Analysis of Wheat Prolamins, the Causative Agents of Celiac Sprue, Using Reversed Phase High Performance Liquid Chromatography (RP-HPLC) and Matrix-Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry (MALDI-TOF-MS)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Plant Material
2.3. Protein Extraction
2.4. Polyacrylamide Gel Electrophoresis (PAGE)
2.4.1. Acid-PAGE
2.4.2. Sodium Dodecyl Sulfate-PAGE (SDS-PAGE)
2.5. Reversed-Phase High-Performance Liquid Chromatography (RP-HPLC)
2.6. Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF-MS)
2.7. Relative-Quantification of Gliadin and Glutenin Fractions
2.7.1. Standard Curve Development Using Densitometry
2.7.2. Standard Curve Development Using HPLC
3. Results and Discussion
3.1. Parameter Optimization for RP-HPLC
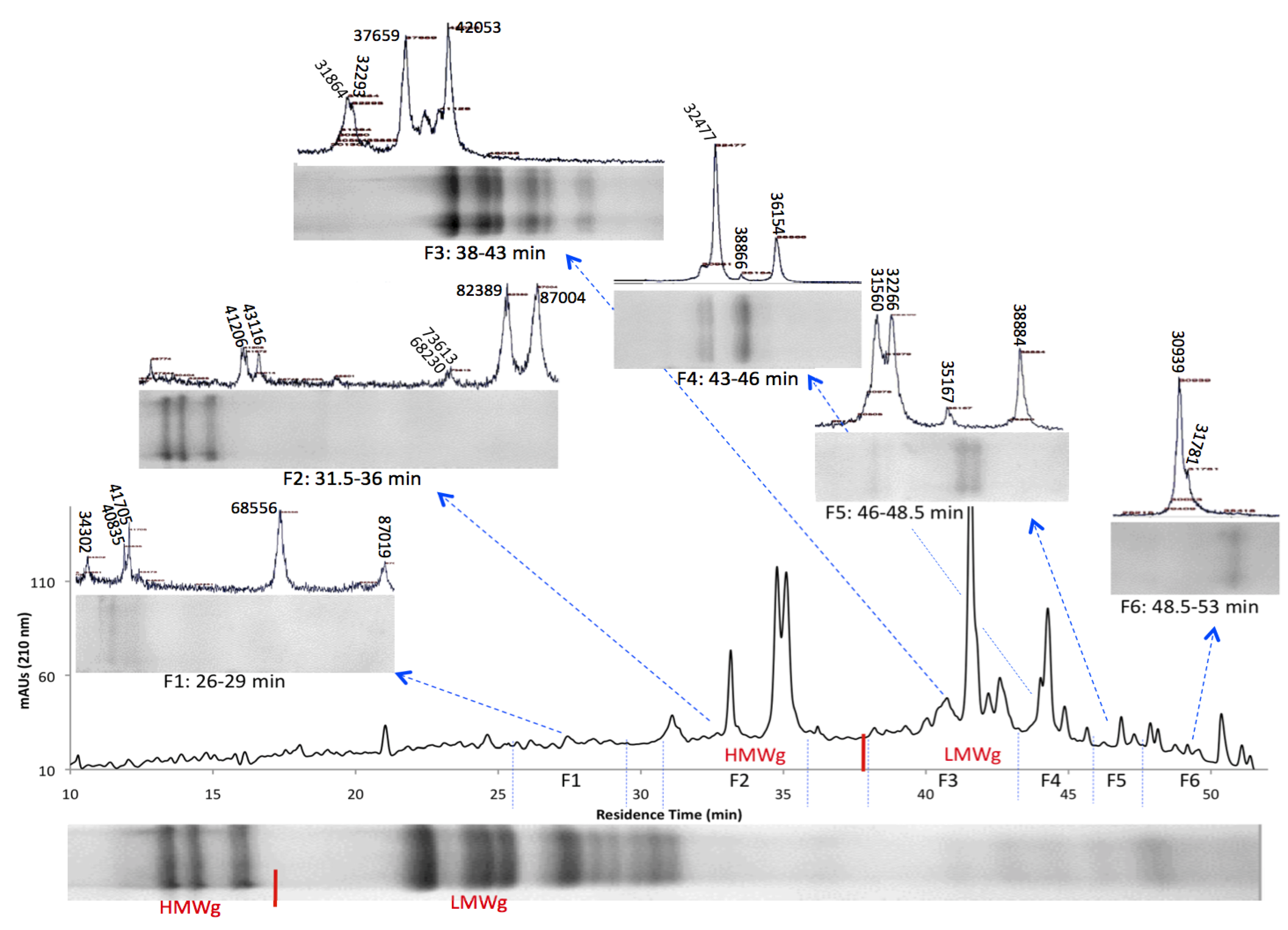
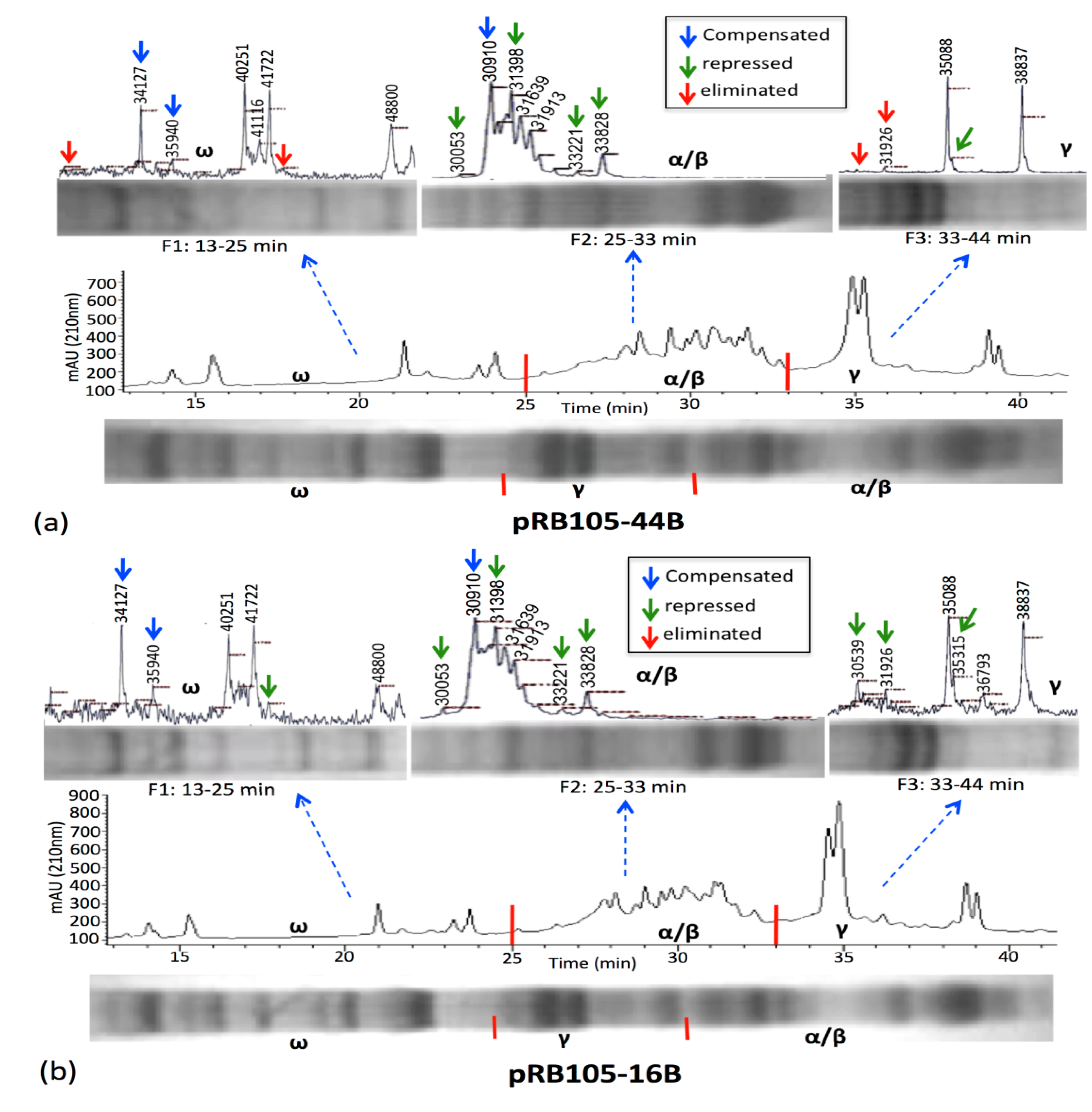
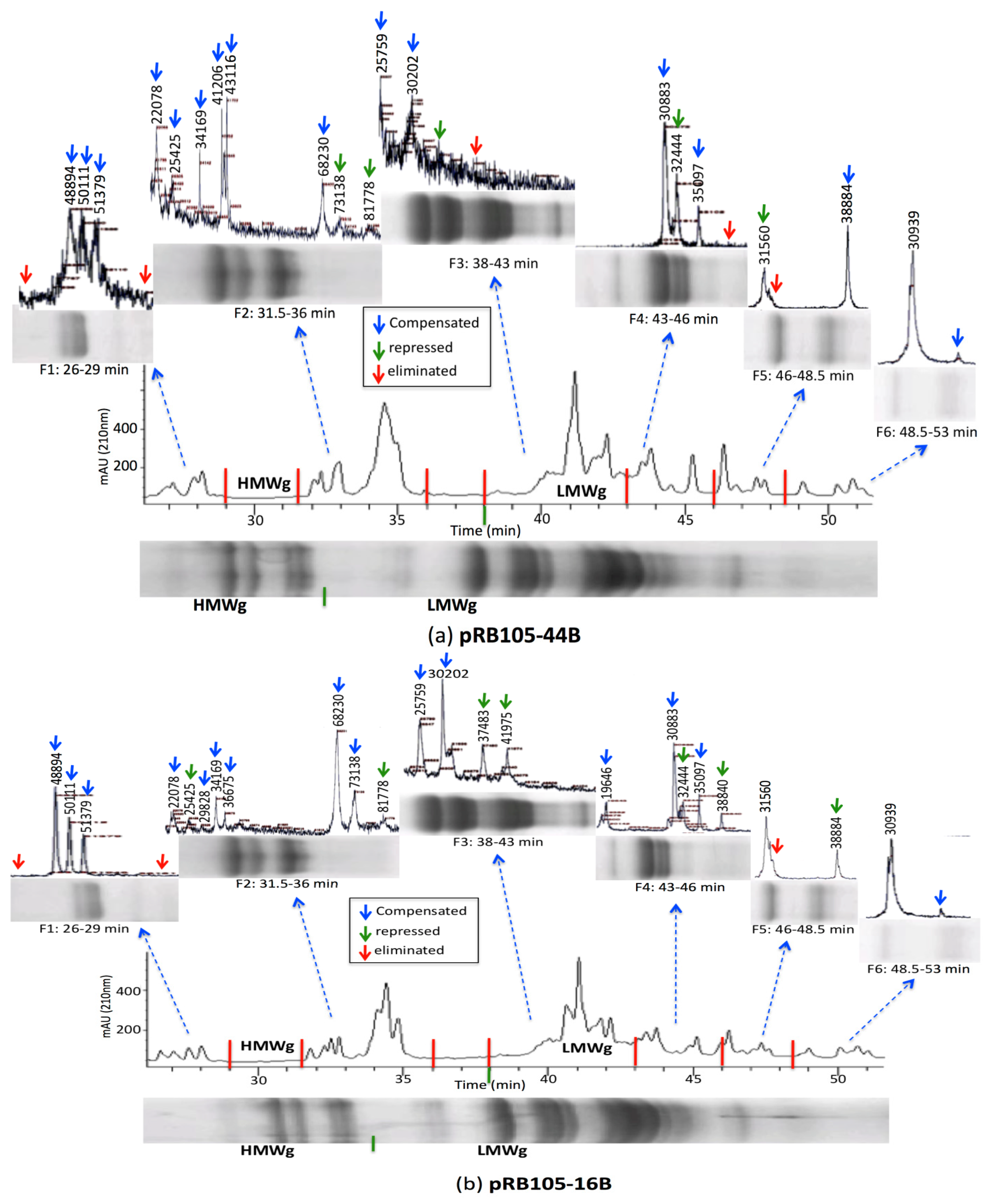
3.2. HPLC Fractionation and Mass Spectrometry for High-Resolution Analysis of Wheat Prolamins
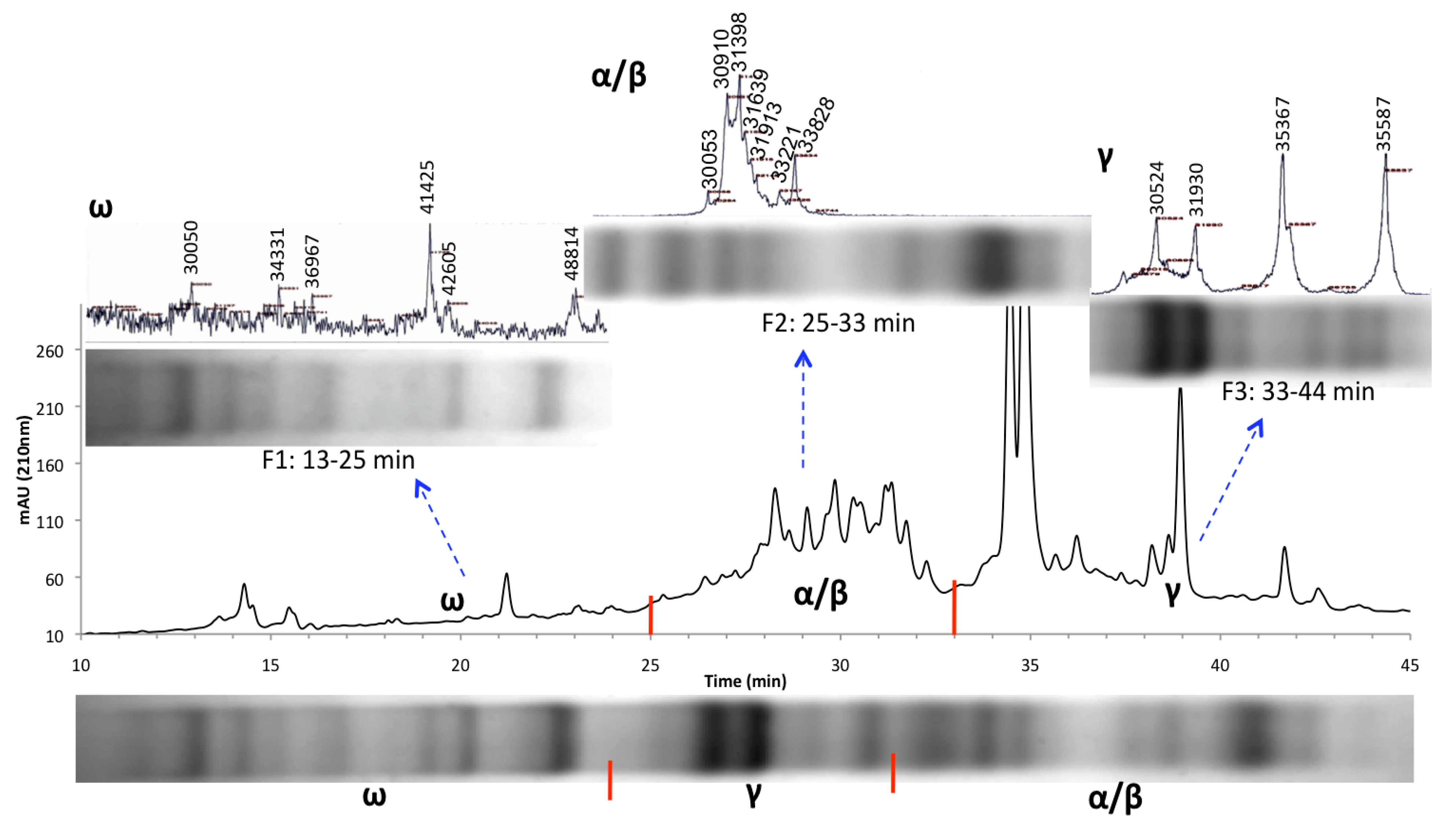
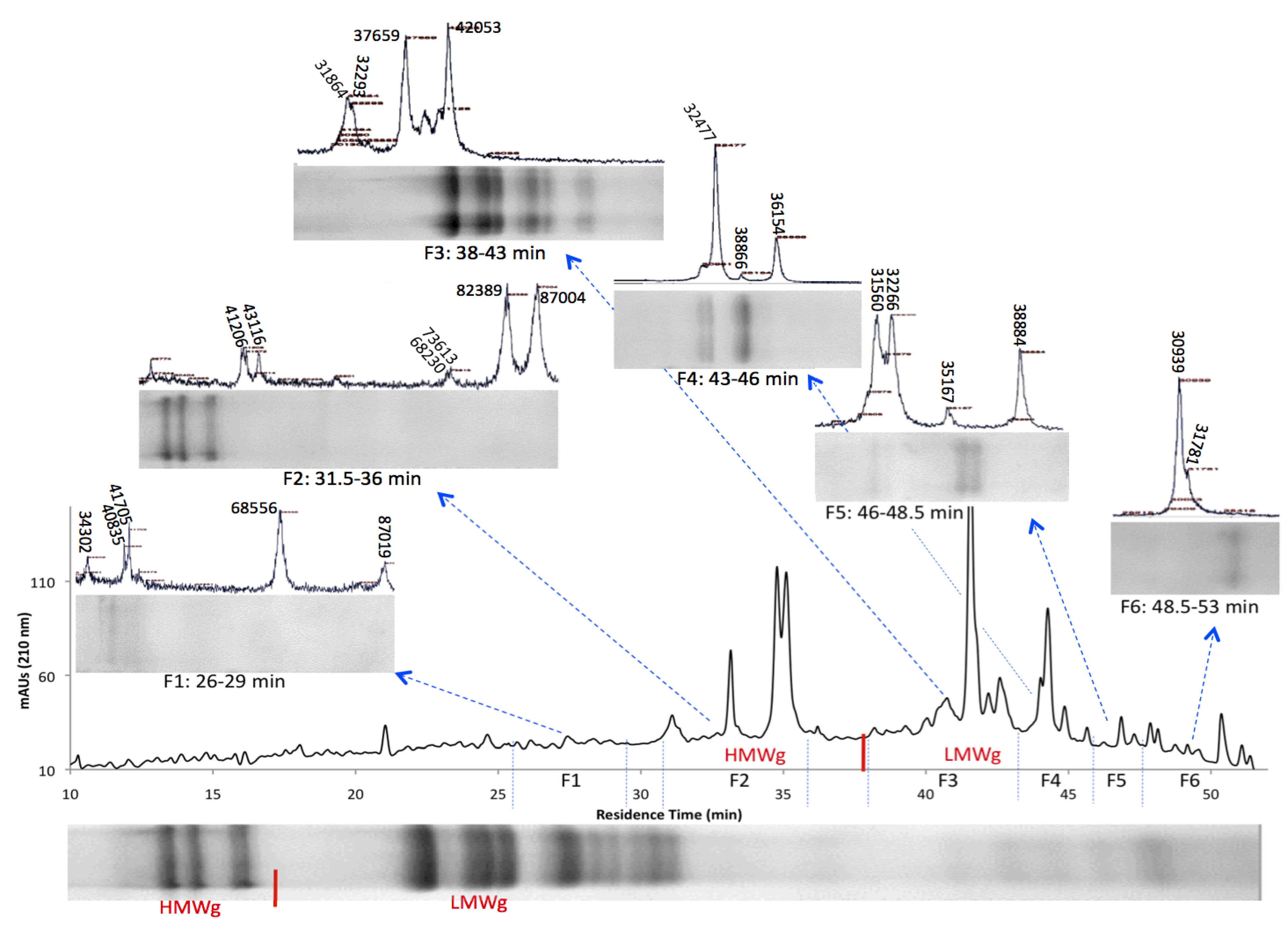
3.3. Relative Gliadin and Glutenin Concentrations Calculated Using Two Semi-Quantitative Methods
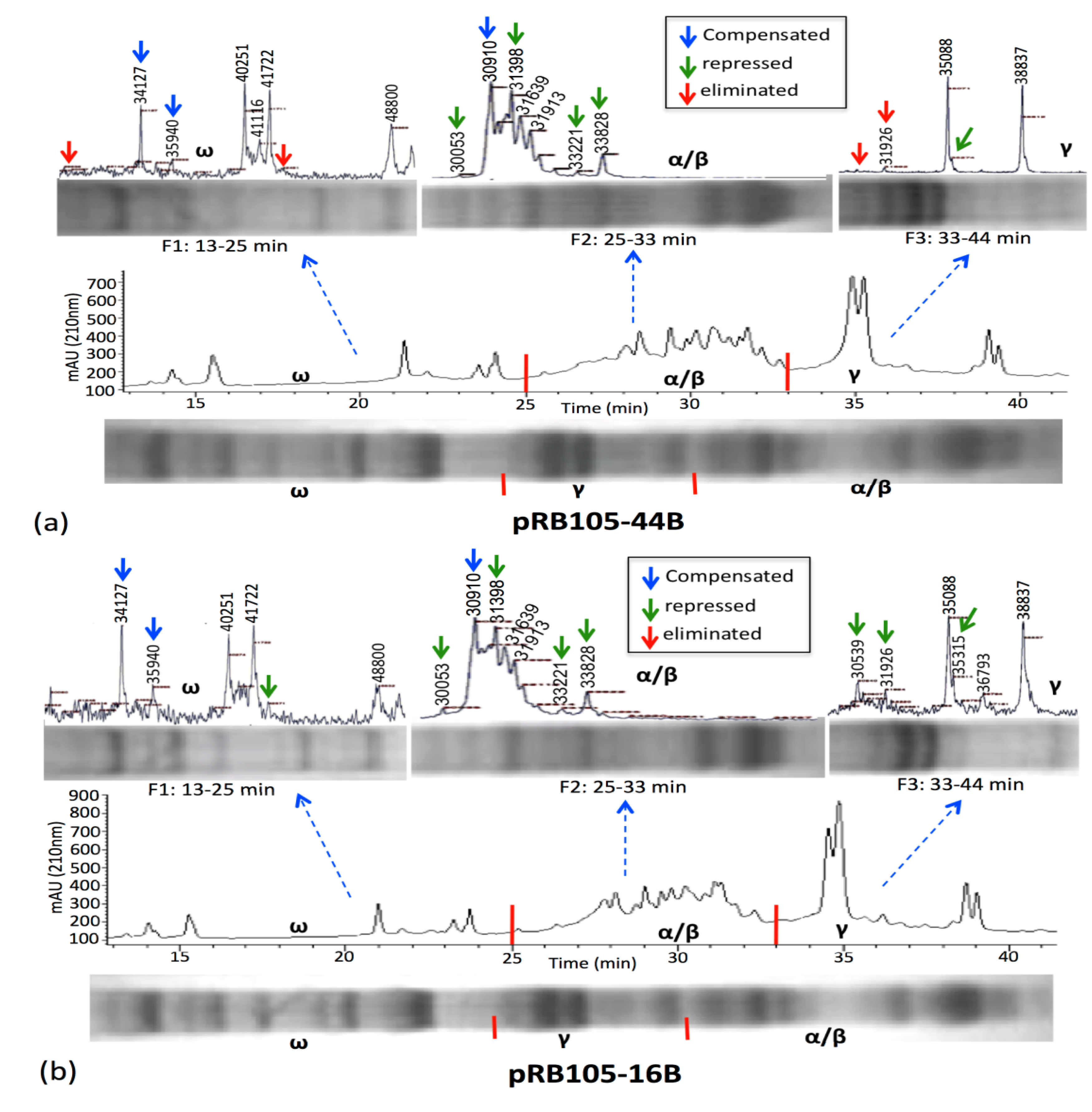
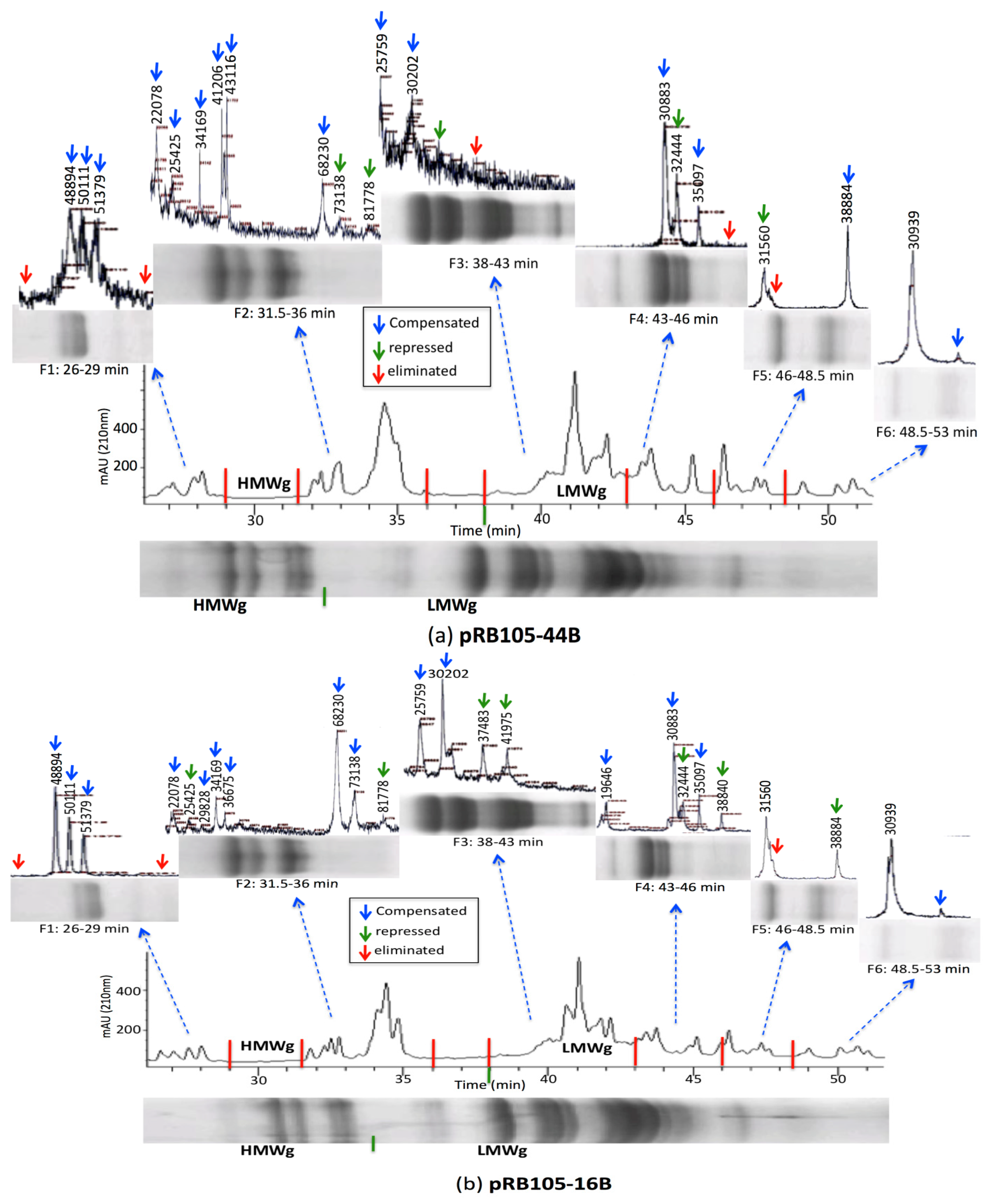
4. Conclusions
Abbreviations
| RP-HPLC | reversed phase high performance liquid chromatography |
| LMWg | low molecular weight glutenin |
| HMWg | high molecular weight glutenin |
| MALDI-TOF-MS | matrix assisted laser desorption ionization time of flight mass spectrometry |
Supplementary Files
Acknowledgments
Conflicts of Interest
References
- Brenchley, R.; Spannagl, M.; Pfeifer, M.; Barker, G.L.; D’Amore, R.; Allen, A.M.; McKenzie, N.; Kramer, M.; Kerhornou, A.; Bolser, D.; et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 2012, 491, 705–710. [Google Scholar]
