Non-Alcoholic Fatty Liver Disease (NAFLD) and Its Connection with Insulin Resistance, Dyslipidemia, Atherosclerosis and Coronary Heart Disease


Abstract
:1. Introduction
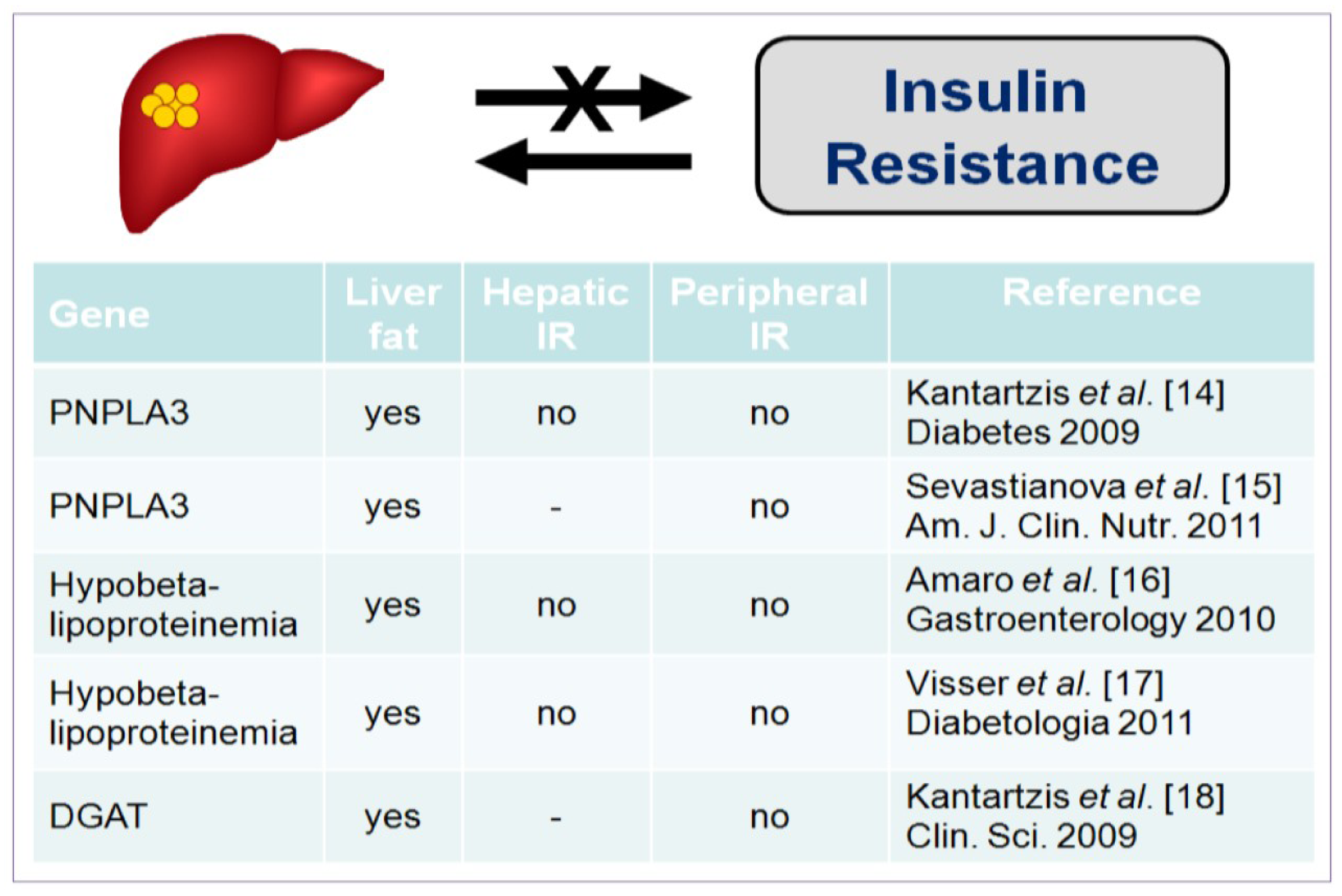
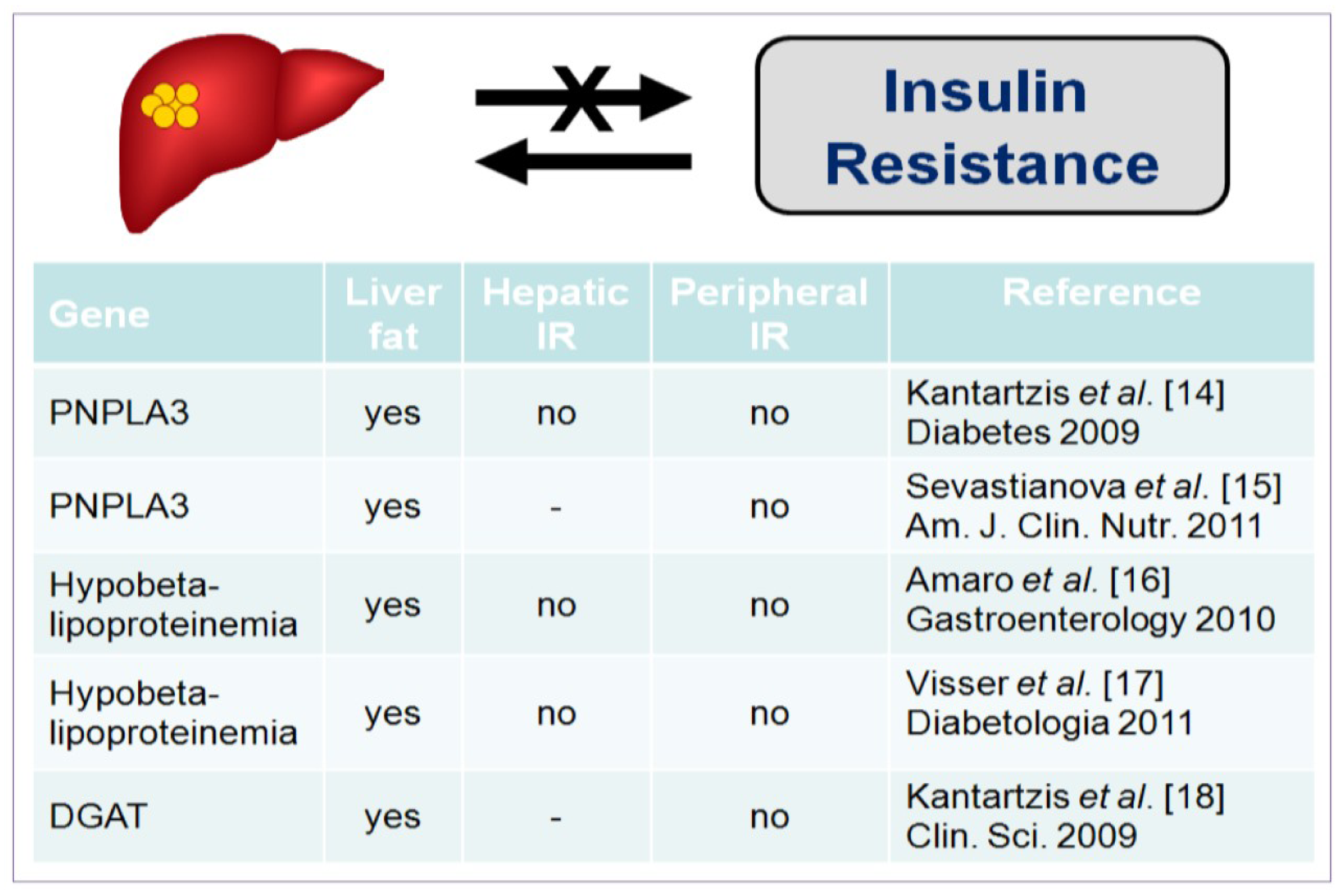
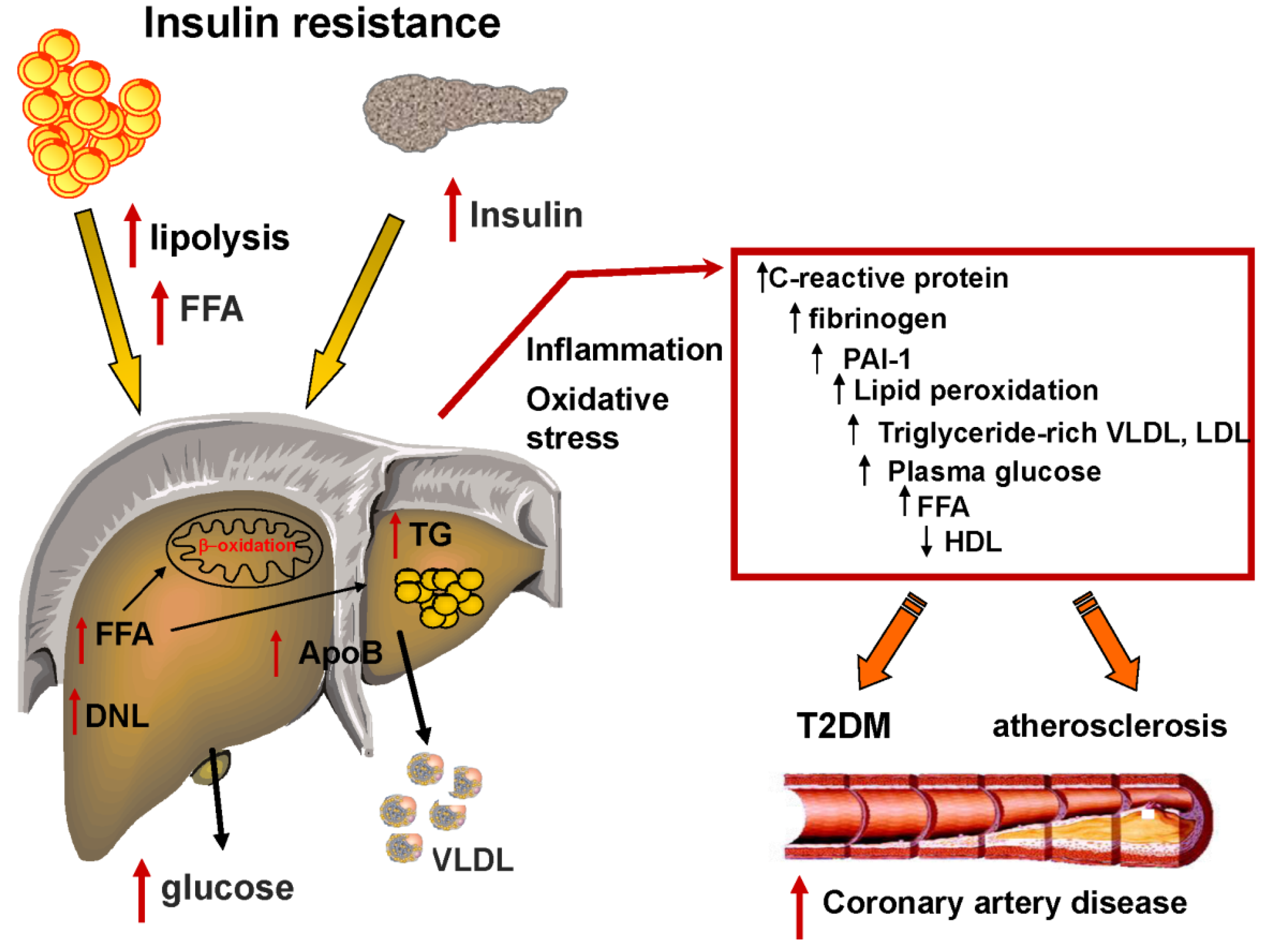
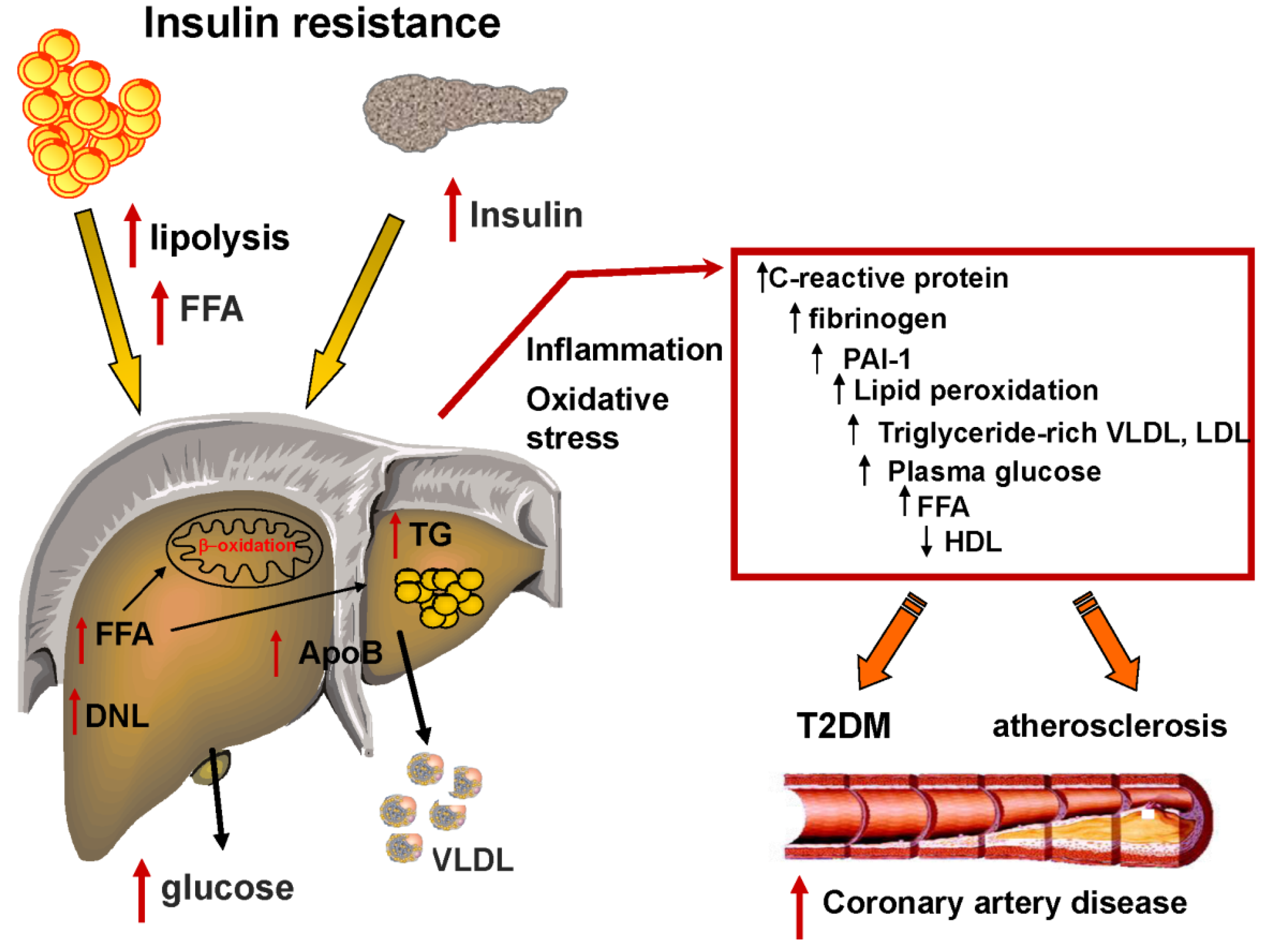
2. Insulin Resistance and the Development of NAFLD
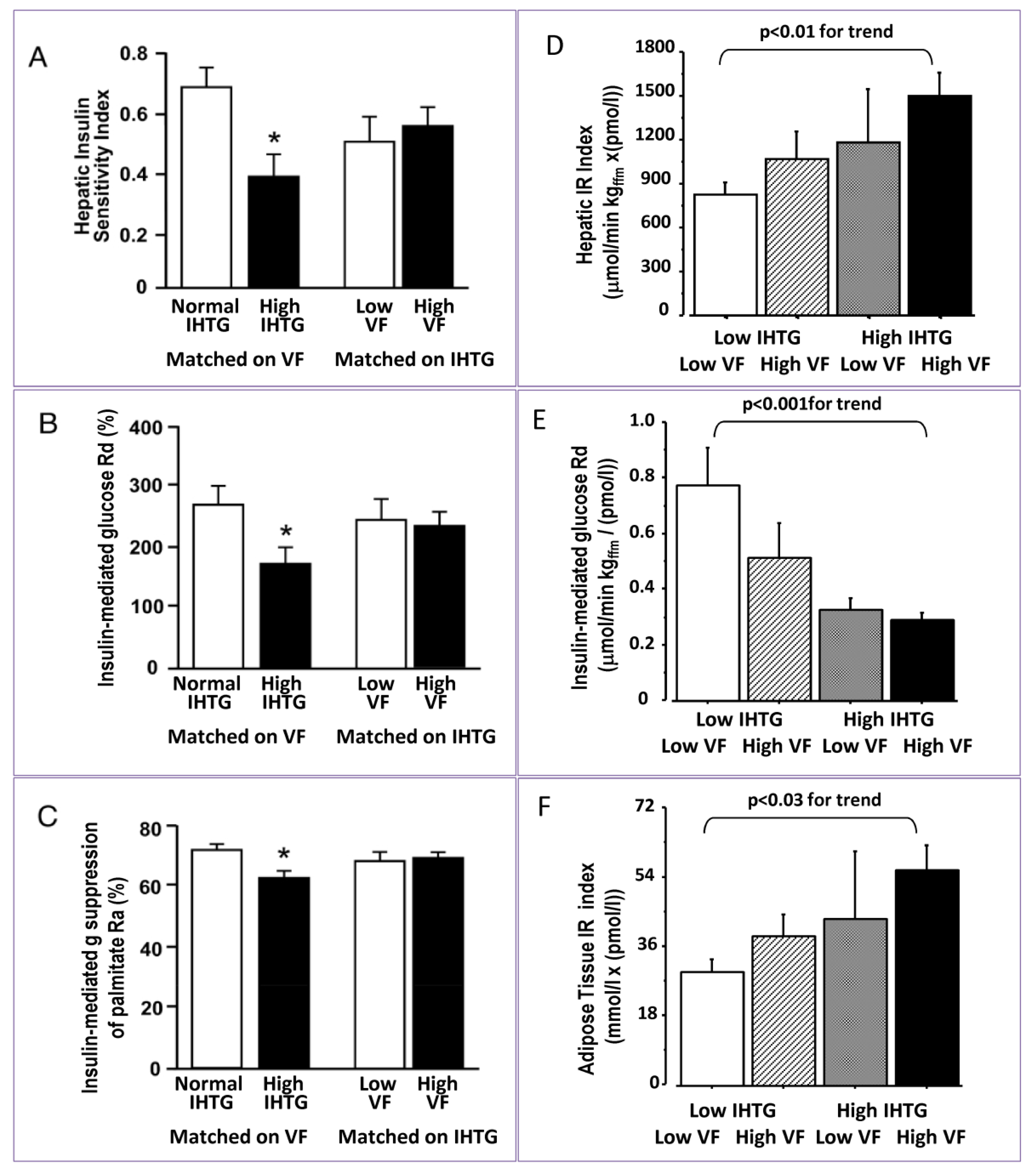
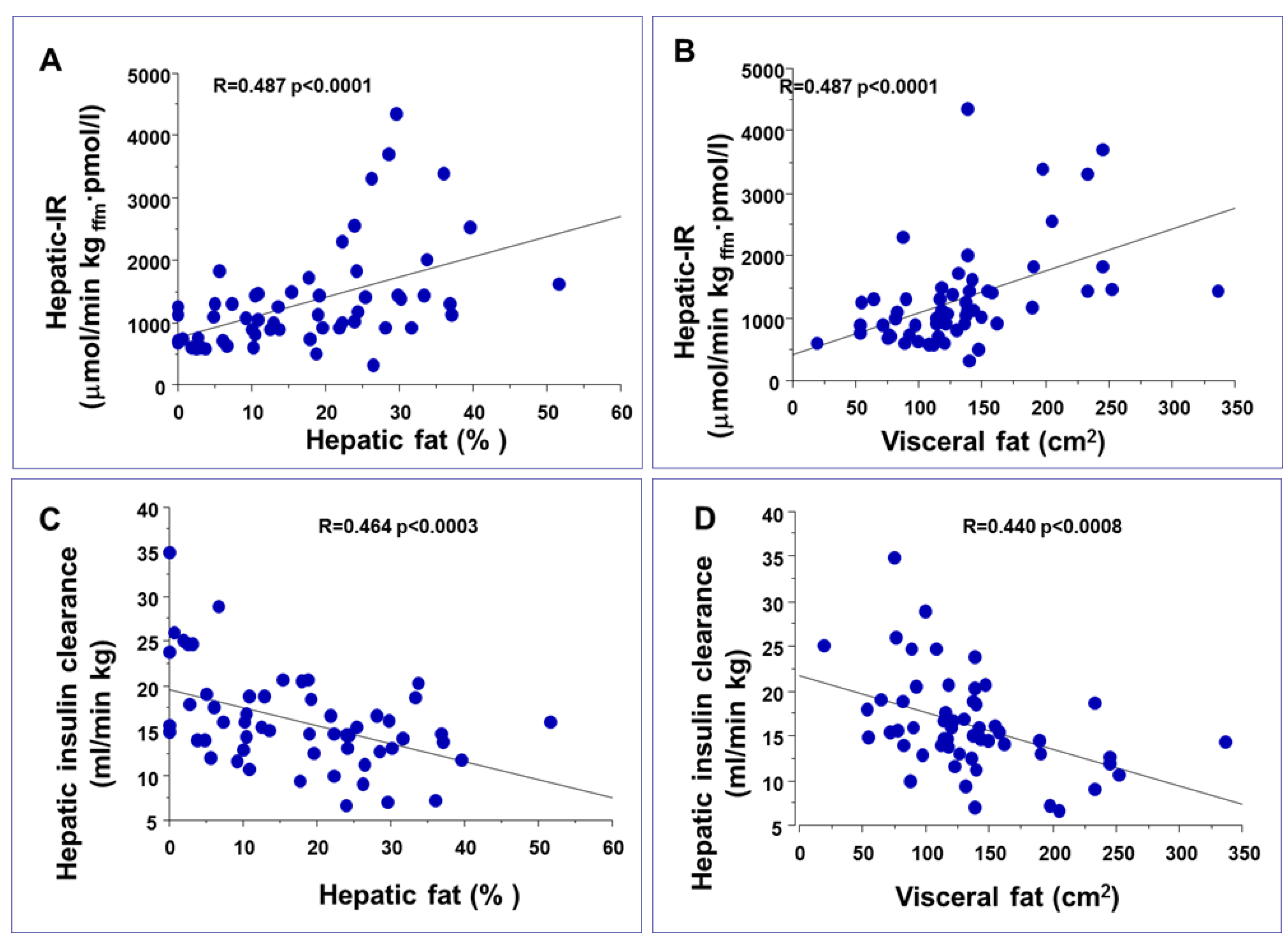
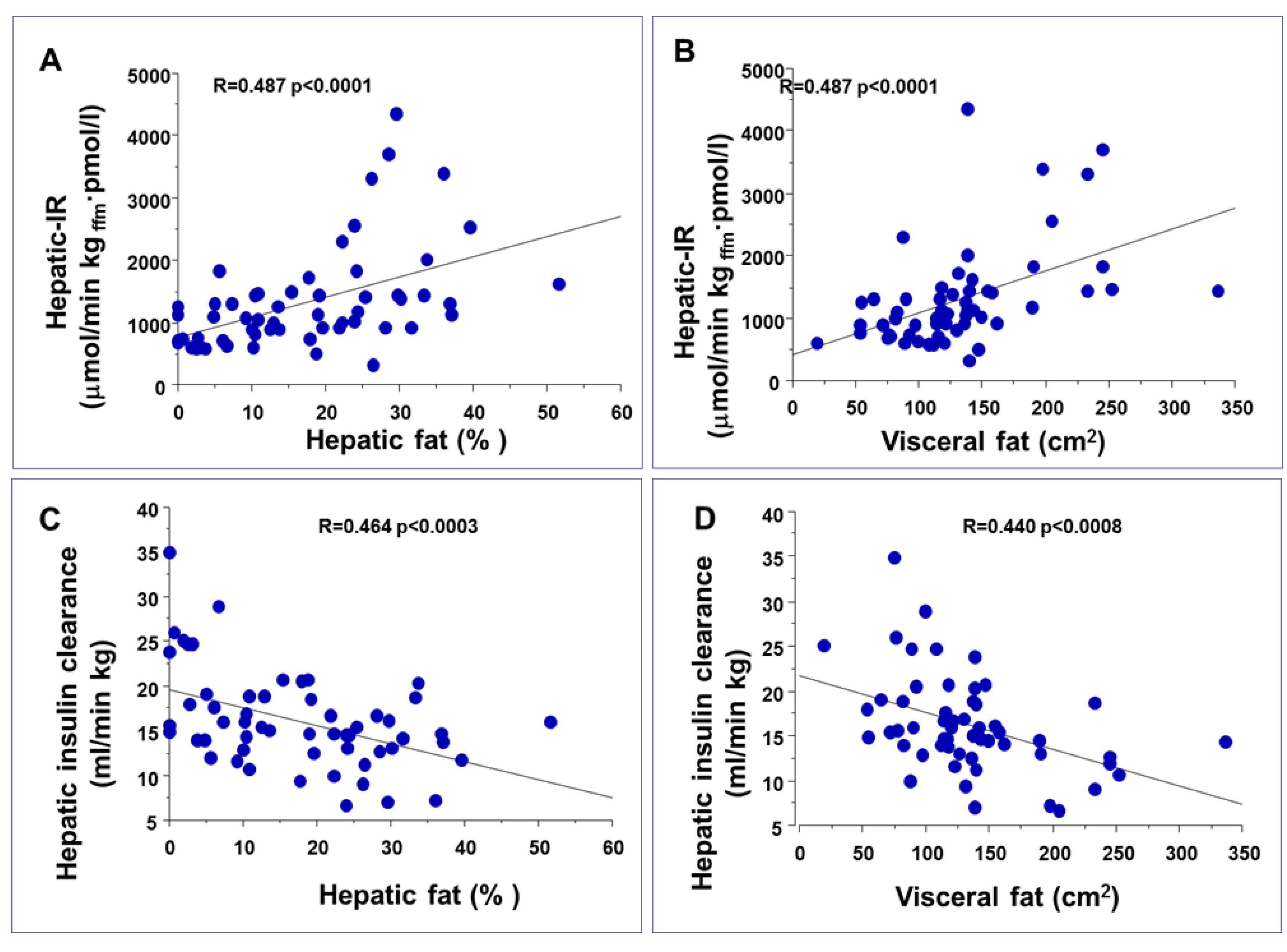
3. Impact of Hepatic and Visceral Fat Accumulation on Metabolic Profile


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of subjects | IHTG (%) | VF | Reference | |
|---|---|---|---|---|
| Matched on VAT | Normal IHTG | 3.6 ± 0.5 | 1.29 ± 0.24 kg | [12] |
| High IHTG | 25.3 ± 3.5 | 1.34 ± 0.18 kg | ||
| Matched on IHTG | Low VF | 13.2 ± 3.5 | 0.76 ± 0.08 kg | [12] |
| High VF | 13.2 ± 3.3 | 1.94 ± 0.32 kg | ||
| Low IH-TG | Low VF | 2.1 ± 0.6 | 70 ± 8 cm2 | [7] |
| High VF | 12.9 ± 1.9 | 84 ± 5 cm2 | ||
| High IH-TG | Low VF | 3.2 ± 1.6 | 165 ± 41 cm2 | [7] |
| High VF | 23.3 ± 1.7 | 159 ± 9 cm2 | ||
4. Fatty Liver, Dyslipidemia and the Metabolic Syndrome
Fatty Liver and Metabolic Syndrome
5. NAFLD and Cardiovascular Disease
5.1. Fatty Liver and Atherosclerosis
5.2. Fatty Liver and Endothelial Dysfunction
5.3. Fatty Liver and Coronary Artery Disease (CAD)
6. Conclusions
Acknowledgments
Conflict of Interest
References
- Szczepaniak, L.S.; Babcock, E.E.; Schick, F.; Dobbins, R.L.; Garg, A.; Burns, D.K.; McGarry, J.D.; Stein, D.T. Measurement of intracellular triglyceride stores by H spectroscopy: validation in vivo. Am. J. Physiol. 1999, 276, E977–E989. [Google Scholar]
- Browning, J.D.; Szczepaniak, L.S.; Dobbins, R.; Nuremberg, P.; Horton, J.D.; Cohen, J.C.; Grundy, S.M.; Hobbs, H.H. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 2004, 40, 1387–1395. [Google Scholar] [CrossRef]
- Day, C.P. Pathogenesis of steatohepatitis. Best Pract. Res. Clin. Gastroenterol. 2002, 16, 663–678. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012, 142, 1592–1609. [Google Scholar] [CrossRef]
- Fracanzani, A.L.; Valenti, L.; Bugianesi, E.; Andreoletti, M.; Colli, A.; Vanni, E.; Bertelli, C.; Fatta, E.; Bignamini, D.; Marchesini, G.; et al. Risk of severe liver disease in nonalcoholic fatty liver disease with normal aminotransferase levels: A role for insulin resistance and diabetes. Hepatology 2008, 48, 792–798. [Google Scholar]
- Kotronen, A.; Juurinen, L.; Hakkarainen, A.; Westerbacka, J.; Corner, A.; Bergholm, R.; Yki-Jarvinen, H. Liver fat is increased in type 2 diabetic patients and underestimated by serum alanine aminotransferase compared with equally obese nondiabetic subjects. Diabetes Care 2008, 31, 165–169. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Cusi, K.; Pettiti, M.; Hardies, J.; Miyazaki, Y.; Berria, R.; Buzzigoli, E.; Sironi, A.M.; Cersosimo, E.; Ferrannini, E.; et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology 2007, 133, 496–506. [Google Scholar] [CrossRef]
