The Relationship between Selenoprotein P and Glucose Metabolism in Experimental Studies

Abstract
:
{kind=link}
{kind=link}
1. Introduction
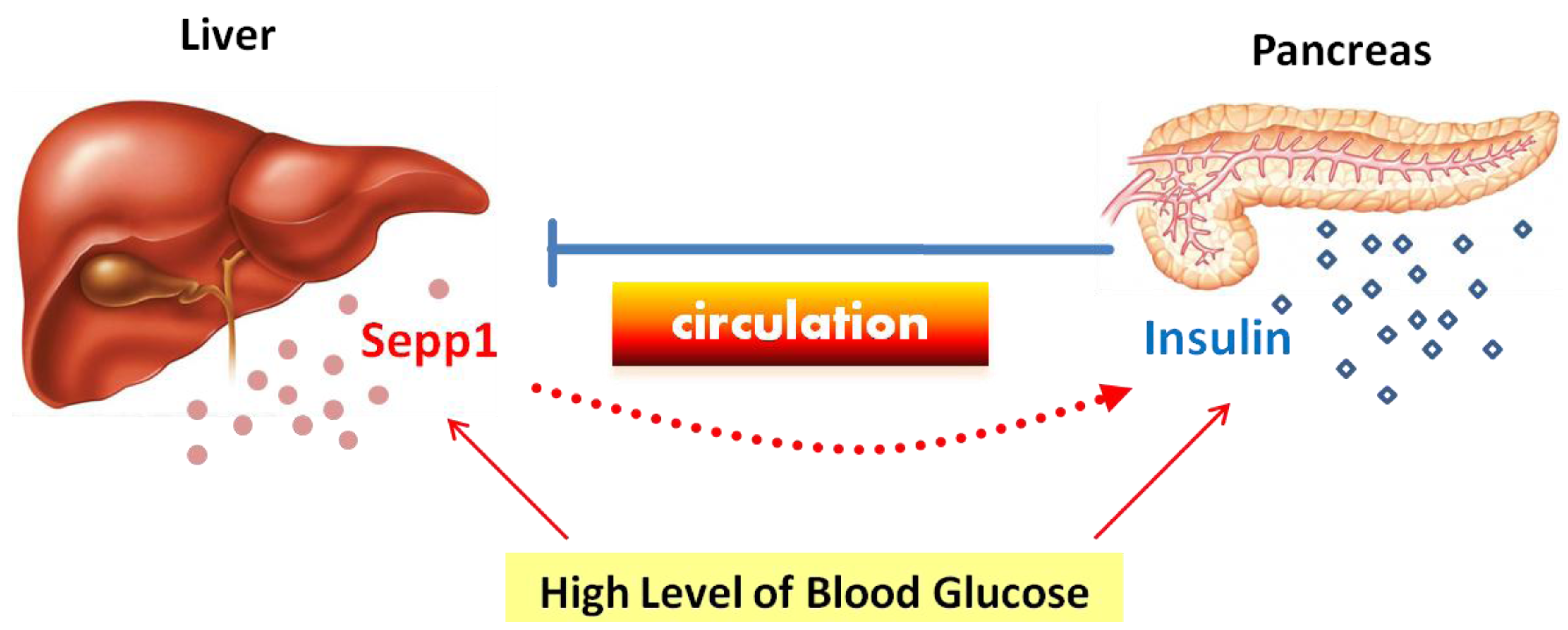
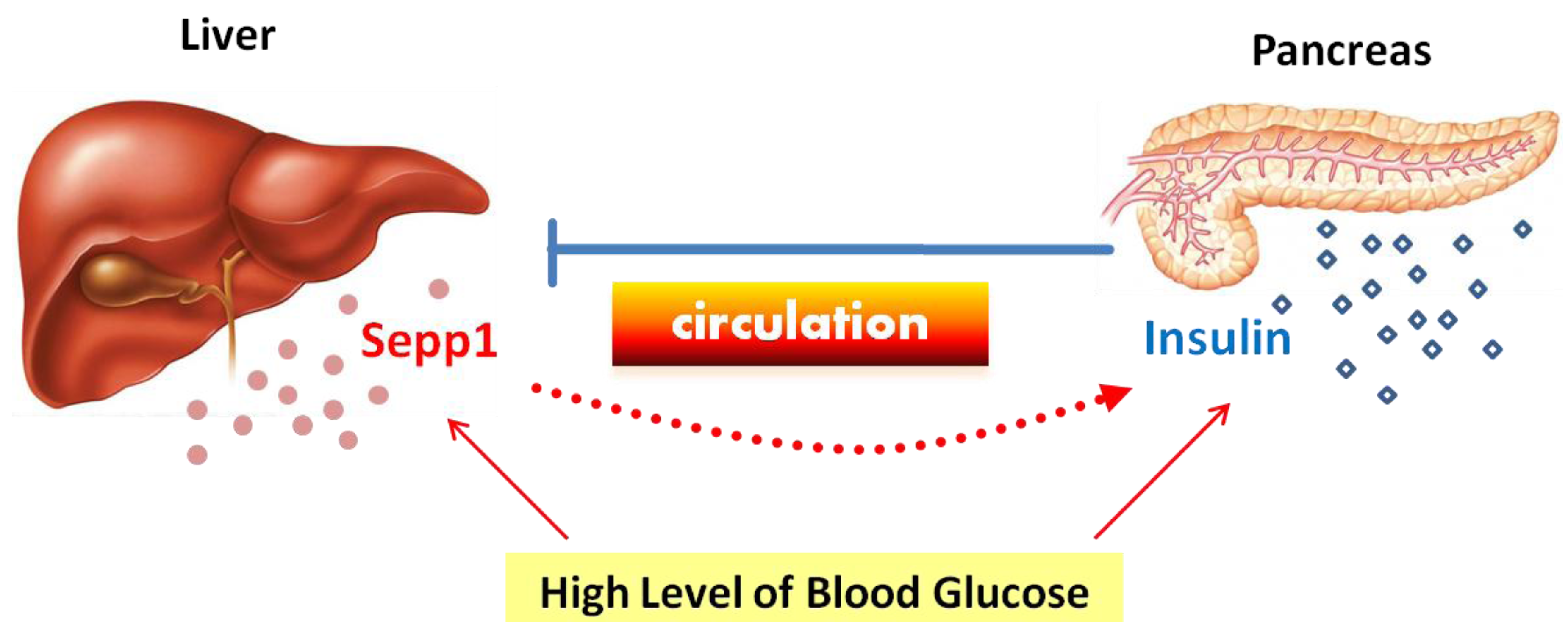
2. Expression of Sepp1 in Liver, Pancreas and Adipose Tissue
3. The Change of Sepp1 Expression with Glucose and Insulin in Models without Gene Modification
4. The Change of Sepp1 Expression with Glucose and Insulin in Models of Targeted Sepp1 Depletion
5. Regulation of Hepatic Sepp1 Expression by Factors Related to Glucose Metabolism
6. Sepp1 and Energy Metabolism
7. Conclusions
Acknowledgments
Conflict of Interest
References
- Schweizer, U.; Brauer, A.U.; Kohrle, J.; Nitsch, R.; Savaskan, N.E. Selenium and brain function: A poorly recognized liaison. Brain Res. Rev. 2004, 45, 164–178. [Google Scholar] [CrossRef]
- Boitani, C.; Puglisi, R. Selenium, a key element in spermatogenesis and male fertility. Adv. Exp. Med. Biol. 2008, 636, 65–73. [Google Scholar] [CrossRef]
- Huang, Z.; Rose, A.H.; Hoffmann, P.R. The role of selenium in inflammation and immunity: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2012, 16, 705–743. [Google Scholar] [CrossRef]
- Schomburg, L. Selenium, selenoproteins and the thyroid gland: Interactions in health and disease. Nat. Rev. Endocrinol. 2012, 8, 160–171. [Google Scholar] [CrossRef]
- Muecke, R.; Schomburg, L.; Buentzel, J.; Kisters, K.; Micke, O. Selenium or no selenium—That is the question in tumor patients: A new controversy. Integr. Cancer Ther. 2010, 9, 136–141. [Google Scholar] [CrossRef]
- Ezaki, O. The insulin-like effects of selenate in rat adipocytes. J. Biol. Chem. 1990, 265, 1124–1128. [Google Scholar]
- Roden, M.; Prskavec, M.; Furnsinn, C.; Elmadfa, I.; Konig, J.; Schneider, B.; Wagner, O.; Waldhausl, W. Metabolic effect of sodium selenite: Insulin-like inhibition of glucagon-stimulated glycogenolysis in the isolated perfused rat liver. Hepatology 1995, 22, 169–174. [Google Scholar]
- Mueller, A.S.; Pallauf, J. Compendium of the antidiabetic effects of supranutritional selenate doses. In vivo and in vitro investigations with type ii diabetic db/db mice. J. Nutr. Biochem. 2006, 17, 548–560. [Google Scholar] [CrossRef]
- Czernichow, S.; Couthouis, A.; Bertrais, S.; Vergnaud, A.C.; Dauchet, L.; Galan, P.; Hercberg, S. Antioxidant supplementation does not affect fasting plasma glucose in the supplementation with antioxidant vitamins and minerals (su.Vi.Max) study in france: Association with dietary intake and plasma concentrations. Am. J. Clin. Nutr. 2006, 84, 395–399. [Google Scholar]