- Western Organization of Resource Councils (WORC) Website. Available online: http://www.worc.org/userfiles/WorldWheatFacts.pdf (accessed 1 November 2013).
- Tatham, A.S.; Shewry, P.R. Allergens to wheat and related cereals. Clin. Exp. Allergy 2008, 38, 1712–1726. [Google Scholar]
- Shewry, P.R. Wheat. J. Exp. Bot. 2009, 60, 1537–1553. [Google Scholar] [CrossRef]
- Hadjivassiliou, M.; Sanders, D.S.; Grünewald, R.A.; Woodroofe, N.; Boscolo, S.; Aeschlimann, D. Gluten sensitivity: From gut to brain. Lancet Neurol. 2010, 9, 318–330. [Google Scholar] [CrossRef]
- Sapone, A.; Bai, J.C.; Ciacci, C.; Dolinsek, J.; Green, P.H.; Hadjivassiliou, M.; Kaukinen, K.; Rostami, K.; Sanders, D.S.; Schumann, M.; et al. Spectrum of gluten-related disorders: Consensus on new nomenclature and classification. BMC Med. 2012, 10. [Google Scholar] [CrossRef]
- Abadie, V.; Sollid, L.M.; Barreiro, L.B.; Jabri, B. Integration of genetic and immunological insights into a model of celiac disease pathogenesis. Annu. Rev. Immunol. 2011, 29, 493–525. [Google Scholar] [CrossRef]
- Sollid, L.M. Coeliac disease: Dissecting a complex inflammatory disorder. Nat. Rev. Immunol. 2002, 2, 647–655. [Google Scholar] [CrossRef]
- Osorio, C.; Wen, N.; Gemini, R.; Zemetra, R.; von Wettstein, D.; Rustgi, S. Targeted modification of wheat grain protein to reduce the content of celiac causing epitopes. Funct. Integr. Genomics 2012, 12, 417–438. [Google Scholar] [CrossRef]
- Wieser, H. Chemistry of gluten proteins. Food Microbiol. 2007, 24, 115–119. [Google Scholar] [CrossRef]