- Bellentani, S.; Saccoccio, G.; Masutti, F.; Croce, L.S.; Brandi, G.; Sasso, F.; Cristanini, G.; Tiribelli, C. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann. Intern. Med. 2000, 132, 112–117. [Google Scholar]
- Bhatia, L.S.; Curzen, N.P.; Calder, P.C.; Byrne, C.D. Non-alcoholic fatty liver disease: a new and important cardiovascular risk factor? Eur. Heart J. 2012, 33, 1190–1200. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef]
- Yki-Jarvinen, H. Liver fat in the pathogenesis of insulin resistance and type 2 diabetes. Dig. Dis. 2010, 28, 203–209. [Google Scholar] [CrossRef]
- Fabbrini, E.; Magkos, F.; Mohammed, B.S.; Pietka, T.; Abumrad, N.A.; Patterson, B.W.; Okunade, A.; Klein, S. Intrahepatic fat, not visceral fat, is linked with metabolic complications of obesity. Proc. Natl. Acad. Sci. USA 2009, 106, 15430–15435. [Google Scholar] [CrossRef]
- Bugianesi, E.; Gastaldelli, A.; Vanni, E.; Gambino, R.; Cassader, M.; Baldi, S.; Ponti, V.; Pagano, G.; Ferrannini, E.; Rizzetto, M. Insulin resistance in non-diabetic patients with non-alcoholic fatty liver disease: Sites and mechanisms. Diabetologia 2005, 48, 634–642. [Google Scholar] [CrossRef]
- Kantartzis, K.; Peter, A.; Machicao, F.; Machann, J.; Wagner, S.; Konigsrainer, I.; Konigsrainer, A.; Schick, F.; Fritsche, A.; Haring, H.U.; et al. Dissociation between fatty liver and insulin resistance in humans carrying a variant of the patatin-like phospholipase 3 gene. Diabetes 2009, 58, 2616–2623. [Google Scholar] [CrossRef]
- Sevastianova, K.; Kotronen, A.; Gastaldelli, A.; Perttila, J.; Hakkarainen, A.; Lundbom, J.; Suojanen, L.; Orho-Melander, M.; Lundbom, N.; Ferrannini, E.; et al. Genetic variation in PNPLA3 (adiponutrin) confers sensitivity to weight loss-induced decrease in liver fat in humans. Am. J. Clin. Nutr. 2011, 94, 104–111. [Google Scholar] [CrossRef]
- Amaro, A.; Fabbrini, E.; Kars, M.; Yue, P.; Schechtman, K.; Schonfeld, G.; Klein, S. Dissociation between intrahepatic triglyceride content and insulin resistance in familial hypobetalipoproteinemia. Gastroenterology 2010, 139, 149–153. [Google Scholar] [CrossRef]
- Visser, M.E.; Lammers, N.M.; Nederveen, A.J.; van der Graaf, M.; Heerschap, A.; Ackermans, M.T.; Sauerwein, H.P.; Stroes, E.S.; Serlie, M.J. Hepatic steatosis does not cause insulin resistance in people with familial hypobetalipoproteinaemia. Diabetologia 2011, 54, 2113–2121. [Google Scholar] [CrossRef]
- Kantartzis, K.; Machicao, F.; Machann, J.; Schick, F.; Fritsche, A.; Haring, H.U.; Stefan, N. The DGAT2 gene is a candidate for the dissociation between fatty liver and insulin resistance in humans. Clin. Sci. (Lond.) 2009, 116, 531–537. [Google Scholar] [CrossRef]
- Monetti, M.; Levin, M.C.; Watt, M.J.; Sajan, M.P.; Marmor, S.; Hubbard, B.K.; Stevens, R.D.; Bain, J.R.; Newgard, C.B.; Farese, R.V., Sr.; et al. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 2007, 6, 69–78. [Google Scholar] [CrossRef]
- Targher, G.; Bertolini, L.; Padovani, R.; Rodella, S.; Tessari, R.; Zenari, L.; Day, C.; Arcaro, G. Prevalence of nonalcoholic fatty liver disease and its association with cardiovascular disease among type 2 diabetic patients. Diabetes Care 2007, 30, 1212–1218. [Google Scholar] [CrossRef]
