Peculiarities of One-Carbon Metabolism in the Strict Carnivorous Cat and the Role in Feline Hepatic Lipidosis



Abstract
:1. Introduction
2. Feline-Specific Metabolic Features

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amount/kg BW0.75 | Cat (1) | Mink (2) | Dog (3) | Rat (4) | Human Being (5) | |
|---|---|---|---|---|---|---|
| Male | Female | |||||
| Protein (g) | 4.48 | 7.81 | 3.28 | 1.47 | 2.31 | 1.90 |
| Arginine (g) | 0.17 | NA | 0.11 | 0.13 † | NE | NE |
| Methionine (g) | 0.038 | NA | 0.11 | NA | NA | NA |
| Methionine + Cysteine (g) | 0.076 | NA | 0.21 | 0.068 | 0.055 | 0.055 |
| Taurine (g) | 0.009 | NA | NE | NE | NE | NE |
| Cobalamin (μg) | 0.50 | 0.98 * | 1.15 | 1.47 † | 0.099 | 0.099 |
| Choline (mg) | 57.0 | NA | 56.0 | 22.1 † | 22.7 | 17.6 |
| Folate (μg) | 16.8 | 15.0 * | 8.9 | 29. 47 † | 16.5 | 16.5 |
| Pyridoxine (mg) | 0.056 | 0.048 * | 0.049 | 0. 177 † | 0.054 | 0.054 |
2.1. Dietary Protein Requirement
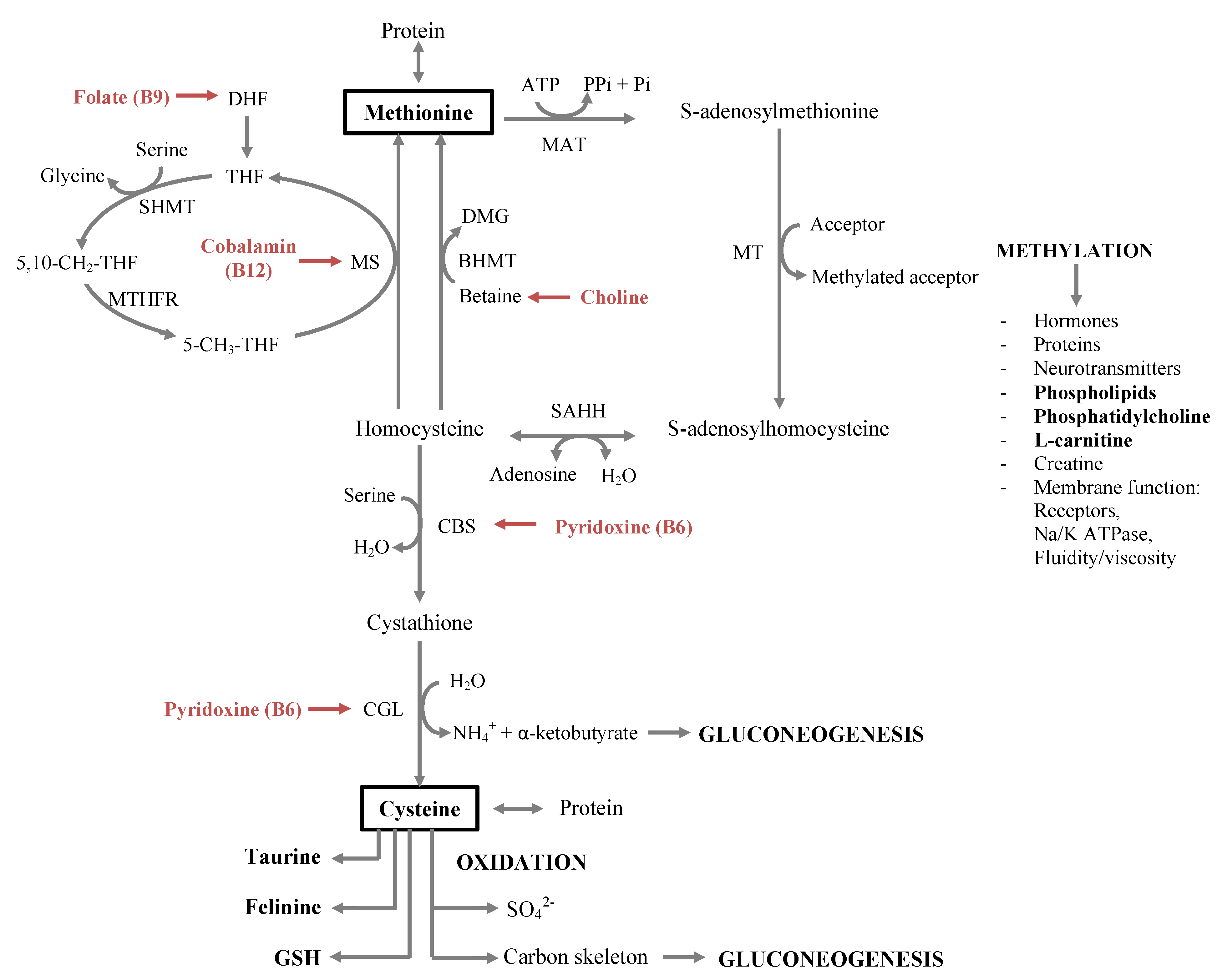
2.2. One-Carbon Metabolism
2.2.1. Essential Amino Acids

2.2.2. B-Vitamins Involved in One-Carbon Metabolism
2.3. Essential Fatty Acid Requirement
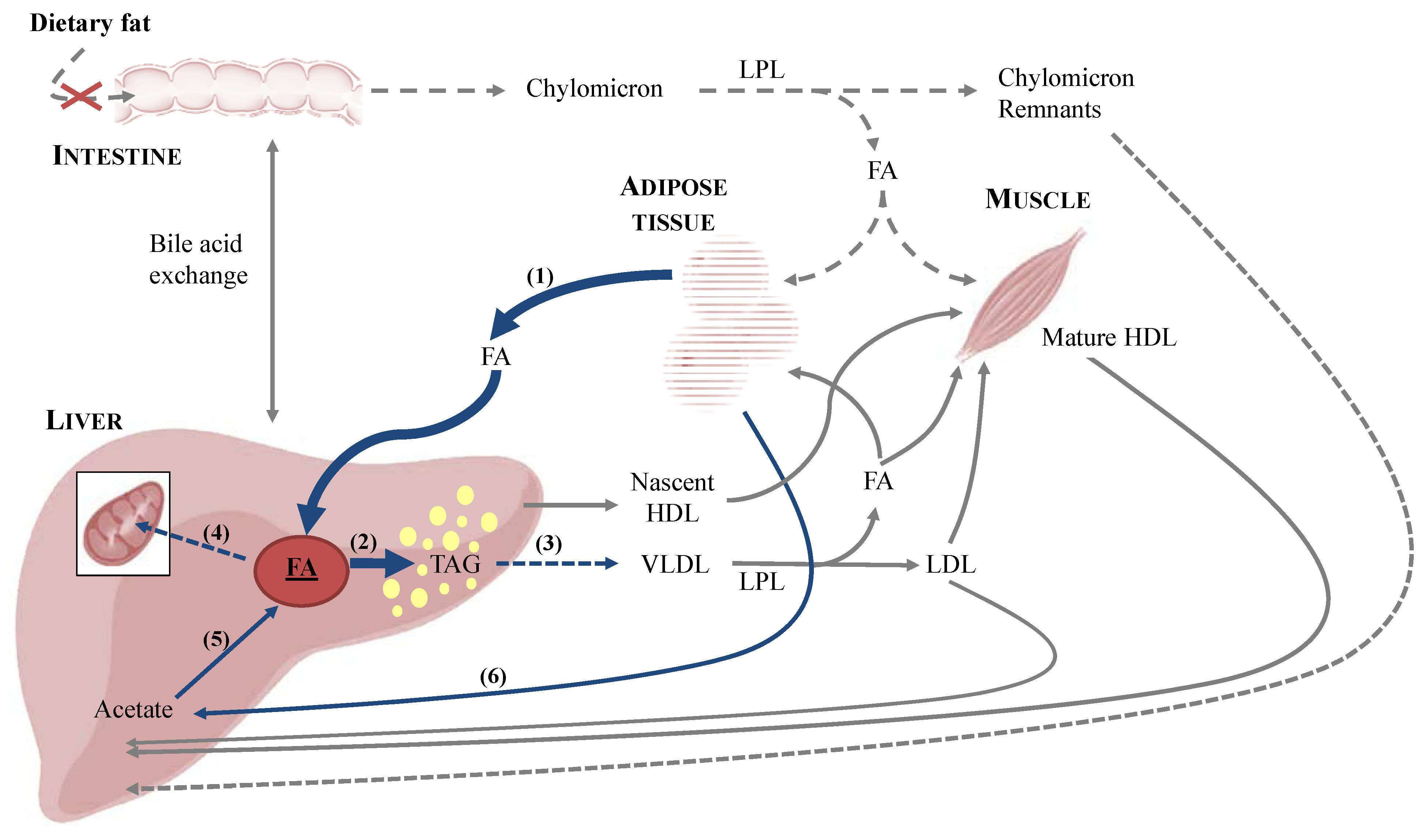
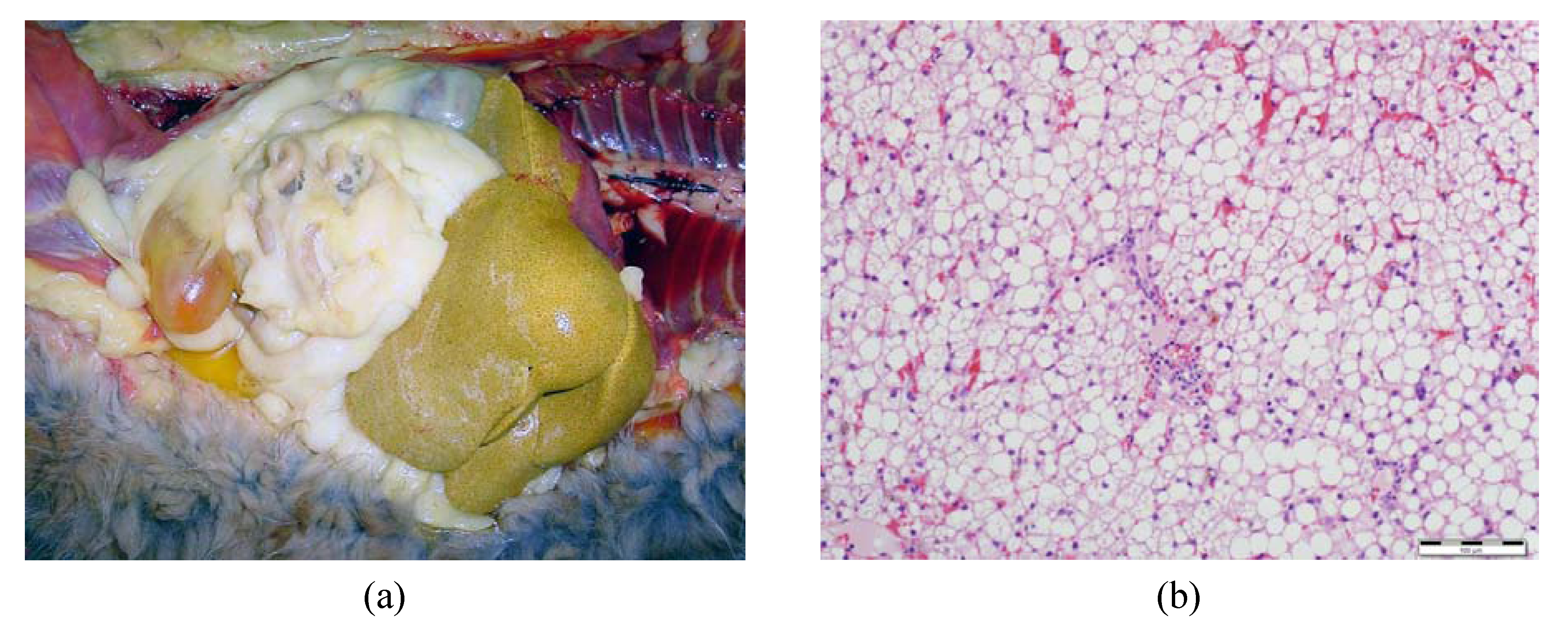
3. Implications for Feline Health—Feline Hepatic Lipidosis

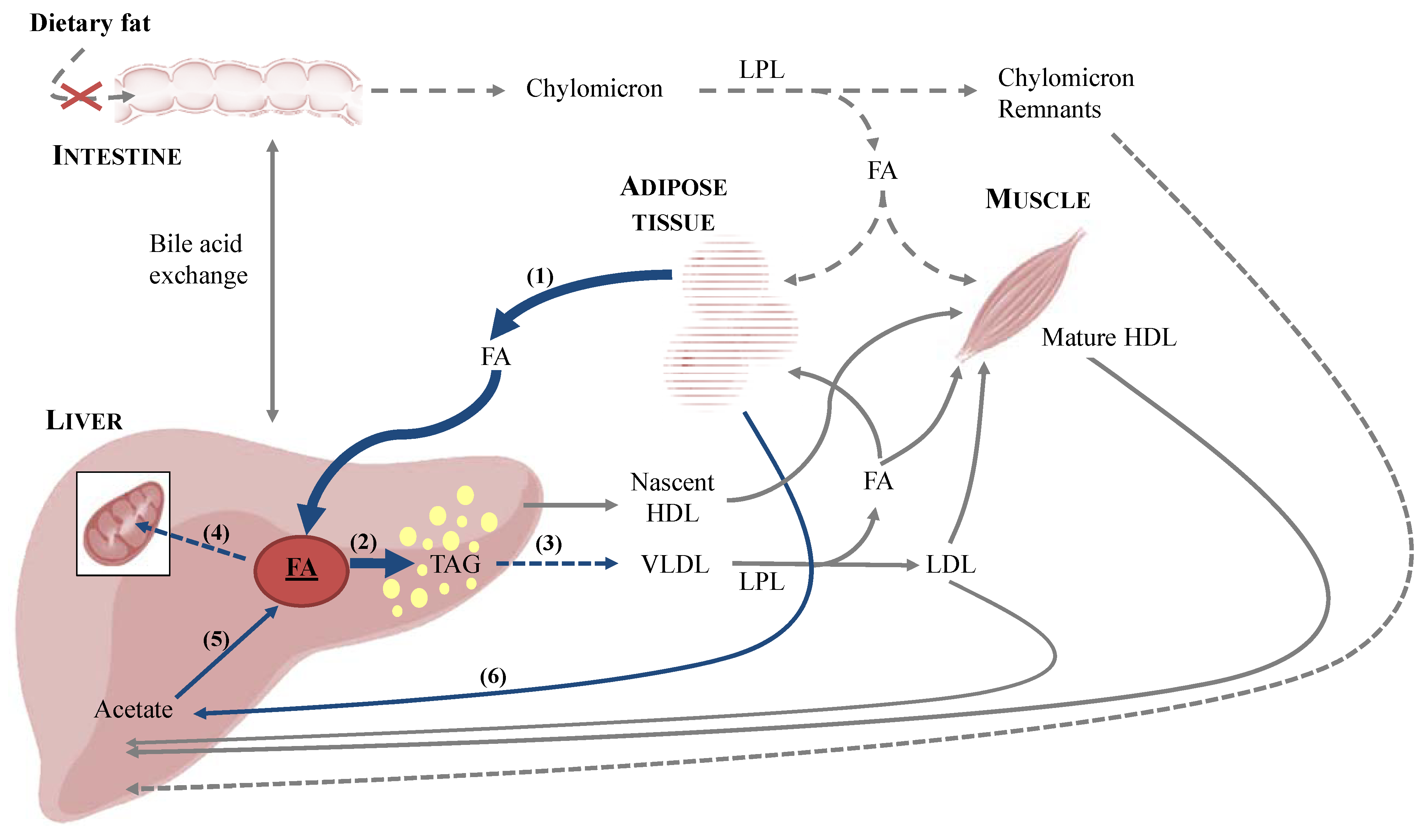
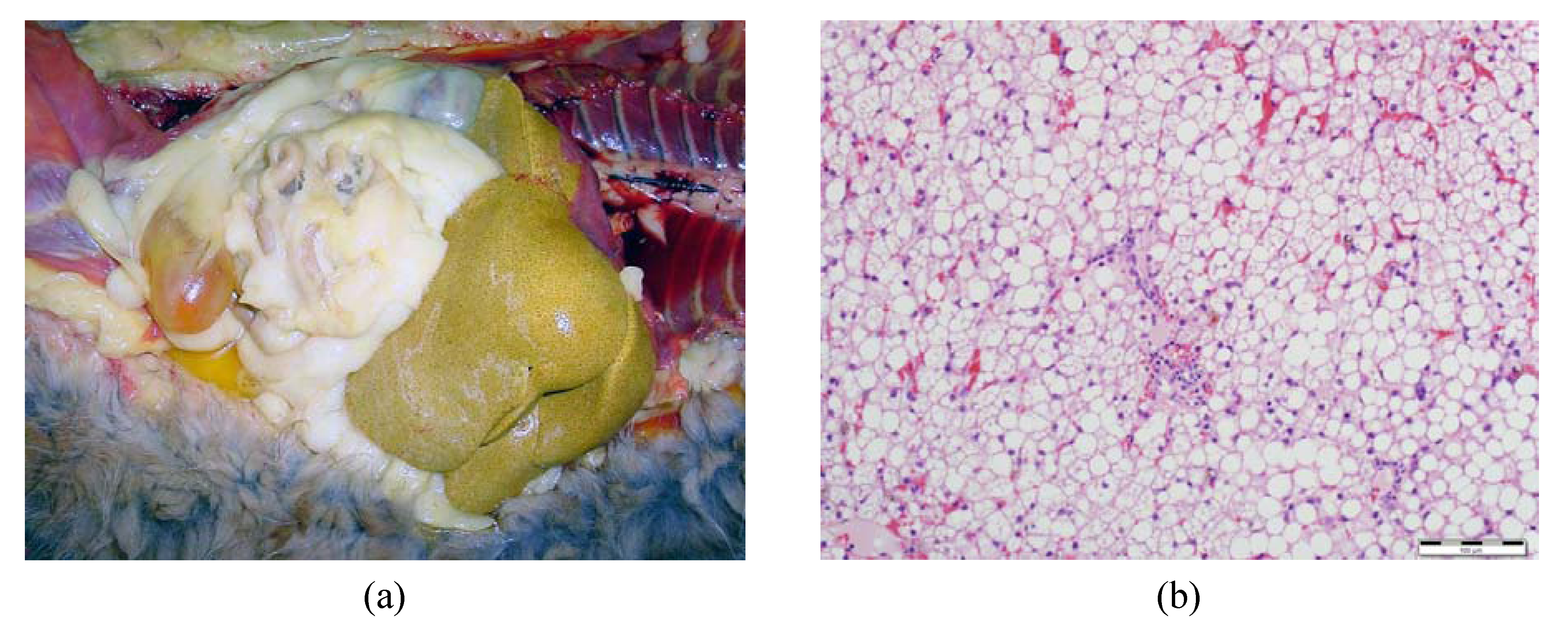
3.1. Onset of Feline Hepatic Lipidosis
3.1.1. Anorexia and Insulin Resistance
3.1.2. De Novo Synthesis of Fatty Acids
3.2. Metabolic Aspects of Feline Hepatic Lipidosis
3.2.1. Protein Malnutrition, Arginine and Taurine Deficiency
3.2.2. Importance of Methionine, SAMe and Carnitine for Proper Lipid Metabolism
3.2.3. B-Vitamin Deficiency
3.2.4. Essential Fatty Acid Deficiency
4. Conclusions
Acknowledgements
Conflict of Interest
References
- Angulo, P.; Lindor, K.D. Treatment of non-alcoholic steatohepatitis. Best Pract. Res. Clin. Gastroenterol. 2002, 16, 797–810. [Google Scholar] [CrossRef]
- Reddy, J.K.; Rao, M.S. Lipid metabolism and liver inflammation. II. Fatty liver disease and fatty acid oxidation. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G852–G858. [Google Scholar] [CrossRef]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion 2006, 6, 1–28. [Google Scholar]
- Larter, C.Z.; Yeh, M.M. Animal models of NASH: Getting both pathology and metabolic context right. J. Gastroenterol. Hepatol. 2008, 23, 1635–1648. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Goldin, R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int. J. Exp. Pathol. 2006, 87, 1–16. [Google Scholar] [CrossRef]
- Ogasawara, M.; Hirose, A.; Ono, M.; Aritake, K.; Nozaki, Y.; Takahashi, M.; Okamoto, N.; Sakamoto, S.; Iwasaki, S.; Asanuma, T.; et al. A novel and comprehensive mouse model of human non-alcoholic steatohepatitis with the full range of dysmetabolic and histological abnormalities induced by gold thioglucose and a high-fat diet. Liver Int. 2011, 31, 542–551. [Google Scholar]
- Hoenig, M. The cat as a model for human nutrition and disease. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 584–588. [Google Scholar] [CrossRef]
- Henson, M.S.; O’Brien, T.D. Feline models of type 2 diabetes mellitus. ILAR J. 2006, 47, 234–242. [Google Scholar] [CrossRef]
- German, A.J. The growing problem of obesity in dogs and cats. J. Nutr. 2006, 136, 1940S–1946S. [Google Scholar]
- Scarlett, J.M.; Donoghue, S. Associations between body condition and disease in cats. J. Am. Vet. Med. Assoc. 1998, 212, 1725–1731. [Google Scholar]