- Delcour, J.A.; Joye, I.J.; Pareyt, B.; Wilderjans, E.; Brijs, K.; Lagrain, B. Wheat gluten functionality as a quality determinant in cereal-based food products. Annu. Rev. Food Sci. Technol. 2012, 3, 469–492. [Google Scholar] [CrossRef]
- Wen, S.; Wen, N.; Pang, J.; Langen, G.; Brew-Appiah, R.A.; Mejias, J.H.; Osorio, C.; Yang, M.; Gemini, R.; Moehs, C.P.; et al. Structural genes of wheat and barley 5-methylcytosine DNA glycosylases and their potential applications for human health. Proc. Natl. Acad. Sci. USA 2012, 109, 20543–20548. [Google Scholar] [CrossRef]
- Van den Broeck, H.C.; America, A.H.P.; Smulders, M.J.M.; Bosch, D.; Hamer, R.J.; Gilissen, L.J.; van der Meer, I.M. A modified extraction protocol enables detection and quantification of celiac disease-related gluten proteins from wheat. J. Chromatogr. 2009, 877, 975–982. [Google Scholar] [CrossRef]
- Cunsolo, V.; Foti, S.; Saletti, R. Mass spectrometry in the characterization of cereal seed proteins. Eur. J. Mass Spectrom. 2004, 10, 359–370. [Google Scholar] [CrossRef]
- Stern, M.; Ciclitira, P.J.; van Eckert, R.; Feighery, C.; Janssen, F.W.; Méndez, E.; Mothes, T.; Troncone, R.; Wieser, H. Analysis and clinical effects of gluten in coeliac disease. Eur. J. Gastroenterol. Hepatol. 2001, 13, 741–747. [Google Scholar] [CrossRef]
- Piper, J.L.; Gray, G.M.; Khosla, C. High selectivity of human tissue transglutaminase for immunoactive gliadin peptides: Implications for celiac sprue. Biochemistry 2002, 41, 386–393. [Google Scholar] [CrossRef]
- Piper, J.L.; Gray, G.M.; Khosla, C. Effect of prolyl endopeptidase on digestive-resistant gliadin peptides in vivo. J. Pharmacol. Exp. Ther. 2004, 311, 213–219. [Google Scholar] [CrossRef]
