.; Ringman, J.M.; Jicha, G.A.; Cotman, C.; Cottrell, B.; Montine, T.J.; Thomas, R.G.; Aisen, P. Antioxidants for Alzheimer disease: A randomized clinical trial with cerebrospinal fluid biomarker measures. Arch. Neurol. 2012, 69, 836–841. [Google Scholar]
- Spector, R.; Johanson, C.E. Sustained choroid plexus function in human elderly and Alzheimer’s disease patients. Fluids Barriers CNS 2013, 10, 28. [Google Scholar] [CrossRef]
- Polidori, M.C.; Pientka, L. Bridging the pathophysiology of Alzheimer’s disease with vascular pathology: the feed-back, the feed-forward, and oxidative stress. J. Alzheimer’s Dis. (JAD) 2012, 28, 1–9. [Google Scholar]
- Polidori, M.C.; Pientka, L.; Mecocci, P. A review of the major vascular risk factors related to Alzheimer’s disease. J. Alzheimer’s Dis. (JAD) 2012, 32, 521–530. [Google Scholar]
- de la Torre, J.C. Cerebral hemodynamics and vascular risk factors: setting the stage for Alzheimer’s disease. J. Alzheimer’s Dis. (JAD) 2012, 32, 553–567. [Google Scholar]
- Aguero-Torres, H.; Kivipelto, M.; von Strauss, E. Rethinking the dementia diagnoses in a population-based study: What is Alzheimer’s disease and what is vascular dementia? A study from the kungsholmen project. Dement. Geriatr. Cogn. Disord. 2006, 22, 244–249. [Google Scholar] [CrossRef]
- de la Torre, J.C.; Stefano, G.B. Evidence that Alzheimer’s disease is a microvascular disorder: The role of constitutive nitric oxide. Brain Res. Brain Res. Rev. 2000, 34, 119–136. [Google Scholar] [CrossRef]
- Polidori, M.C.; Mecocci, P.; Frei, B. Plasma vitamin C levels are decreased and correlated with brain damage in patients with intracranial hemorrhage or head trauma. Stroke 2001, 32, 898–902. [Google Scholar] [CrossRef]
- Knopman, D.; Boland, L.L.; Mosley, T.; Howard, G.; Liao, D.; Szklo, M.; McGovern, P.; Folsom, A.R. Cardiovascular risk factors and cognitive decline in middle-aged adults. Neurology 2001, 56, 42–48. [Google Scholar] [CrossRef]
- Polidori, M.C.; Pratico, D.; Savino, K.; Rokach, J.; Stahl, W.; Mecocci, P. Increased F2 isoprostane plasma levels in patients with congestive heart failure are correlated with antioxidant status and disease severity. J. Card. Fail. 2004, 10, 334–338. [Google Scholar] [CrossRef]
- Polidori, M.C.; Pratico, D.; Ingegni, T.; Mariani, E.; Spazzafumo, L.; Del Sindaco, P.; Cecchetti, R.; Yao, Y.; Ricci, S.; Cherubini, A.; et al. Effects of vitamin C and aspirin in ischemic stroke-related lipid peroxidation: Results of the AVASAS (Aspirin Versus Ascorbic acid plus Aspirin in Stroke) Study. Biofactors 2005, 24, 265–274. [Google Scholar] [CrossRef]
- Wendell, C.R.; Zonderman, A.B.; Metter, E.J.; Najjar, S.S.; Waldstein, S.R. Carotid intimal medial thickness predicts cognitive decline among adults without clinical vascular disease. Stroke 2009, 40, 3180–3185. [Google Scholar]
- Frei, B. To C or not to C, that is the question! J. Am. Coll. Cardiol. 2003, 42, 253–255. [Google Scholar] [CrossRef]
- Polidori, M.C.; Mecocci, P.; Reimann, A.; Cherubini, A.; Cecchetti, R.; Briviba, K.; Stahl, W.; Sies, H.; Senin, U. Plasma lipid peroxidation and vitamin C status in healthy centenarians. J. Am. Geriatr. Soc. 1999, 47, 1038–1039. [Google Scholar]
- Hunter, D.C.; Skinner, M.A.; Wolber, F.M.; Booth, C.L.; Loh, J.M.; Wohlers, M.; Stevenson, L.M.; Kruger, M.C. Consumption of gold kiwifruit reduces severity and duration of selected upper respiratory tract infection symptoms and increases plasma vitamin C concentration in healthy older adults. Br. J. Nutr. 2012, 108, 1235–1245. [Google Scholar] [CrossRef]