- Blanchard, G.; Paragon, B.M.; Serougne, C.; Ferezou, J.; Milliat, F.; Lutton, C. Plasma lipids, lipoprotein composition and profile during induction and treatment of hepatic lipidosis in cats and the metabolic effect of one daily meal in healthy cats. J. Anim. Physiol. Anim. Nutr. 2004, 88, 73–87. [Google Scholar] [CrossRef]
- Armstrong, P.J.; Blanchard, G. Hepatic lipidosis in cats. Vet. Clin. N. Am. Small Anim. Pract. 2009, 39, 599–616. [Google Scholar] [CrossRef]
- Center, S.A. Feline hepatic lipidosis. Vet. Clin. N. Am. Small Anim. Pract. 2005, 35, 225–269. [Google Scholar] [CrossRef]
- Griffin, B. Feline hepatic lipidosis: Pathophysiology, clinical signs, and diagnosis. Comp. Cont. Educ. Pract. 2000, 22, 847–856. [Google Scholar]
- MacDonald, M.L.; Rogers, Q.R.; Morris, J.G. Nutrition of the domestic cat, a mammalian carnivore. Annu. Rev. Nutr. 1984, 4, 521–562. [Google Scholar] [CrossRef]
- Morris, J.G. Idiosyncratic nutrient requirements of cats appear to be diet-induced evolutionary adaptations. Nutr. Res. Rev. 2002, 15, 153–168. [Google Scholar] [CrossRef]
- Zoran, D.L. The carnivore connection to nutrition in cats. J. Am. Vet. Med. Assoc. 2002, 221, 1559–1567. [Google Scholar] [CrossRef]
- Verbrugghe, A.; Hesta, M.; Daminet, S.; Janssens, G.P. Nutritional modulation of insulin resistance in the true carnivorous cat: A review. Crit. Rev. Food Sci. Nutr. 2012, 52, 172–182. [Google Scholar] [CrossRef]
- Eisert, R. Hypercarnivory and the brain: Protein requirements of cats reconsidered. J. Comp. Physiol. 2011, 181, 1–17. [Google Scholar]
- National Research Council, Nutrient Requirements and Dietary Nutrient Concentrations. In Nutrient Requirements of Dogs and Cats; The National Academies Press: Washington, DC, USA, 2006; pp. 354–370.
- National Research Council, Nutrient Requirements of Mink and Foxes, 2nd ed.; The National Academies Press: Washington, DC, USA, 1982; pp. 33–66.
- National Research Council, Nutrient Requirements of the Laboratory Rat. In Nutrient Requirements of Laboratory Animals, 4th ed.; The National Academies Press: Washington, DC, USA, 1995; pp. 11–79.
- Food and Nutrition Board, Institute of Medicine, Protein and Amino Acids. In Dietary Reference Intakes for Energy, Carbohydrates, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids; The National Academies Press: Washington, DC, USA, 2005; pp. 589–768.
- Food and Nutrition Board, Institute of Medicine, Summary Tables, Dietary Reference Intakes. In Dietary Reference Intakes for Energy, Carbohydrates, Fiber, Fat, Fatty Acids, Cholesterol, Protein and Amino Acids; The National Academies Press: Washington, DC, USA, 2005; pp. 1319–1332.
- Schimke, R.T. Differential effects of fasting and protein-free diets on levels of urea cycle enzymes in rat liver. J. Biol. Chem. 1962, 237, 1921–1924. [Google Scholar]
- Schimke, R.T. Studies on factors affecting the levels of urea cycle enzymes in rat liver. J. Biol. Chem. 1963, 238, 1012–1018. [Google Scholar]
- Payne, E.; Morris, J.G. The effect of protein content of the diet on the rate of urea formation in sheep liver. Biochem. J. 1969, 113, 659–662. [Google Scholar]
- Rosebrough, R.W.; Steele, N.C.; McMurtry, J.P. Effect of protein level and supplemental lysine on growth and urea cycle enzyme activity in the pig. Growth 1983, 47, 348–360. [Google Scholar]
- Rogers, Q.R.; Morris, J.G.; Freedland, R.A. Lack of hepatic enzymatic adaptation to low and high levels of dietary protein in the adult cat. Enzyme 1977, 22, 348–356. [Google Scholar]