- Bleys, J.; Navas-Acien, A.; Guallar, E. Serum selenium and diabetes in U.S. Adults. Diabetes Care 2007, 30, 829–834. [Google Scholar] [CrossRef]
- Laclaustra, M.; Navas-Acien, A.; Stranges, S.; Ordovas, J.M.; Guallar, E. Serum selenium concentrations and diabetes in U.S. Adults: National health and nutrition examination survey (nhanes) 2003–2004. Environ. Health Perspect. 2009, 117, 1409–1413. [Google Scholar]
- Stranges, S.; Galletti, F.; Farinaro, E.; D’Elia, L.; Russo, O.; Iacone, R.; Capasso, C.; Carginale, V.; de Luca, V.; Della Valle, E.; et al. Associations of selenium status with cardiometabolic risk factors: An 8-year follow-up analysis of the olivetti heart study. Atherosclerosis 2011, 217, 274–278. [Google Scholar] [CrossRef]
- Rajpathak, S.; Rimm, E.; Morris, J.S.; Hu, F. Toenail selenium and cardiovascular disease in men with diabetes. J. Am. College Nutr. 2005, 24, 250–256. [Google Scholar]
- Akbaraly, T.N.; Arnaud, J.; Rayman, M.P.; Hininger-Favier, I.; Roussel, A.M.; Berr, C.; Fontbonne, A. Plasma selenium and risk of dysglycemia in an elderly french population: Results from the prospective epidemiology of vascular ageing study. Nutr. Metabol. 2010, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Park, K.; Rimm, E.B.; Siscovick, D.S.; Spiegelman, D.; Manson, J.E.; Morris, J.S.; Hu, F.B.; Mozaffarian, D. Toenail selenium and incidence of type 2 diabetes in U.S. men and women. Diabetes Care 2012, 35, 1544–1551. [Google Scholar] [CrossRef]
- Rayman, M.P.; Blundell-Pound, G.; Pastor-Barriuso, R.; Guallar, E.; Steinbrenner, H.; Stranges, S. A randomized trial of selenium supplementation and risk of type-2 diabetes, as assessed by plasma adiponectin. PLoS One 2012, 7, e45269. [Google Scholar]
- Lippman, S.M.; Klein, E.A.; Goodman, P.J.; Lucia, M.S.; Thompson, I.M.; Ford, L.G.; Parnes, H.L.; Minasian, L.M.; Gaziano, J.M.; Hartline, J.A.; et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: The selenium and vitamin e cancer prevention trial (select). JAMA 2009, 301, 39–51. [Google Scholar] [CrossRef]
- Klein, E.A.; Thompson, I.M., Jr.; Tangen, C.M.; Crowley, J.J.; Lucia, M.S.; Goodman, P.J.; Minasian, L.M.; Ford, L.G.; Parnes, H.L.; Gaziano, J.M.; et al. Vitamin E and the risk of prostate cancer: The selenium and vitamin e cancer prevention trial (select). JAMA 2011, 306, 1549–1556. [Google Scholar] [CrossRef]