- Lomonaco, R.; Ortiz-Lopez, C.; Orsak, B.; Webb, A.; Hardies, J.; Darland, C.; Finch, J.; Gastaldelli, A.; Harrison, S.; Tio, F.; et al. Effect of adipose tissue insulin resistance on metabolic parameters and liver histology in obese patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 1389–1397. [Google Scholar] [CrossRef]
- Arner, P. Insulin resistance in type 2 diabetes: Role of fatty acids. Diabetes Metab. Res. Rev. 2002, 18 (Suppl. 2), S5–S9. [Google Scholar] [CrossRef]
- Korenblat, K.M.; Fabbrini, E.; Mohammed, B.S.; Klein, S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology 2008, 134, 1369–1375. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Harrison, S.A.; Belfort-Aguilar, R.; Hardies, L.J.; Balas, B.; Schenker, S.; Cusi, K. Importance of changes in adipose tissue insulin resistance to histological response during thiazolidinedione treatment of patients with nonalcoholic steatohepatitis. Hepatology 2009, 50, 1087–1093. [Google Scholar] [CrossRef]
- Fabbrini, E.; Mohammed, B.S.; Magkos, F.; Korenblat, K.M.; Patterson, B.W.; Klein, S. Alterations in adipose tissue and hepatic lipid kinetics in obese men and women with nonalcoholic fatty liver disease. Gastroenterology 2008, 134, 424–431. [Google Scholar]
- Adiels, M.; Taskinen, M.R.; Packard, C.; Caslake, M.J.; Soro-Paavonen, A.; Westerbacka, J.; Vehkavaara, S.; Hakkinen, A.; Olofsson, S.O.; Yki-Jarvinen, H.; et al. Overproduction of large VLDL particles is driven by increased liver fat content in man. Diabetologia 2006, 49, 755–765. [Google Scholar] [CrossRef]
- Miles, J.M.; Nelson, R.H. Contribution of triglyceride-rich lipoproteins to plasma free fatty acids. Horm. Metab. Res. 2007, 39, 726–729. [Google Scholar] [CrossRef]
- Machado, M.V.; Ferreira, D.M.; Castro, R.E.; Silvestre, A.R.; Evangelista, T.; Coutinho, J.; Carepa, F.; Costa, A.; Rodrigues, C.M.; Cortez-Pinto, H. Liver and muscle in morbid obesity: The interplay of fatty liver and insulin resistance. PLoS One 2012, 7, e31738. [Google Scholar] [CrossRef]
- Hwang, J.H.; Stein, D.T.; Barzilai, N.; Cui, M.H.; Tonelli, J.; Kishore, P.; Hawkins, M. Increased intrahepatic triglyceride is associated with peripheral insulin resistance: in vivo MR imaging and spectroscopy studies. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1663–E1669. [Google Scholar] [CrossRef]
- Choi, S.S.; Diehl, A.M. Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Curr. Opin. Lipidol. 2008, 19, 295–300. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Nontriglyceride hepatic lipotoxicity: the new paradigm for the pathogenesis of NASH. Curr. Gastroenterol. Rep. 2010, 12, 49–56. [Google Scholar] [CrossRef]
- Heni, M.; Machann, J.; Staiger, H.; Schwenzer, N.F.; Peter, A.; Schick, F.; Claussen, C.D.; Stefan, N.; Haring, H.U.; Fritsche, A. Pancreatic fat is negatively associated with insulin secretion in individuals with impaired fasting glucose and/or impaired glucose tolerance: A nuclear magnetic resonance study. Diabetes Metab. Res. Rev. 2010, 26, 200–205. [Google Scholar] [CrossRef]