- Silva, S.V.P.S.; Mercer, J.R. Effect of protein intake on amino acid catabolism and gluconeogensis by isolated hepatocytes from cats (Felis domestica). Comp. Biochem. Physiol. 1985, 80, 603–607. [Google Scholar]
- Russell, K.; Lobley, G.E.; Rawlings, J.; Millward, D.J.; Harper, E.J. Urea kinetics of a carnivore, Felissilvestris catus. Br. J. Nutr. 2000, 84, 597–604. [Google Scholar]
- Russell, K.; Murgatroyd, P.R.; Batt, R.M. Net protein oxidation is adapted to dietary protein intake in domestic cats (Felissilvestris catus). J. Nutr. 2002, 132, 456–460. [Google Scholar]
- Green, A.S.; Ramsey, J.J.; Villaverde, C.; Asami, D.K.; Wei, A.; Fascetti, A.J. Cats are able to adapt protein oxidation to protein intake provided their requirement for dietary protein is met. J. Nutr. 2008, 138, 1053–1060. [Google Scholar]
- Russell, K.; Lobley, G.E.; Millward, D.J. Whole-body protein turnover of a carnivore, Felissilvestris catus. Br. J. Nutr. 2003, 89, 29–37. [Google Scholar] [CrossRef]
- Washizu, T.; Tanaka, A.; Sako, T.; Washizu, M.; Arai, T. Comparison of the activities of enzymes related to glycolysis and gluconeogenesis in the liver of dogs and cats. Res. Vet. Sci. 1999, 67, 205–206. [Google Scholar] [CrossRef]
- Tanaka, A.; Inoue, A.; Takeguchi, A.; Washizu, T.; Bonkobara, M.; Arai, T. Comparison of expression of glucokinase gene and activities of enzymes related to glucose metabolism in livers between dog and cat. Vet. Res. Commun. 2005, 29, 477–485. [Google Scholar] [CrossRef]
- Kettelhut, I.C.; Foss, M.C.; Migliorini, R.H. Glucose homeostasis in a carnivorous animal (Cat) and in rats fed a high-protein diet. Am. J. Physiol. 1980, 239, R437–R444. [Google Scholar]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
- Brosnan, J.T.; Brosnan, M.E. The sulfur-containing amino acids: An overview. J. Nutr. 2006, 136, 1636S–1640S. [Google Scholar]
- Mudd, S.H.; Brosnan, J.T.; Brosnan, M.E.; Jacobs, R.L.; Stabler, S.P.; Allen, R.H.; Vance, D.E.; Wagner, C. Methyl balance and transmethylation fluxes in humans. Am. J. Clin. Nutr. 2007, 85, 19–25. [Google Scholar]
- Stead, L.M.; Brosnan, J.T.; Brosnan, M.E.; Vance, D.E.; Jacobs, R.L. Is it time to reevaluate methyl balance in humans? Am. J. Clin. Nutr. 2006, 83, 5–10. [Google Scholar]
- Vaz, F.M.; Wanders, R.J. Carnitine biosynthesis in mammals. Biochem. J. 2002, 361, 417–429. [Google Scholar] [CrossRef]
- Teeter, R.G.; Baker, D.H.; Corbin, J.E. Methionine and cystine requirements of the cat. J. Nutr. 1978, 108, 291–295. [Google Scholar]
- Ashmore, H.; Uttley, M. Measurement of the rate of growth of rodent hair using cystine labelled with sulphur35. Nature 1965, 206, 108–109. [Google Scholar] [CrossRef]
- Hendriks, W.H.; Rutherfurd, S.M.; Rutherfurd, K.J. Importance of sulfate, cysteine and methionine as precursors to felinine synthesis by domestic cats (Felis catus). Comp. Biochem. Physiol. C 2001, 129, 211–216. [Google Scholar]
- Hendriks, W.H.; Rutherfurd-Markwick, K.J.; Weidgraaf, K.; Hugh Morton, R.; Rogers, Q.R. Urinary felinine excretion in intact male cats is increased by dietary cystine. Br. J. Nutr. 2008, 100, 801–809. [Google Scholar]
- Hendriks, W.H.; Moughan, P.J.; Tarttelin, M.F.; Woolhouse, A.D. Felinine: A urinary amino acid of felidae. Comp. Biochem. Physiol. B 1995, 112, 581–588. [Google Scholar] [CrossRef]
- Oomori, S.; Mizuhara, S. Structure of a new sulfur-containing amino acid. Arch. Biochem. Biophys. 1962, 96, 179–185. [Google Scholar] [CrossRef]