- Stranges, S.; Marshall, J.R.; Natarajan, R.; Donahue, R.P.; Trevisan, M.; Combs, G.F.; Cappuccio, F.P.; Ceriello, A.; Reid, M.E. Effects of long-term selenium supplementation on the incidence of type 2 diabetes: A randomized trial. Ann. Intern. Med. 2007, 147, 217–223. [Google Scholar] [CrossRef]
- Hill, K.E.; Wu, S.; Motley, A.K.; Stevenson, T.D.; Winfrey, V.P.; Capecchi, M.R.; Atkins, J.F.; Burk, R.F. Production of selenoprotein P (sepp1) by hepatocytes is central to selenium homeostasis. J. Biol. Chem. 2012, 287, 40414–40424. [Google Scholar] [CrossRef]
- Yang, S.J.; Hwang, S.Y.; Choi, H.Y.; Yoo, H.J.; Seo, J.A.; Kim, S.G.; Kim, N.H.; Baik, S.H.; Choi, D.S.; Choi, K.M. Serum selenoprotein P levels in patients with type 2 diabetes and prediabetes: Implications for insulin resistance, inflammation, and atherosclerosis. J. Clin. Endocrinol. Metabol. 2011, 96, E1325–E1329. [Google Scholar] [CrossRef]
- Misu, H.; Ishikura, K.; Kurita, S.; Takeshita, Y.; Ota, T.; Saito, Y.; Takahashi, K.; Kaneko, S.; Takamura, T. Inverse correlation between serum levels of selenoprotein P and adiponectin in patients with type 2 diabetes. PLoS One 2012, 7, e34952. [Google Scholar] [CrossRef]
- Saito, Y.; Sato, N.; Hirashima, M.; Takebe, G.; Nagasawa, S.; Takahashi, K. Domain structure of bi-functional selenoprotein P. Biochem. J. 2004, 381, 841–846. [Google Scholar] [CrossRef]
- Burk, R.F.; Hill, K.E.; Motley, A.K. Plasma selenium in specific and non-specific forms. BioFactors 2001, 14, 107–114. [Google Scholar] [CrossRef]
- Akesson, B.; Bellew, T.; Burk, R.F. Purification of selenoprotein P from human plasma. Biochim. Biophys. Acta 1994, 1204, 243–249. [Google Scholar] [CrossRef]
- Meplan, C.; Crosley, L.K.; Nicol, F.; Beckett, G.J.; Howie, A.F.; Hill, K.E.; Horgan, G.; Mathers, J.C.; Arthur, J.R.; Hesketh, J.E. Genetic polymorphisms in the human selenoprotein P gene determine the response of selenoprotein markers to selenium supplementation in a gender-specific manner (the selgen study). FASEB J. 2007, 21, 3063–3074. [Google Scholar] [CrossRef]
- Xia, Y.; Hill, K.E.; Byrne, D.W.; Xu, J.; Burk, R.F. Effectiveness of selenium supplements in a low-selenium area of china. Am. J. Clin. Nutr. 2005, 81, 829–834. [Google Scholar]
- Combs, G.F., Jr.; Jackson, M.I.; Watts, J.C.; Johnson, L.K.; Zeng, H.; Idso, J.; Schomburg, L.; Hoeg, A.; Hoefig, C.S.; Chiang, E.C.; et al. Differential responses to selenomethionine supplementation by sex and genotype in healthy adults. Br. J. Nutr. 2012, 107, 1514–1525. [Google Scholar] [CrossRef]
- Hoffmann, P.R.; Hoge, S.C.; Li, P.A.; Hoffmann, F.W.; Hashimoto, A.C.; Berry, M.J. The selenoproteome exhibits widely varying, tissue-specific dependence on selenoprotein P for selenium supply. Nucl. Acids Res. 2007, 35, 3963–3973. [Google Scholar] [CrossRef]