- Hausch, F.; Shan, L.; Santiago, N.A.; Gray, G.M.; Khosla, C. Intestinal digestive resistance of immunodominant gliadin peptides. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G996–G1003. [Google Scholar]
- Shan, L.; Molberg, O.; Parrot, I.; Hausch, F.; Filiz, F.; Gray, G.M.; Sollid, L.M.; Khosla, C. Structural basis for gluten intolerance in celiac sprue. Science 2002, 297, 2275–2279. [Google Scholar] [CrossRef]
- Nilsen, E.M.; Lundin, K.E.A.; Krajci, P.; Scott, H.; Sollid, L.M.; Brandtzaeg, P. Gluten specific, HLA-DQ restricted T-cells from celiac mucosa produce cytokines with Thl or Th0 profile dominated by interferon γ. Gut 1995, 37, 766–776. [Google Scholar]
- Van de Wal, Y.; Kooy, Y.M.C.; Drijfhout, J.W.; Amons, R.; Koning, F. Peptide binding characteristics of the coeliac disease-associated DQ(α1*0501, β1*0201) molecule. Immunogenetics 1996, 44, 246–253. [Google Scholar] [CrossRef]
- Arentz-Hansen, H.; Körner, R.; Molberg, O.; Quarsten, H.; Vader, W.; Kooy, Y.M.; Lundin, K.E.; Koning, F.; Roepstorff, P.; Sollid, L.M.; et al. The intestinal T cell response to α-gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J. Exp. Med. 2000, 191, 603–612. [Google Scholar] [CrossRef]
- Molberg, O.; McAdam, S.; Lundin, K.E.A.; Kristiansen, C.; Arentz-Hansen, H.; Kett, K.; Sollid, L.M. T cells from celiac disease lesions recognize gliadin epitopes deamidated in situ by endogenous tissue transglutaminase. Eur. J. Immunol. 2001, 31, 1317–1323. [Google Scholar]
- Wieser, H.; Antes, S.; Seilmeier, W. Quantitative determination of gluten protein types in wheat flour by reversed-phase high-performance liquid chromatography. Cereal. Chem. 1998, 75, 644–650. [Google Scholar] [CrossRef]