- Tarttelin, M.F.; Hendriks, W.H.; Moughan, P.J. Relationship between plasma testosterone and urinary felinine in the growing kitten. Physiol. Behav. 1998, 65, 83–87. [Google Scholar] [CrossRef]
- Hendriks, W.H.; Tarttelin, M.F.; Moughan, P.J. Twenty-four hour felinine excretion patterns in entire and castrated cats. Physiol. Behav. 1995, 58, 467–469. [Google Scholar] [CrossRef]
- Knopf, K.; Sturman, J.A.; Armstrong, M.; Hayes, K.C. Taurine: An essential nutrient for the cat. J. Nutr. 1978, 108, 773–778. [Google Scholar]
- Rabin, B.; Nicolosi, R.J.; Hayes, K.C. Dietary influence on bile acid conjugation in the cat. J. Nutr. 1976, 106, 1241–1246. [Google Scholar]
- Washizu, T.; Ikenaga, H.; Washizu, M.; Ishida, T.; Tomoda, I.; Kaneko, J.J. Bile acid composition of dog and cat gall-bladder bile. Nihon Juigaku Zasshi 1990, 52, 423–425. [Google Scholar] [CrossRef]
- Hepner, G.W.; Sturman, J.A.; Hofmann, A.F.; Thomas, P.J. Metabolism of steroid and amino acid moieties of conjugated bile acids in man. 3. Cholyltaurine (taurocholic acid). J. Clin. Investig. 1973, 52, 433–440. [Google Scholar] [CrossRef]
- Morris, J.G.; Rogers, Q.R.; Pacioretty, L. Taurine: An essential nutrient for cats. J. Small Anim. Pract. 1990, 31, 502–509. [Google Scholar] [CrossRef]
- Hickman, M.A.; Rogers, Q.R.; Morris, J.G. Effect of processing on fate of dietary 14C-taurine in cats. J. Nutr. 1990, 120, 995–1000. [Google Scholar]
- Hansen, S.H. The role of taurine in diabetes and the development of diabetic complications. Diabetes Metab. Res. Rev. 2001, 17, 330–346. [Google Scholar] [CrossRef]
- Huxtable, R.J. Physiological actions of taurine. Physiol. Rev. 1992, 72, 101–163. [Google Scholar]
- Jacobsen, J.G.; Smith, L.H. Biochemistry and physiology of taurine and taurine derivatives. Physiol. Rev. 1968, 48, 424–511. [Google Scholar]
- Hayes, K.C.; Sturman, J.A. Taurine in metabolism. Annu. Rev. Nutr. 1981, 1, 401–425. [Google Scholar] [CrossRef]
- Stapleton, P.P.; O’Flaherty, L.; Redmond, H.P.; Bouchier-Hayes, D.J. Host defense—A role for the amino acid taurine? J. Parenter. Enter. Nutr. 1998, 22, 42–48. [Google Scholar]
- Hayes, K.C.; Carey, R.E.; Schmidt, S.Y. Retinal degeneration associated with taurine deficiency in the cat. Science 1975, 188, 949–951. [Google Scholar]
- Schmidt, S.Y.; Berson, E.L.; Hayes, K.C. Retinal degeneration in the taurine-deficient cat. Trans. Sect. Ophthalmol. Am. Acad. Ophthalmol. Otolaryngol. 1976, 81, OP687–OP693. [Google Scholar]
- Sturman, J.A. Nutritional taurine and central nervous system development. Ann. N. Y. Acad. Sci. 1986, 477, 196–213. [Google Scholar] [CrossRef]
- Sturman, J.A.; Messing, J.M. Dietary taurine content and feline reproduction and outcome. J. Nutr. 1991, 121, 1195–1203. [Google Scholar]
- Pion, P.D.; Kittleson, M.D.; Rogers, Q.R.; Morris, J.G. Myocardial failure in cats associated with low plasma taurine: A reversible cardiomyopathy. Science 1987, 237, 764–768. [Google Scholar]
- Cantafora, A.; Blotta, I.; Rossi, S.S.; Hofmann, A.F.; Sturman, J.A. Dietary taurine content changes liver lipids in cats. J. Nutr. 1991, 121, 1522–1528. [Google Scholar]
- Fau, D.; Morris, J.G.; Rogers, Q.R. Effects of high dietary methionine on activities of selected enzymes in the liver of kittens (Felis domesticus). Comp. Biochem. Physiol. B 1987, 88, 551–555. [Google Scholar]
- Morris, J.G.; Rogers, Q.R. Ammonia intoxication in the near-adult cat as a result of a dietary deficiency of arginine. Science 1978, 199, 431–432. [Google Scholar]