- Kato, T.; Read, R.; Rozga, J.; Burk, R.F. Evidence for intestinal release of absorbed selenium in a form with high hepatic extraction. Am. J. Physiol. 1992, 262, G854–G858. [Google Scholar]
- Schweizer, U.; Streckfuss, F.; Pelt, P.; Carlson, B.A.; Hatfield, D.L.; Kohrle, J.; Schomburg, L. Hepatically derived selenoprotein P is a key factor for kidney but not for brain selenium supply. Biochem. J. 2005, 386, 221–226. [Google Scholar] [CrossRef]
- Steinbrenner, H.; Hotze, A.L.; Speckmann, B.; Pinto, A.; Sies, H.; Schott, M.; Ehlers, M.; Scherbaum, W.A.; Schinner, S. Localization and regulation of pancreatic selenoprotein P. J. Mol. Endocrinol. 2013, 50, 31–42. [Google Scholar]
- Lee, S.R.; Yon, J.M.; Baek, I.J.; Kim, M.R.; Park, C.G.; Lee, B.J.; Yun, Y.W.; Nam, S.Y. Spatiotemporal expression of the selenoprotein P gene in postimplantational mouse embryos. Int. J. Dev. Biol. 2008, 52, 1005–1011. [Google Scholar] [CrossRef]
- Niwa, H.; Harrison, L.C.; DeAizpurua, H.J.; Cram, D.S. Identification of pancreatic β cell-related genes by representational difference analysis. Endocrinology 1997, 138, 1419–1426. [Google Scholar] [CrossRef]
- Yang, X.; Jansson, P.A.; Nagaev, I.; Jack, M.M.; Carvalho, E.; Sunnerhagen, K.S.; Cam, M.C.; Cushman, S.W.; Smith, U. Evidence of impaired adipogenesis in insulin resistance. Biochem. Biophys. Res. Commun. 2004, 317, 1045–1051. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, X. Reducing selenoprotein P expression suppresses adipocyte differentiation as a result of increased preadipocyte inflammation. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E77–E85. [Google Scholar] [CrossRef]
- Clark, L.C.; Combs, G.F., Jr.; Turnbull, B.W.; Slate, E.H.; Chalker, D.K.; Chow, J.; Davis, L.S.; Glover, R.A.; Graham, G.F.; Gross, E.G.; et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. Nutritional prevention of cancer study group. JAMA 1996, 276, 1957–1963. [Google Scholar] [CrossRef]
- Duffield-Lillico, A.J.; Dalkin, B.L.; Reid, M.E.; Turnbull, B.W.; Slate, E.H.; Jacobs, E.T.; Marshall, J.R.; Clark, L.C. Selenium supplementation, baseline plasma selenium status and incidence of prostate cancer: An analysis of the complete treatment period of the nutritional prevention of cancer trial. BJU Int. 2003, 91, 608–612. [Google Scholar] [CrossRef]