- Kotronen, A.; Yki-Jarvinen, H.; Sevastianova, K.; Bergholm, R.; Hakkarainen, A.; Pietilainen, K.H.; Juurinen, L.; Lundbom, N.; Sorensen, T.I. Comparison of the relative contributions of intra-abdominal and liver fat to components of the metabolic syndrome. Obesity (Silver Spring) 2011, 19, 23–28. [Google Scholar] [CrossRef]
- Wald, D.; Teucher, B.; Dinkel, J.; Kaaks, R.; Delorme, S.; Boeing, H.; Seidensaal, K.; Meinzer, H.P.; Heimann, T. Automatic quantification of subcutaneous and visceral adipose tissue from whole-body magnetic resonance images suitable for large cohort studies. J. Magn. Reson. Imaging 2012, 36, 1421–1434. [Google Scholar] [CrossRef]
- Kotronen, A.; Juurinen, L.; Tiikkainen, M.; Vehkavaara, S.; Yki-Jarvinen, H. Increased liver fat, impaired insulin clearance, and hepatic and adipose tissue insulin resistance in type 2 diabetes. Gastroenterology 2008, 135, 122–130. [Google Scholar] [CrossRef]
- Gastaldelli, A. Role of beta-cell dysfunction, ectopic fat accumulation and insulin resistance in the pathogenesis of type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2011, 93 (Suppl. 1), S60–S65. [Google Scholar] [CrossRef]
- Fabbrini, E.; Tamboli, R.A.; Magkos, F.; Marks-Shulman, P.A.; Eckhauser, A.W.; Richards, W.O.; Klein, S.; Abumrad, N.N. Surgical removal of omental fat does not improve insulin sensitivity and cardiovascular risk factors in obese adults. Gastroenterology 2010, 139, 448–455. [Google Scholar] [CrossRef]
- Albu, J.B.; Curi, M.; Shur, M.; Murphy, L.; Matthews, D.E.; Pi-Sunyer, F.X. Systemic resistance to the antilipolytic effect of insulin in black and white women with visceral obesity. Am. J. Physiol. 1999, 277, E551–E560. [Google Scholar]
- Wajchenberg, B.L. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr. Rev. 2000, 21, 697–738. [Google Scholar] [CrossRef]
- Jensen, M.D. Role of body fat distribution and the metabolic complications of obesity. J. Clin. Endocrinol. Metab. 2008, 93, S57–S63. [Google Scholar] [CrossRef]
- Moore, M.C.; Connolly, C.C.; Cherrington, A.D. Autoregulation of hepatic glucose production. Eur. J. Endocrinol. 1998, 138, 240–248. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Baldi, S.; Pettiti, M.; Toschi, E.; Camastra, S.; Natali, A.; Landau, B.R.; Ferrannini, E. Influence of obesity and type 2 diabetes on gluconeogenesis and glucose output in humans: a quantitative study. Diabetes 2000, 49, 1367–1373. [Google Scholar] [CrossRef]
- Jenssen, T.; Nurjhan, N.; Consoli, A.; Gerich, J.E. Failure of substrate-induced gluconeogenesis to increase overall glucose appearance in normal humans. Demonstration of hepatic autoregulation without a change in plasma glucose concentration. J. Clin. Invest. 1990, 86, 489–497. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Kozakova, M.; Hojlund, K.; Flyvbjerg, A.; Favuzzi, A.; Mitrakou, A.; Balkau, B. Fatty liver is associated with insulin resistance, risk of coronary heart disease, and early atherosclerosis in a large European population. Hepatology 2009, 49, 1537–1544. [Google Scholar] [CrossRef]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Miyazaki, Y.; Pettiti, M.; Matsuda, M.; Mahankali, S.; Santini, E.; DeFronzo, R.A.; Ferrannini, E. Metabolic effects of visceral fat accumulation in type 2 diabetes. J. Clin. Endocrinol. Metab. 2002, 87, 5098–5103. [Google Scholar] [CrossRef]