- Moretti, M.; Colla, A.; de Oliveira Balen, G.; dos Santos, D.B.; Budni, J.; de Freitas, A.E.; Farina, M.; Severo Rodrigues, A.L. Ascorbic acid treatment, similarly to fluoxetine, reverses depressive-like behavior and brain oxidative damage induced by chronic unpredictable stress. J. Psychiatr. Res. 2012, 46, 331–340. [Google Scholar] [CrossRef]
- Simon, J.A.; Hudes, E.S.; Tice, J.A. Relation of serum ascorbic acid to mortality among US adults. J. Am. Coll. Nutr. 2001, 20, 255–263. [Google Scholar] [CrossRef]
- Boekholdt, S.M.; Meuwese, M.C.; Day, N.E.; Luben, R.; Welch, A.; Wareham, N.J.; Khaw, K.T. Plasma concentrations of ascorbic acid and C-reactive protein, and risk of future coronary artery disease, in apparently healthy men and women: the EPIC-Norfolk prospective population study. Br. J. Nutr. 2006, 96, 516–522. [Google Scholar]
- Khaw, K.T.; Bingham, S.; Welch, A.; Luben, R.; Wareham, N.; Oakes, S.; Day, N. Relation between plasma ascorbic acid and mortality in men and women in EPIC-Norfolk prospective study: a prospective population study. European Prospective Investigation into Cancer and Nutrition. Lancet 2001, 357, 657–663. [Google Scholar] [CrossRef]
- Ellingsen, I.; Seljeflot, I.; Arnesen, H.; Tonstad, S. Vitamin C consumption is associated with less progression in carotid intima media thickness in elderly men: A 3-year intervention study. Nutr. Metab. Cardiovasc. Dis. (NMCD) 2009, 19, 8–14. [Google Scholar] [CrossRef]
- Hodis, H.N.; Mack, W.J.; LaBree, L.; Mahrer, P.R.; Sevanian, A.; Liu, C.R.; Liu, C.H.; Hwang, J.; Selzer, R.H.; Azen, S.P. Alpha-tocopherol supplementation in healthy individuals reduces low-density lipoprotein oxidation but not atherosclerosis: The Vitamin E Atherosclerosis Prevention Study (VEAPS). Circulation 2002, 106, 1453–1459. [Google Scholar] [CrossRef]
- Zureik, M.; Galan, P.; Bertrais, S.; Mennen, L.; Czernichow, S.; Blacher, J.; Ducimetiere, P.; Hercberg, S. Effects of long-term daily low-dose supplementation with antioxidant vitamins and minerals on structure and function of large arteries. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1485–1491. [Google Scholar] [CrossRef]
- Luzzi, S.; Vella, L.; Bartolini, M.; Provinciali, L.; Silvestrini, M. Atherosclerosis in the evolution of Alzheimer’s disease: Can treatment reduce cognitive decline? J. Alzheimer’s Dis. (JAD) 2010, 20, 893–901. [Google Scholar]
- Terpstra, M.; Marjanska, M.; Henry, P.G.; Tkac, I.; Gruetter, R. Detection of an antioxidant profile in the human brain in vivo via double editing with MEGA-PRESS. Magn. Reson. Med. 2006, 56, 1192–1199. [Google Scholar] [CrossRef]
- Emir, U.E.; Raatz, S.; McPherson, S.; Hodges, J.S.; Torkelson, C.; Tawfik, P.; White, T.; Terpstra, M. Noninvasive quantification of ascorbate and glutathione concentration in the elderly human brain. NMR Biomed. 2011, 24, 888–894. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Harrison, F.E.; Bowman, G.L.; Polidori, M.C. Ascorbic Acid and the Brain: Rationale for the Use against Cognitive Decline. Nutrients 2014, 6, 1752-1781. https://doi.org/10.3390/nu6041752
Harrison FE, Bowman GL, Polidori MC. Ascorbic Acid and the Brain: Rationale for the Use against Cognitive Decline. Nutrients. 2014; 6(4):1752-1781. https://doi.org/10.3390/nu6041752
Chicago/Turabian StyleHarrison, Fiona E., Gene L. Bowman, and Maria Cristina Polidori. 2014. "Ascorbic Acid and the Brain: Rationale for the Use against Cognitive Decline" Nutrients 6, no. 4: 1752-1781. https://doi.org/10.3390/nu6041752