- Angstwurm, M.W.; Engelmann, L.; Zimmermann, T.; Lehmann, C.; Spes, C.H.; Abel, P.; Strauss, R.; Meier-Hellmann, A.; Insel, R.; Radke, J.; et al. Selenium in Intensive Care (SIC): Results of a prospective randomized, placebo-controlled, multiple-center study in patients with severe systemic inflammatory response syndrome, sepsis, and septic shock. Crit. Care Med. 2007, 35, 118–126. [Google Scholar] [CrossRef]
- Campbell, S.C.; Aldibbiat, A.; Marriott, C.E.; Landy, C.; Ali, T.; Ferris, W.F.; Butler, C.S.; Shaw, J.A.; Macfarlane, W.M. Selenium stimulates pancreatic beta-cell gene expression and enhances islet function. FEBS Lett. 2008, 582, 2333–2337. [Google Scholar] [CrossRef]
- Speckmann, B.; Sies, H.; Steinbrenner, H. Attenuation of hepatic expression and secretion of selenoprotein P by metformin. Biochem. Biophys. Res. Commun. 2009, 387, 158–163. [Google Scholar] [CrossRef]
- Misu, H.; Takamura, T.; Takayama, H.; Hayashi, H.; Matsuzawa-Nagata, N.; Kurita, S.; Ishikura, K.; Ando, H.; Takeshita, Y.; Ota, T.; et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab. 2010, 12, 483–495. [Google Scholar] [CrossRef]
- Puigserver, P.; Rhee, J.; Donovan, J.; Walkey, C.J.; Yoon, J.C.; Oriente, F.; Kitamura, Y.; Altomonte, J.; Dong, H.; Accili, D.; et al. Insulin-regulated hepatic gluconeogenesis through foxo1-pgc-1α interaction. Nature 2003, 423, 550–555. [Google Scholar] [CrossRef]
- Speckmann, B.; Walter, P.L.; Alili, L.; Reinehr, R.; Sies, H.; Klotz, L.O.; Steinbrenner, H. Selenoprotein P expression is controlled through interaction of the coactivator pgc-1α with foxo1a and hepatocyte nuclear factor 4α transcription factors. Hepatology 2008, 48, 1998–2006. [Google Scholar] [CrossRef]
- Yoon, J.C.; Puigserver, P.; Chen, G.; Donovan, J.; Wu, Z.; Rhee, J.; Adelmant, G.; Stafford, J.; Kahn, C.R.; Granner, D.K.; et al. Control of hepatic gluconeogenesis through the transcriptional coactivator pgc-1. Nature 2001, 413, 131–138. [Google Scholar] [CrossRef]
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef]
- Burk, R.F.; Hill, K.E. Selenoprotein P—expression, functions, and roles in mammals. Biochim. Biophys. Acta 2009, 1790, 1441–1447. [Google Scholar] [CrossRef]
- Kurokawa, S.; Hill, K.E.; McDonald, W.H.; Burk, R.F. Long isoform mouse selenoprotein P (sepp1) supplies rat myoblast l8 cells with selenium via endocytosis mediated by heparin binding properties and apolipoprotein e receptor-2 (apoer2). J. Biol. Chem. 2012, 287, 28717–28726. [Google Scholar]
- Olson, G.E.; Winfrey, V.P.; Nagdas, S.K.; Hill, K.E.; Burk, R.F. Apolipoprotein e receptor-2 (apoer2) mediates selenium uptake from selenoprotein P by the mouse testis. J. Biol. Chem. 2007, 282, 12290–12297. [Google Scholar]