- Boden, G.; Chen, X.; Capulong, E.; Mozzoli, M. Effects of free fatty acids on gluconeogenesis and autoregulation of glucose production in type 2 diabetes. Diabetes 2001, 50, 810–816. [Google Scholar] [CrossRef]
- Wajngot, A.; Chandramouli, V.; Schumann, W.C.; Ekberg, K.; Jones, P.K.; Efendic, S.; Landau, B.R. Quantitative contributions of gluconeogenesis to glucose production during fasting in type 2 diabetes mellitus. Metabolism 2001, 50, 47–52. [Google Scholar] [CrossRef]
- Souza, M.R.; Diniz Mde, F.; Medeiros-Filho, J.E.; Araujo, M.S. Metabolic syndrome and risk factors for non-alcoholic fatty liver disease. Arq. Gastroenterol. 2012, 49, 89–96. [Google Scholar] [CrossRef]
- Barrows, B.R.; Timlin, M.T.; Parks, E.J. Spillover of dietary fatty acids and use of serum nonesterified fatty acids for the synthesis of VLDL-triacylglycerol under two different feeding regimens. Diabetes 2005, 54, 2668–2673. [Google Scholar] [CrossRef]
- Chapman, M.J.; Ginsberg, H.N.; Amarenco, P.; Andreotti, F.; Boren, J.; Catapano, A.L.; Descamps, O.S.; Fisher, E.; Kovanen, P.T.; Kuivenhoven, J.A.; et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur. Heart J. 2011, 32, 1345–1361. [Google Scholar] [CrossRef]
- Bugianesi, E.; Pagotto, U.; Manini, R.; Vanni, E.; Gastaldelli, A.; de Iasio, R.; Gentilcore, E.; Natale, S.; Cassader, M.; Rizzetto, M.; et al. Plasma adiponectin in nonalcoholic fatty liver is related to hepatic insulin resistance and hepatic fat content, not to liver disease severity. J. Clin. Endocrinol. Metab. 2005, 90, 3498–3504. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Harrison, S.; Belfort-Aguiar, R.; Hardies, J.; Balas, B.; Schenker, S.; Cusi, K. Pioglitazone in the treatment of NASH: the role of adiponectin. Aliment. Pharmacol. Ther. 2010, 32, 769–775. [Google Scholar] [CrossRef]
- Yamauchi, T.; Kamon, J.; Minokoshi, Y.; Ito, Y.; Waki, H.; Uchida, S.; Yamashita, S.; Noda, M.; Kita, S.; Ueki, K.; et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat. Med. 2002, 8, 1288–1295. [Google Scholar] [CrossRef]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef]
- Aygun, C.; Kocaman, O.; Sahin, T.; Uraz, S.; Eminler, A.T.; Celebi, A.; Senturk, O.; Hulagu, S. Evaluation of metabolic syndrome frequency and carotid artery intima-media thickness as risk factors for atherosclerosis in patients with nonalcoholic fatty liver disease. Dig. Dis. Sci. 2007, 53, 1352–1357. [Google Scholar]
- Marchesini, G.; Bugianesi, E.; Forlani, G.; Cerrelli, F.; Lenzi, M.; Manini, R.; Natale, S.; Vanni, E.; Villanova, N.; Melchionda, N.; et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology 2003, 37, 917–923. [Google Scholar] [CrossRef]