- Morris, J.G.; Rogers, Q.R. Arginine: An essential amino acid for the cat. J. Nutr. 1978, 108, 1944–1953. [Google Scholar]
- Burns, R.A.; Milner, J.A.; Corbin, J.E. Arginine: An indispensable amino acid for mature dogs. J. Nutr. 1981, 111, 1020–1024. [Google Scholar]
- Rogers, Q.R.; Phang, J.M. Deficiency of pyrroline-5-carboxylate synthase in the intestinal mucosa of the cat. J. Nutr. 1985, 115, 146–150. [Google Scholar]
- Morris, J.G. Nutritional and metabolic responses to arginine deficiency in carnivores. J. Nutr. 1985, 115, 524–531. [Google Scholar]
- Morris, J.G.; Rogers, Q.R.; Winterrowd, D.L.; Kamikawa, E.M. The utilization of ornithine and citrulline by the growing kitten. J. Nutr. 1979, 109, 724–729. [Google Scholar]
- Morrisett, J.D.; Jackson, R.L.; Gotto, A.M., Jr. Lipid-protein interactions in the plasma lipoproteins. Biochim. Biophys. Acta 1977, 472, 93–133. [Google Scholar]
- Ruaux, C.G.; Steiner, J.M.; Williams, D.A. Metabolism of amino acids in cats with severe cobalamin deficiency. Am. J. Vet. Res. 2001, 62, 1852–1858. [Google Scholar] [CrossRef]
- Ruaux, C.G.; Steiner, J.M.; Williams, D.A. Relationships between low serum cobalamin concentrations and methlymalonic acidemia in cats. J. Vet. Intern. Med. 2009, 23, 472–475. [Google Scholar] [CrossRef]
- Da Silva, A.C.; de Angelis, R.C.; Pontes, M.A.; Guerios, M.F. The domestic cat as a laboratory animal for experimental nutrition studies. IV. Folic acid deficiency. J. Nutr. 1955, 56, 199–213. [Google Scholar]
- Yu, S.; Schultze, E.; Morris, J.G. Plasma homocysteine concentration is affected by folate status and sex of cats. FASEB J. 1999, 13, A229–A229. [Google Scholar]
- Brattstrom, L.; Israelsson, B.; Norrving, B.; Bergqvist, D.; Thorne, J.; Hultberg, B.; Hamfelt, A. Impaired homocysteine metabolism in early-onset cerebral and peripheral occlusive arterial disease. Effects of pyridoxine and folic acid treatment. Atherosclerosis 1990, 81, 51–60. [Google Scholar] [CrossRef]
- Selhub, J.; Jacques, P.F.; Bostom, A.G.; D’Agostino, R.B.; Wilson, P.W.; Belanger, A.J.; O’Leary, D.H.; Wolf, P.A.; Schaefer, E.J.; Rosenberg, I.H. Association between plasma homocysteine concentrations and extracranial carotid-artery stenosis. N. Engl. J. Med. 1995, 332, 286–291. [Google Scholar] [CrossRef]
- Selhub, J. Homocysteine metabolism. Annu. Rev. Nutr. 1999, 19, 217–246. [Google Scholar] [CrossRef]
- McMichael, M.A.; Freeman, L.M.; Selhub, J.; Rozanski, E.A.; Brown, D.J.; Nadeau, M.R.; Rush, J.E. Plasma homocysteine, B vitamins, and amino acid concentrations in cats with cardiomyopathy and arterial thromboembolism. J. Vet. Intern. Med. 2000, 14, 507–512. [Google Scholar] [CrossRef]
- National Research Council, Vitamins. In Nutrient Requirements of Dogs and Cats; The National Academies Press: Washington, DC, USA, 2006; pp. 193–245.
- Da Silva, A.C.; Guerios, M.F.; Monsao, S.R. The domestic cat as a laboratory animal for experimental nutrition studies. VI. Choline deficiency. J. Nutr. 1959, 67, 537–547. [Google Scholar]
- Anderson, P.A.; Baker, D.H.; Sherry, P.A.; Corbin, J.E. Choline-methionine interrelationship in feline nutrition. J. Anim. Sci. 1979, 49, 522–527. [Google Scholar]
- Schaeffer, M.C.; Rogers, Q.R.; Morris, J.G. The choline requirement of the growing kitten in the presence of just adequate dietary methionine. Nutr. Res. 1982, 2, 289–299. [Google Scholar] [CrossRef]
- Sinclair, A.J.; McLean, J.G.; Monger, E.A. Metabolism of linoleic acid in the cat. Lipids 1979, 14, 932–936. [Google Scholar] [CrossRef]