- Mamone, G.; Addeo, F.; Chianese, L.; di Luccia, A.; de Martino, A.; Nappo, A.; Formisano, A.; de Vivo, P.; Ferranti, P. Characterization of wheat gliadin proteins by combined two-dimensional gel electrophoresis and tandem mass spectrometry. Proteomics 2005, 5, 2859–2865. [Google Scholar] [CrossRef]
- Dupont, F.M.; Vensel, W.H.; Tanaka, C.K.; Hurkman, W.J.; Altenbach, S.B. Deciphering the complexities of the wheat flour proteome using quantitative two-dimensional electrophoresis, three proteases and tandem mass spectrometry. Proteome Sci. 2011, 9. [Google Scholar] [CrossRef]
- Rombouts, I.; Lagrain, B.; Brunnbauer, M.; Delcour, J.A.; Koehler, P. Improved identification of wheat gluten proteins through alkylation of cysteine residues and peptide-based mass spectrometry. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef]
- Lagrain, B.; Brunnbauer, M.; Rombouts, I.; Koehler, P. Identification of intact high molecular weight glutenin subunits from the wheat proteome using combined liquid chromatography-electrospray ionization mass spectrometry. PLoS One 2013, 8, e58682. [Google Scholar]
- Stephen, L. Gluten detection and speciation by liquid chromatography mass spectrometry (LC-MS/MS). Foods 2014, 3, 13–29. [Google Scholar]
- Vensel, W.H.; Tanaka, C.K.; Altenbach, S.B. Protein composition of wheat gluten polymer fractions determined by quantitative two-dimensional gel electrophoresis and tandem mass spectrometry. Proteome Sci. 2014, 12. [Google Scholar] [CrossRef]
- Bradford, M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Khan, K.; Hamada, A.S.; Patek, J. Polyacrylamide gel electrophoresis for wheat variety identification: Effect of variables on gel properties. Cereal. Chem. 1985, 62, 310–313. [Google Scholar]