- Kantartzis, K.; Rittig, K.; Cegan, A.; Machann, J.; Schick, F.; Balletshofer, B.; Fritsche, A.; Schleicher, E.; Haring, H.U.; Stefan, N. Fatty liver is independently associated with alterations in circulating HDL2 and HDL3 subfractions. Diabetes Care 2008, 31, 366–368. [Google Scholar]
- Toledo, F.G.; Sniderman, A.D.; Kelley, D.E. Influence of hepatic steatosis (fatty liver) on severity and composition of dyslipidemia in type 2 diabetes. Diabetes Care 2006, 29, 1845–1850. [Google Scholar] [CrossRef]
- Sung, K.C.; Wild, S.H.; Kwag, H.J.; Byrne, C.D. Fatty liver, insulin resistance, and features of metabolic syndrome: relationships with coronary artery calcium in 10,153 people. Diabetes Care 2012, 35, 2359–2364. [Google Scholar] [CrossRef]
- Vanni, E.; Bugianesi, E.; Kotronen, A.; De Minicis, S.; Yki-Jarvinen, H.; Svegliati-Baroni, G. From the metabolic syndrome to NAFLD or vice versa? Dig. Liver Dis. 2010, 42, 320–330. [Google Scholar] [CrossRef]
- Gastaldelli, A. Fatty liver disease: the hepatic manifestation of metabolic syndrome. Hypertens. Res. 2010, 33, 546–547. [Google Scholar] [CrossRef]
- Ribeireiro, T.; Swain, J.; Sarr, M.; Kendrick, M.; Que, F.; Sanderson, S.; Krishnan, A.; Viker, K.; Watt, K.; Charlton, M. NAFLD and insulin resistance do not increase the risk of postoperative complications among patients undergoing bariatric surgery––A prospective analysis. Obes. Surg. 2011, 21, 310–315. [Google Scholar] [CrossRef]
- Huang, H.L.; Lin, W.Y.; Lee, L.T.; Wang, H.H.; Lee, W.J.; Huang, K.C. Metabolic syndrome is related to nonalcoholic steatohepatitis in severely obese subjects. Obes. Surg. 2007, 17, 1457–1463. [Google Scholar] [CrossRef]
- Sironi, A.M.; Sicari, R.; Folli, F.; Gastaldelli, A. Ectopic fat storage, insulin resistance, and hypertension. Curr. Pharm. Des. 2011, 17, 3074–3080. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Basta, G. Ectopic fat and cardiovascular disease: What is the link? Nutr. Metab. Cardiovasc. Dis. 2010, 20, 481–490. [Google Scholar] [CrossRef]
- Ghouri, N.; Preiss, D.; Sattar, N. Liver enzymes, nonalcoholic fatty liver disease, and incident cardiovascular disease: A narrative review and clinical perspective of prospective data. Hepatology 2010, 52, 1156–1161. [Google Scholar] [CrossRef]
- Ortiz-Lopez, C.; Lomonaco, R.; Orsak, B.; Finch, J.; Chang, Z.; Kochunov, V.G.; Hardies, J.; Cusi, K. Prevalence of prediabetes and diabetes and metabolic profile of patients with nonalcoholic fatty liver disease (NAFLD). Diabetes Care 2012, 35, 873–878. [Google Scholar] [CrossRef]
- Targher, G.; Zoppini, G.; Day, C.P. Risk of all-cause and cardiovascular mortality in patients with chronic liver disease. Gut 2011, 60, 1602–1603; author reply 1603–1604. [Google Scholar] [CrossRef]
- Targher, G.; Bertolini, L.; Padovani, R.; Poli, F.; Scala, L.; Tessari, R.; Zenari, L.; Falezza, G. Increased prevalence of cardiovascular disease in Type 2 diabetic patients with non-alcoholic fatty liver disease. Diabet. Med. 2006, 23, 403–409. [Google Scholar] [CrossRef]