- Rivers, J.P.; Sinclair, A.J.; Craqford, M.A. Inability of the cat to desaturate essential fatty acids. Nature 1975, 258, 171–173. [Google Scholar] [CrossRef]
- Hassam, A.G.; Rivers, J.P.; Crawford, M.A. The failure of the cat to desaturate linoleic acid; its nutritional implications. Nutr. Metab. 1977, 21, 321–328. [Google Scholar] [CrossRef]
- Frankel, T.L.; Rivers, J.P. The nutritional and metabolic impact of gamma-linolenic acid (18:3omega6) on cats deprived of animal lipid. Br. J. Nutr. 1978, 39, 227–231. [Google Scholar]
- Pawlosky, R.; Barnes, A.; Salem, N., Jr. Essential fatty acid metabolism in the feline: Relationship between liver and brain production of long-chain polyunsaturated fatty acids. J. Lipid Res. 1994, 35, 2032–2040. [Google Scholar]
- Trevizan, L.; de Mello Kessler, A.; Brenna, J.T.; Lawrence, P.; Waldron, M.K.; Bauer, J.E. Maintenance of arachidonic acid and evidence of ∆5 desaturation in cats fed γ-linolenic and linoleic acid enriched diets. Lipids 2012, 47, 413–423. [Google Scholar] [CrossRef]
- Barsanti, J.A.; Jones, B.D.; Spano, J.S.; Taylor, H.W. Prolonged anorexia associated with hepatic lipidosis in 3 cats. Feline Pract. 1977, 7, 52–57. [Google Scholar]
- Hall, J.A.; Barstad, L.A.; Connor, W.E. Lipid composition of hepatic and adipose tissues from normal cats and from cats with idiopathic hepatic lipidosis. J. Vet. Intern. Med. 1997, 11, 238–242. [Google Scholar] [CrossRef]
- Dimski, D.S.; Taboada, J. Feline idiopathic hepatic lipidosis. Vet. Clin. N. Am. Small Anim. Pract. 1995, 25, 357–373. [Google Scholar]
- Postic, C.; Girard, J. The role of the lipogenic pathway in the development of hepatic steatosis. Diabetes Metab. 2008, 34, 643–648. [Google Scholar] [CrossRef]
- Biourge, V.C.; Groff, J.M.; Munn, R.J.; Kirk, C.A.; Nyland, T.G.; Madeiros, V.A.; Morris, J.G.; Rogers, Q.R. Experimental induction of hepatic lipidosis in cats. Am. J. Vet. Res. 1994, 55, 1291–1302. [Google Scholar]
- Nieminen, P.; Mustonen, A.M.; Karja, V.; Asikainen, J.; Rouvinen-Watt, K. Fatty acid composition and development of hepatic lipidosis during food deprivation—Mustelids as a potential animal model for liver steatosis. Exp. Biol. Med. 2009, 234, 278–286. [Google Scholar] [CrossRef]
- Mustonen, A.M.; Puukka, M.; Rouvinen-Watt, K.; Aho, J.; Asikainen, J.; Nieminen, P. Response to fasting in an unnaturally obese carnivore, the captive european polecat mustela putorius. Exp. Biol. Med. 2009, 234, 1287–1295. [Google Scholar] [CrossRef]
- Mustonen, A.M.; Pyykonen, T.; Paakkonen, T.; Ryokkynen, A.; Asikainen, J.; Aho, J.; Mononen, J.; Nieminen, P. Adaptations to fasting in the american mink (Mustela vison): Carbohydrate and lipid metabolism. Comp. Biochem. Physiol. A 2005, 140, 195–202. [Google Scholar] [CrossRef]
- Rouvinen-Watt, K.; Harris, L.; Dick, M.; Pal, C.; Lei, S.; Mustonen, A.M.; Nieminen, P. Role of hepatic de novo lipogenesis in the development of fasting-induced fatty liver in the american mink (Neovison vison). Br. J. Nutr. 2012, 108, 1360–1370. [Google Scholar] [CrossRef]
- Dimski, D.S.; Buffington, C.A.; Johnson, S.E.; Sherding, R.G.; Rosol, T.J. Serum lipoprotein concentrations and hepatic lesions in obese cats undergoing weight loss. Am. J. Vet. Res. 1992, 53, 1259–1262. [Google Scholar]
- Hoenig, M.; Thomaseth, K.; Waldron, M.; Ferguson, D.C. Insulin sensitivity, fat distribution, and adipocytokine response to different diets in lean and obese cats before and after weight loss. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R227–R234. [Google Scholar]