- Fling, S.P.; Gregerson, D.S. Peptide and protein molecular weight determination by electrophoresis using a high-molarity Tris buffer system without urea. Anal. Biochem. 1986, 155, 83–88. [Google Scholar] [CrossRef]
- Neuhoff, V.; Arold, N.; Taube, D.; Ehrhardt, W. Improved staining of proteins in polyacrylamide gels including isoelectric focusing gels with clear background at nanogram sensitivity using Coomassie Brilliant Blue G-250 and R-250. Electrophoresis 1988, 9, 255–262. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to Image J: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Long, W. Scale-up: Development of preparative HPLC separation from analytical HPLC. Sep. Times 2003, 16, 15–16. [Google Scholar]
- Shewry, P.R.; Halford, N.G. Cereal seed storage proteins: Structures, properties and role in grain utilization. J. Exp. Bot. 2002, 53, 947–958. [Google Scholar] [CrossRef]
- Triboï, E.; Martre, P.; Triboï-Blondel, A.M. Environmentally-induced changes in protein composition in developing grains of wheat are related to changes in total protein content. J. Exp. Bot. 2003, 54, 1731–1742. [Google Scholar] [CrossRef]
- Shewry, P.R.; Halford, N.G.; Lafiandra, D. Genetics of wheat gluten proteins. Adv. Genet. 2003, 49, 111–184. [Google Scholar] [CrossRef]
- Bietz, J.A. HPLC of Cereal Endosperm Storage Proteins. In HPLC of Biological Macro-Molecules; Gooding, K.M., Regnier, F.E., Eds.; Marcel Dekker: New York, NY, USA, 2002; pp. 547–587. [Google Scholar]
- Ricker, R.D.; Woodward, C.B., III; Forrer, K.; Permar, B.J.; Chen, W. Options for rapid analysis of peptides and proteins, using wide-pore, superficially porous, high-performance liquid chromatography particles with unique bonded-phase ligands. J. Chromatogr. Sci. 2008, 46, 261–268. [Google Scholar] [CrossRef]