- Yki-Jarvinen, H. Fat in the liver and insulin resistance. Ann. Med. 2005, 37, 347–356. [Google Scholar] [CrossRef]
- Auberger, P.; Falquerho, L.; Contreres, J.O.; Pages, G.; Le Cam, G.; Rossi, B.; Le Cam, A. Characterization of a natural inhibitor of the insulin receptor tyrosine kinase: cDNA cloning, purification, and anti-mitogenic activity. Cell 1989, 58, 631–640. [Google Scholar] [CrossRef]
- Pal, D.; Dasgupta, S.; Kundu, R.; Maitra, S.; Das, G.; Mukhopadhyay, S.; Ray, S.; Majumdar, S.S.; Bhattacharya, S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 2012, 18, 1279–1285. [Google Scholar] [CrossRef]
- Hennige, A.M.; Staiger, H.; Wicke, C.; Machicao, F.; Fritsche, A.; Haring, H.U.; Stefan, N. Fetuin-A induces cytokine expression and suppresses adiponectin production. PLoS One 2008, 3, e1765. [Google Scholar] [CrossRef]
- Dogru, T.; Genc, H.; Tapan, S.; Aslan, F.; Ercin, C.N.; Ors, F.; Kara, M.; Sertoglu, E.; Karslioglu, Y.; Bagci, S.; et al. Plasma fetuin-A is associated with endothelial dysfunction and subclinical atherosclerosis in subjects with nonalcoholic fatty liver disease. Clin. Endocrinol. (Oxf.) 2013, 78, 712–717. [Google Scholar] [CrossRef]
- Rittig, K.; Thamer, C.; Haupt, A.; Machann, J.; Peter, A.; Balletshofer, B.; Fritsche, A.; Haring, H.U.; Stefan, N. High plasma fetuin-A is associated with increased carotid intima-media thickness in a middle-aged population. Atherosclerosis 2009, 207, 341–342. [Google Scholar] [CrossRef]
- Weikert, C.; Stefan, N.; Schulze, M.B.; Pischon, T.; Berger, K.; Joost, H.G.; Haring, H.U.; Boeing, H.; Fritsche, A. Plasma fetuin-a levels and the risk of myocardial infarction and ischemic stroke. Circulation 2008, 118, 2555–2562. [Google Scholar] [CrossRef]
- Stefan, N.; Fritsche, A.; Weikert, C.; Boeing, H.; Joost, H.G.; Haring, H.U.; Schulze, M.B. Plasma fetuin-A levels and the risk of type 2 diabetes. Diabetes 2008, 57, 2762–2767. [Google Scholar] [CrossRef]
- Villanova, N.; Moscatiello, S.; Ramilli, S.; Bugianesi, E.; Magalotti, D.; Vanni, E.; Zoli, M.; Marchesini, G. Endothelial dysfunction and cardiovascular risk profile in nonalcoholic fatty liver disease. Hepatology 2005, 42, 473–480. [Google Scholar] [CrossRef]
- Kozakova, M.; Palombo, C.; Eng, M.P.; Dekker, J.; Flyvbjerg, A.; Mitrakou, A.; Gastaldelli, A.; Ferrannini, E. Fatty liver index, gamma-glutamyltransferase, and early carotid plaques. Hepatology 2012, 55, 1406–1415. [Google Scholar] [CrossRef]
- Bonapace, S.; Perseghin, G.; Molon, G.; Canali, G.; Bertolini, L.; Zoppini, G.; Barbieri, E.; Targher, G. Nonalcoholic fatty liver disease is associated with left ventricular diastolic dysfunction in patients with type 2 diabetes. Diabetes Care 2012, 35, 389–395. [Google Scholar] [CrossRef]
- Mofrad, P.; Contos, M.J.; Haque, M.; Sargeant, C.; Fisher, R.A.; Luketic, V.A.; Sterling, R.K.; Shiffman, M.L.; Stravitz, R.T.; Sanyal, A.J. Clinical and histologic spectrum of nonalcoholic fatty liver disease associated with normal ALT values. Hepatology 2003, 37, 1286–1292. [Google Scholar] [CrossRef]