- Olson, G.E.; Winfrey, V.P.; Hill, K.E.; Burk, R.F. Megalin mediates selenoprotein P uptake by kidney proximal tubule epithelial cells. J. Biol. Chem. 2008, 283, 6854–6860. [Google Scholar]
- Erranz, B.; Miquel, J.F.; Argraves, W.S.; Barth, J.L.; Pimentel, F.; Marzolo, M.P. Megalin and cubilin expression in gallbladder epithelium and regulation by bile acids. J. Lipid Res. 2004, 45, 2185–2198. [Google Scholar] [CrossRef]
- Lei, X.G.; Vatamaniuk, M.Z. Two tales of antioxidant enzymes on beta cells and diabetes. Antioxid. Redox Signal. 2011, 14, 489–503. [Google Scholar] [CrossRef]
- Walter, P.L.; Steinbrenner, H.; Barthel, A.; Klotz, L.O. Stimulation of selenoprotein P promoter activity in hepatoma cells by foxo1a transcription factor. Biochem. Biophys. Res. Commun. 2008, 365, 316–321. [Google Scholar] [CrossRef]
- Seale, L.A.; Hashimoto, A.C.; Kurokawa, S.; Gilman, C.L.; Seyedali, A.; Bellinger, F.P.; Raman, A.V.; Berry, M.J. Disruption of the selenocysteine lyase-mediated selenium recycling pathway leads to metabolic syndrome in mice. Mol. Cell. Biol. 2012, 32, 4141–4154. [Google Scholar] [CrossRef]
- Collins, R.; Johansson, A.L.; Karlberg, T.; Markova, N.; van den Berg, S.; Olesen, K.; Hammarstrom, M.; Flores, A.; Schuler, H.; Schiavone, L.H.; et al. Biochemical discrimination between selenium and sulfur 1: A single residue provides selenium specificity to human selenocysteine lyase. PLoS One 2012, 7, e30581. [Google Scholar] [CrossRef]
- Mihara, H.; Kurihara, T.; Watanabe, T.; Yoshimura, T.; Esaki, N. Cdna cloning, purification, and characterization of mouse liver selenocysteine lyase. Candidate for selenium delivery protein in selenoprotein synthesis. J. Biol. Chem. 2000, 275, 6195–6200. [Google Scholar]
- Schomburg, L.; Schweizer, U. Hierarchical regulation of selenoprotein expression and sex-specific effects of selenium. Biochim. Biophys. Acta 2009, 1790, 1453–1462. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mao, J.; Teng, W. The Relationship between Selenoprotein P and Glucose Metabolism in Experimental Studies. Nutrients 2013, 5, 1937-1948. https://doi.org/10.3390/nu5061937
Mao J, Teng W. The Relationship between Selenoprotein P and Glucose Metabolism in Experimental Studies. Nutrients. 2013; 5(6):1937-1948. https://doi.org/10.3390/nu5061937
Chicago/Turabian StyleMao, Jinyuan, and Weiping Teng. 2013. "The Relationship between Selenoprotein P and Glucose Metabolism in Experimental Studies" Nutrients 5, no. 6: 1937-1948. https://doi.org/10.3390/nu5061937