- Marchylo, B.A.; Hatcher, D.W.; Kruger, J.E.; Kirkland, J.J. Reversed-phase high-performance liquid chromatographic analysis of wheat proteins using a new, highly stable column. Cereal. Chem. 1992, 69, 371–378. [Google Scholar]
- Bietz, J.A.; Cobb, L.A. Improved procedures for rapid wheat varietal identification by reversed-phase high-performance liquid chromatography of gliadin. Cereal. Chem. 1985, 62, 332–339. [Google Scholar]
- Zhu, C.; Goodall, D.M.; Wren, S.A.C. Elevated temperature HPLC: Principles and applications to small molecules and biomolecules. LCGC Asia Pac. 2005, 8, 48–59. [Google Scholar]
- Gupta, P.K.; Rustgi, S. Mass spectrometry: An essential tool for genome and proteome analysis. Ind. J. Biotechnol. 2003, 2, 48–64. [Google Scholar]
- Ferranti, P.; Mamone, G.; Picariello, G.; Addeo, F. Mass spectrometry analysis of gliadins in celiac disease. J. Mass Spectrom. 2007, 42, 1531–1548. [Google Scholar] [CrossRef]
- Geng, T.; Westphal, C.D.; Yeung, J.M. Detection of Gluten by Commercial Test Kits: Effects of Food Matrices and Extraction Procedures. In Food Contaminants Mycotoxins and Food Allergens; Siantar, D.P., Trucksess, M.W., Scott, P.M., Herman, E.M., Eds.; American Chemical Society Symposium Series: Washington, DC, USA, 2008; Volume 1001, pp. 462–475. [Google Scholar]
- Mendez, E.; Camafeita, E.; San Sebastian, J.; Valle, I.; Solis, J.; Mayer-Posner, F.J.; Suckau, D.; Marfisi, C.; Soriano, F. Direct identification of wheat gliadins and related cereal prolamins by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. J. Mass Spectrom. 1995, 199, S123–S128. [Google Scholar]