- Ricci, C.; Longo, R.; Gioulis, E.; Bosco, M.; Pollesello, P.; Masutti, F.; Croce, L.S.; Paoletti, S.; de Bernard, B.; Tiribelli, C.; et al. Noninvasive in vivo quantitative assessment of fat content in human liver. J. Hepatol. 1997, 27, 108–113. [Google Scholar] [CrossRef]
- Tsuneto, A.; Hida, A.; Sera, N.; Imaizumi, M.; Ichimaru, S.; Nakashima, E.; Seto, S.; Maemura, K.; Akahoshi, M. Fatty liver incidence and predictive variables. Hypertens. Res. 2010, 33, 638–643. [Google Scholar] [CrossRef]
- Bedogni, G.; Bellentani, S.; Miglioli, L.; Masutti, F.; Passalacqua, M.; Castiglione, A.; Tiribelli, C. The Fatty Liver Index: A simple and accurate predictor of hepatic steatosis in the general population. BMC Gastroenterol. 2006, 6, 33. [Google Scholar] [CrossRef]
- Targher, G.; Day, C.P.; Bonora, E. Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N. Engl. J. Med. 2010, 363, 1341–1350. [Google Scholar] [CrossRef]
- Hill, J.M.; Zalos, G.; Halcox, J.P.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef]
- Chiang, C.H.; Huang, P.H.; Chung, F.P.; Chen, Z.Y.; Leu, H.B.; Huang, C.C.; Wu, T.C.; Chen, J.W.; Lin, S.J. Decreased circulating endothelial progenitor cell levels and function in patients with nonalcoholic fatty liver disease. PLoS One 2012, 7, e31799. [Google Scholar] [CrossRef]
- Lin, Y.C.; Lo, H.M.; Chen, J.D. Sonographic fatty liver, overweight and ischemic heart disease. World J. Gastroenterol. 2005, 11, 4838–4842. [Google Scholar]
- Bugianesi, E.; Gastaldelli, A. Hepatic and cardiac steatosis: Are they coupled? Heart Fail. Clin. 2012, 8, 663–670. [Google Scholar] [CrossRef]
- Assy, N.; Djibre, A.; Farah, R.; Grosovski, M.; Marmor, A. Presence of coronary plaques in patients with nonalcoholic fatty liver disease. Radiology 2010, 254, 393–400. [Google Scholar] [CrossRef]
- Ebrahimi, M.; Kazemi-Bajestani, S.M.; Ghayour-Mobarhan, M.; Moohebati, M.; Paydar, R.; Azimi-Nezhad, M.; Esmaily, H.O.; Ferns, G.A. Metabolic syndrome may not be a good predictor of coronary artery disease in the Iranian population: population-specific definitions are required. Sci. World J. 2009, 9, 86–96. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gaggini, M.; Morelli, M.; Buzzigoli, E.; DeFronzo, R.A.; Bugianesi, E.; Gastaldelli, A. Non-Alcoholic Fatty Liver Disease (NAFLD) and Its Connection with Insulin Resistance, Dyslipidemia, Atherosclerosis and Coronary Heart Disease. Nutrients 2013, 5, 1544-1560. https://doi.org/10.3390/nu5051544
Gaggini M, Morelli M, Buzzigoli E, DeFronzo RA, Bugianesi E, Gastaldelli A. Non-Alcoholic Fatty Liver Disease (NAFLD) and Its Connection with Insulin Resistance, Dyslipidemia, Atherosclerosis and Coronary Heart Disease. Nutrients. 2013; 5(5):1544-1560. https://doi.org/10.3390/nu5051544
Chicago/Turabian StyleGaggini, Melania, Mariangela Morelli, Emma Buzzigoli, Ralph A. DeFronzo, Elisabetta Bugianesi, and Amalia Gastaldelli. 2013. "Non-Alcoholic Fatty Liver Disease (NAFLD) and Its Connection with Insulin Resistance, Dyslipidemia, Atherosclerosis and Coronary Heart Disease" Nutrients 5, no. 5: 1544-1560. https://doi.org/10.3390/nu5051544