- Ibrahim, W.H.; Bailey, N.; Sunvold, G.D.; Bruckner, G.G. Effects of carnitine and taurine on fatty acid metabolism and lipid accumulation in the liver of cats during weight gain and weight loss. Am. J. Vet. Res. 2003, 64, 1265–1277. [Google Scholar] [CrossRef]
- Kolditz, C.I.; Langin, D. Adipose tissue lipolysis. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 377–381. [Google Scholar] [CrossRef]
- Biourge, V.; Nelson, R.W.; Feldman, E.C.; Willits, N.H.; Morris, J.G.; Rogers, Q.R. Effect of weight gain and subsequent weight loss on glucose tolerance and insulin response in healthy cats. J. Vet. Intern. Med. 1997, 11, 86–91. [Google Scholar] [CrossRef]
- Brown, B.; Mauldin, G.E.; Armstrong, J.; Moroff, S.D.; Mauldin, G.N. Metabolic and hormonal alterations in cats with hepatic lipidosis. J. Vet. Intern. Med. 2000, 14, 20–26. [Google Scholar] [CrossRef]
- Bergen, W.G.; Mersmann, H.J. Comparative aspects of lipid metabolism: Impact on contemporary research and use of animal models. J. Nutr. 2005, 135, 2499–2502. [Google Scholar]
- Richard, M.J.; Holck, J.T.; Beitz, D.C. Lipogenesis in liver and adipose tissue of the domestic cat (Felis domestica). Comp. Biochem. Physiol. B 1989, 93, 561–564. [Google Scholar] [CrossRef]
- Nguyen, P.; Leray, V.; Diez, M.; Serisier, S.; Le Bloc’h, J.; Siliart, B.; Dumon, H. Liver Lipid metabolism. J. Anim. Physiol. Anim. Nutr. 2008, 92, 272–283. [Google Scholar] [CrossRef]
- De Bruijne, J.J. Biochemical obeservations during total starvation in dogs. Int. J. Obes. 1979, 3, 239–247. [Google Scholar]
- Blanchard, G.; Paragon, B.M.; Milliat, F.; Lutton, C. Dietary l-carnitine supplementation in obese cats alters carnitine metabolism and decreases ketosis during fasting and induced hepatic lipidosis. J. Nutr. 2002, 132, 204–210. [Google Scholar]
- Center, S.A.; Crawford, M.A.; Guida, L.; Erb, H.N.; King, J. A retrospective study of 77 cats with severe hepatic lipidosis: 1975–1990. J. Vet. Intern. Med. 1993, 7, 349–359. [Google Scholar] [CrossRef]
- Mustonen, A.M.; Puukka, M.; Pyykonen, T.; Nieminen, P. Adaptations to fasting in the american mink (Mustela vison): Nitrogen metabolism. J. Comp. Physiol. B 2005, 175, 357–363. [Google Scholar] [CrossRef]
- Pazak, H.E.; Bartges, J.W.; Cornelius, L.C.; Scott, M.A.; Gross, K.; Huber, T.L. Characterization of serum lipoprotein profiles of healthy, adult cats and idiopathic feline hepatic lipidosis patients. J. Nutr. 1998, 128, 2747S–2750S. [Google Scholar]
- Biourge, V.; Groff, J.M.; Fisher, C.; Bee, D.; Morris, J.G.; Rogers, Q.R. Nitrogen-balance, plasma-free amino-acid-concentrations and urinary orotic-acid excretion during long-term fasting in cats. J. Nutr. 1994, 124, 1094–1103. [Google Scholar]
- Maugeais, C.; Tietge, U.J.; Tsukamoto, K.; Glick, J.M.; Rader, D.J. Hepatic apolipoprotein E expression promotes very low density lipoprotein-apolipoprotein B production in vivo in mice. J. Lipid Res. 2000, 41, 1673–1679. [Google Scholar]
- Pelech, S.L.; Vance, D.E. Regulation of phosphatidylcholine biosynthesis. Biochem. Biophys. Acta 1984, 779, 217–251. [Google Scholar] [CrossRef]
- Jacobs, G.; Cornelius, L.; Keene, B.; Rakich, P.; Shug, A. Comparison of plasma, liver, and skeletal-muscle carnitine concentrations in cats with idiopathic hepatic lipidosis and in healthy cats. Am. J. Vet. Res. 1990, 51, 1349–1351. [Google Scholar]
- Armstrong, P.J. Feline Hepatic Lipidosis. In Proceeding of the 7th Annual ACVIM Forum, San Diego, CA, USA, May 1989; pp. 335–337.