- Marín, S.; Gil-Humanes, J.; Hernando, A.; Barro, F. Characterization of α/β- and γ-gliadins in commercial varieties and breeding lines of durum wheat using MALDI-TOF and A-PAGE gels. Biochem. Genet. 2011, 49, 735–747. [Google Scholar] [CrossRef]
- Brew-Appiah, R.A.T.; Rustgi, S.; Ankrah, N.; Wen, S.; Claar, M.; Langen, G.; Kogel, K.H.; Weigel, D.; von Wettstein, D. Artificial microRNAs for silencing wheat proteins causing celiac disease. In Proceedings of the Annual Meeting of the American Society of Plant Biologists, Montreal, Canada, 31 July–4 August 2010.
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mejías, J.H.; Lu, X.; Osorio, C.; Ullman, J.L.; Von Wettstein, D.; Rustgi, S. Analysis of Wheat Prolamins, the Causative Agents of Celiac Sprue, Using Reversed Phase High Performance Liquid Chromatography (RP-HPLC) and Matrix-Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry (MALDI-TOF-MS). Nutrients 2014, 6, 1578-1597. https://doi.org/10.3390/nu6041578
Mejías JH, Lu X, Osorio C, Ullman JL, Von Wettstein D, Rustgi S. Analysis of Wheat Prolamins, the Causative Agents of Celiac Sprue, Using Reversed Phase High Performance Liquid Chromatography (RP-HPLC) and Matrix-Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry (MALDI-TOF-MS). Nutrients. 2014; 6(4):1578-1597. https://doi.org/10.3390/nu6041578
Chicago/Turabian StyleMejías, Jaime H., Xiaoqiao Lu, Claudia Osorio, Jeffrey L. Ullman, Diter Von Wettstein, and Sachin Rustgi. 2014. "Analysis of Wheat Prolamins, the Causative Agents of Celiac Sprue, Using Reversed Phase High Performance Liquid Chromatography (RP-HPLC) and Matrix-Assisted Laser Desorption Ionization Time of Flight Mass Spectrometry (MALDI-TOF-MS)" Nutrients 6, no. 4: 1578-1597. https://doi.org/10.3390/nu6041578