- Center, S.A.; Warner, K.L.; Randolph, J.E.; Sunvold, G.D.; Vickers, J.R. Influence of dietary supplementation with l-carnitine on metabolic rate, fatty acid oxidation, body condition, and weight loss in overweight cats. Am. J. Vet. Res. 2012, 73, 1002–1015. [Google Scholar] [CrossRef]
- Center, S.A.; Warner, K.L.; Erb, H.N. Liver glutathione concentrations in dogs and cats with naturally occurring liver disease. Am. J. Vet. Res. 2002, 63, 1187–1197. [Google Scholar] [CrossRef]
- Koplay, M.; Gulcan, E.; Ozkan, F. Association between serum vitamin B12 levels and the degree of steatosis in patients with nonalcoholic fatty liver disease. J. Investig. Med. 2011, 59, 1137–1140. [Google Scholar]
- Brass, E.P.; Allen, R.H.; Ruff, L.J.; Stabler, S.P. Effect of hydroxycobalamin[c-lactam] on propionate and carnitine metabolism in the rat. Biochem. J. 1990, 266, 809–815. [Google Scholar]
- Biourge, V.; Pion, P.; Lewis, J.; Morris, J.G.; Rogers, Q.R. Dietary management of idiopathic feline hepatic lipidosis with a liquid diet supplemented with citrulline and choline. J. Nutr. 1991, 121, 155S–156S. [Google Scholar]
- Ratriyanto, A.; Mosenthin, R.; Bauer, E.; Eklund, M. Metabolic, osmoregulatory and nutritional functions of betaine in monogastric animals. Asian-Aust. J. Anim. Sci. 2009, 22, 1461–1476. [Google Scholar]
- Christensen, K.E.; Wu, Q.; Wang, X.; Deng, L.; Caudill, M.A.; Rozen, R. Steatosis in mice is associated with gender, folate intake, and expression of genes of one-carbon metabolism. J. Nutr. 2010, 140, 1736–1741. [Google Scholar]
- Cordero, P.; Gomez-Uriz, A.M.; Campion, J.; Milagro, F.I.; Martinez, J.A. Dietary supplementation with methyl donors reduces fatty liver and modifies the fatty acid synthase DNA methylation profile in rats fed an obesogenic diet. Genes Nutr. 2013, 8, 105–113. [Google Scholar] [CrossRef]
- MacDonald, M.L.; Rogers, Q.R.; Morris, J.G. Role of linoleate as an essential fatty acid for the cat independent of arachidonate synthesis. J. Nutr. 1983, 113, 1422–1433. [Google Scholar]
- Werner, A.; Havinga, R.; Bos, T.; Bloks, V.W.; Kuipers, F.; Verkade, H.J. Essential fatty acid deficiency in mice is associated with hepatic steatosis and secretion of large VLDL particles. Am. J. Physiol. Gastrointest. Liver Physiol. 2005, 288, G1150–G1158. [Google Scholar] [CrossRef]
- Vu-Dac, N.; Gervois, P.; Jakel, H.; Nowak, M.; Bauge, E.; Dehondt, H.; Staels, B.; Pennacchio, L.A.; Rubin, E.M.; Fruchart-Najib, J.; et al. Apolipoprotein A5, a crucial determinant of plasma triglyceride levels, is highly responsive to peroxisome proliferator-activated receptor alpha activators. J. Biol. Chem. 2003, 278, 17982–17985. [Google Scholar] [CrossRef]
- Yahagi, N.; Shimano, H.; Hasty, A.H.; Amemiya-Kudo, M.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Shionoiri, F.; Ohashi, K.; Osuga, J.; et al. A crucial role of sterol regulatory element-binding protein-1 in the regulation of lipogenic gene expression by polyunsaturated fatty acids. J. Biol. Chem. 1999, 274, 35840–35844. [Google Scholar] [CrossRef]
- Shimano, H.; Yahagi, N.; Amemiya-Kudo, M.; Hasty, A.H.; Osuga, J.; Tamura, Y.; Shionoiri, F.; Iizuka, Y.; Ohashi, K.; Harada, K.; et al. Sterol regulatory element-binding protein-1 as a key transcription factor for nutritional induction of lipogenic enzyme genes. J. Biol. Chem. 1999, 274, 35832–35839. [Google Scholar] [CrossRef]
- El-Badry, A.M.; Graf, R.; Clavien, P.A. Omega 3-Omega 6: What is right for the liver? J. Hepatol. 2007, 47, 718–725. [Google Scholar] [CrossRef]
- Rouvinen-Watt, K.; Mustonen, A.M.; Conway, R.; Pal, C.; Harris, L.; Saarela, S.; Strandberg, U.; Nieminen, P. Rapid development of fasting-induced hepatic lipidosis in the american mink (Neovison vison): Effects of food deprivation and re-alimentation on body fat depots, tissue fatty acid profiles, hematology and endocrinology. Lipids 2010, 45, 111–128. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Verbrugghe, A.; Bakovic, M. Peculiarities of One-Carbon Metabolism in the Strict Carnivorous Cat and the Role in Feline Hepatic Lipidosis. Nutrients 2013, 5, 2811-2835. https://doi.org/10.3390/nu5072811
Verbrugghe A, Bakovic M. Peculiarities of One-Carbon Metabolism in the Strict Carnivorous Cat and the Role in Feline Hepatic Lipidosis. Nutrients. 2013; 5(7):2811-2835. https://doi.org/10.3390/nu5072811
Chicago/Turabian StyleVerbrugghe, Adronie, and Marica Bakovic. 2013. "Peculiarities of One-Carbon Metabolism in the Strict Carnivorous Cat and the Role in Feline Hepatic Lipidosis" Nutrients 5, no. 7: 2811-2835. https://doi.org/10.3390/nu5072811
APA StyleVerbrugghe, A., & Bakovic, M. (2013). Peculiarities of One-Carbon Metabolism in the Strict Carnivorous Cat and the Role in Feline Hepatic Lipidosis. Nutrients, 5(7), 2811-2835. https://doi.org/10.3390/nu5072811