Molecular Link between Vitamin D and Cancer Prevention



Abstract
:1. Vitamin D: Introduction, Function and Metabolism
1.1. Introduction to Vitamin D: History and Physiological Roles

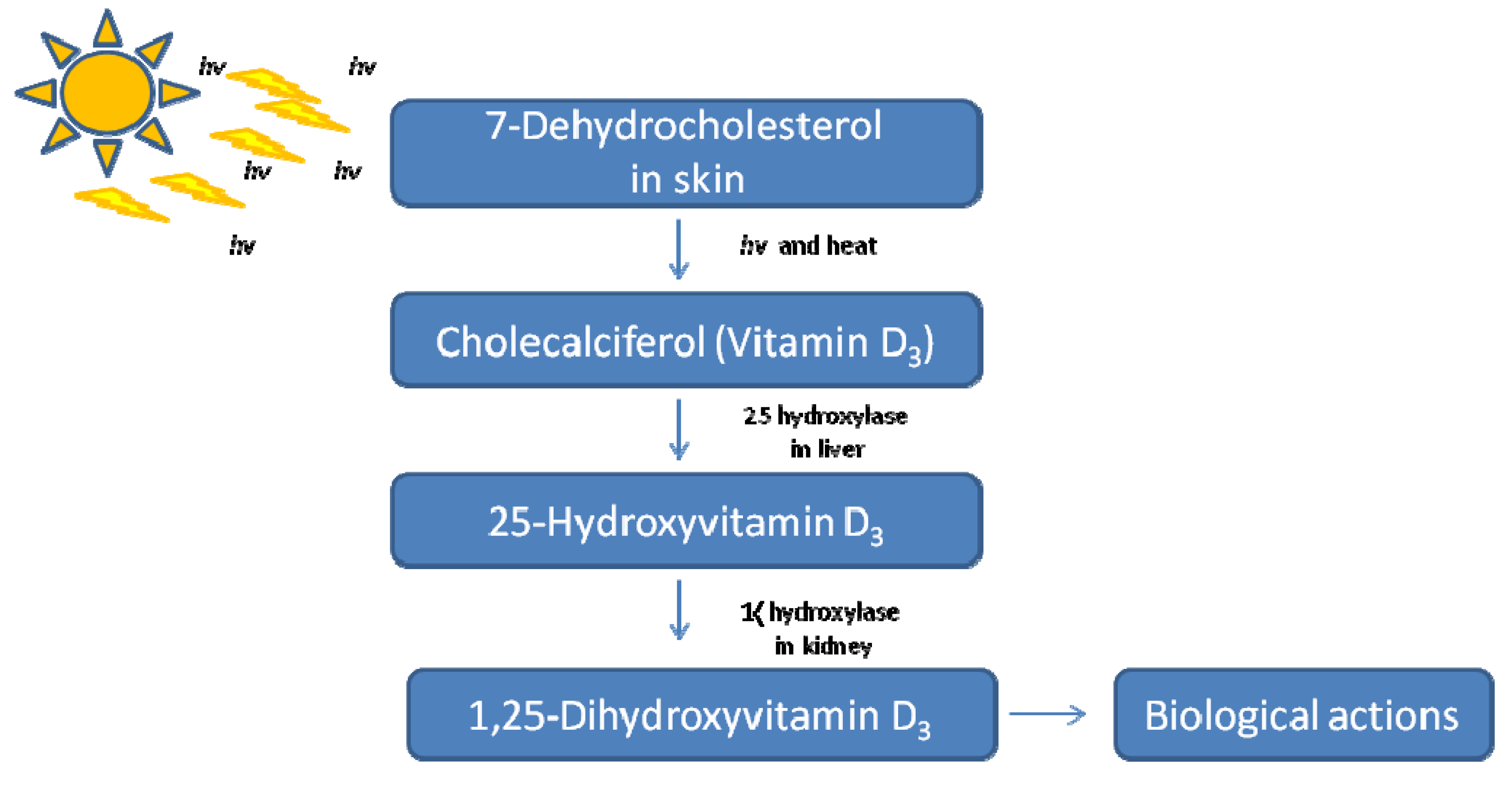{kind=link}
| Organ or system | Target tissue or cell | Specific effects |
|---|---|---|
| Intestine | Duodenum | ↑ Intestinal calcium absorption (TRPV6 intestinal calcium transporters) ↑ Calbindin D28k |
| Jejunum (brush border and basolateral membranes) | ↑ Intestinal phosphate transport | |
| Bone | Osteoblasts (and, in turn, osteoclasts) and chondrocytes | ↑ Bone formation: bone mineralization and matrix formation; ↑ osteocalcin; ↑ osteopontin/SPP1; ↑ RANKL for osteoblasts to activate osteoclasts |
| Parathyroid gland | Chief cells | ↓ PTH |
| Kidneys | Distal tubules (Ca) Proximal Tubules (phosphate) | ↑ Reabsorption of Calcium (↑ TRPV5, calbindin) ↑ Reabsorption of phosphate (↑ NPT1 and NPT2) ↑ Detoxification of 1α,25 dihydroxyvitamin D3 (CYP24A1 OHase) ↑ Calbindin D9k |
| Immune system | Monocytes/macrophages and T lymphocytes (helper type 1) | Suppression of γ-interferon and IL-1–6 |
| Central nervous system | Dorsal root ganglia (glial cells) and hippocampus | ↑ Production of NGF, neurotrophin 3 and leukemia-inhibitory factor |
| Epithelium | Epidermal skin (keratinocyte) | ↑ Differentiation |
| Hair follicle | ↑ Differentiation | |
| Female reproductive tract | Uterine development | |
| Mammary | ↓ Cell growth | |
| Prostate | ↓ Cell growth | |
| Colon | ↓ Cell growth | |
| Endocrine target tissues | Thyroid gland | ↓ TSH |
| Pancreatic β-cells | ↑ Insulin secretion (Calbindin 28K) | |
| Many systems | Diverse cells and cancer cell lines | ↓ Cell growth (↓ c-Fos, ↓ c-Myc) ↑ Differentiation (↑ p21, ↑ p27) ↑ Apoptosis (↓ Bcl-2)↓ Angiogenesis |
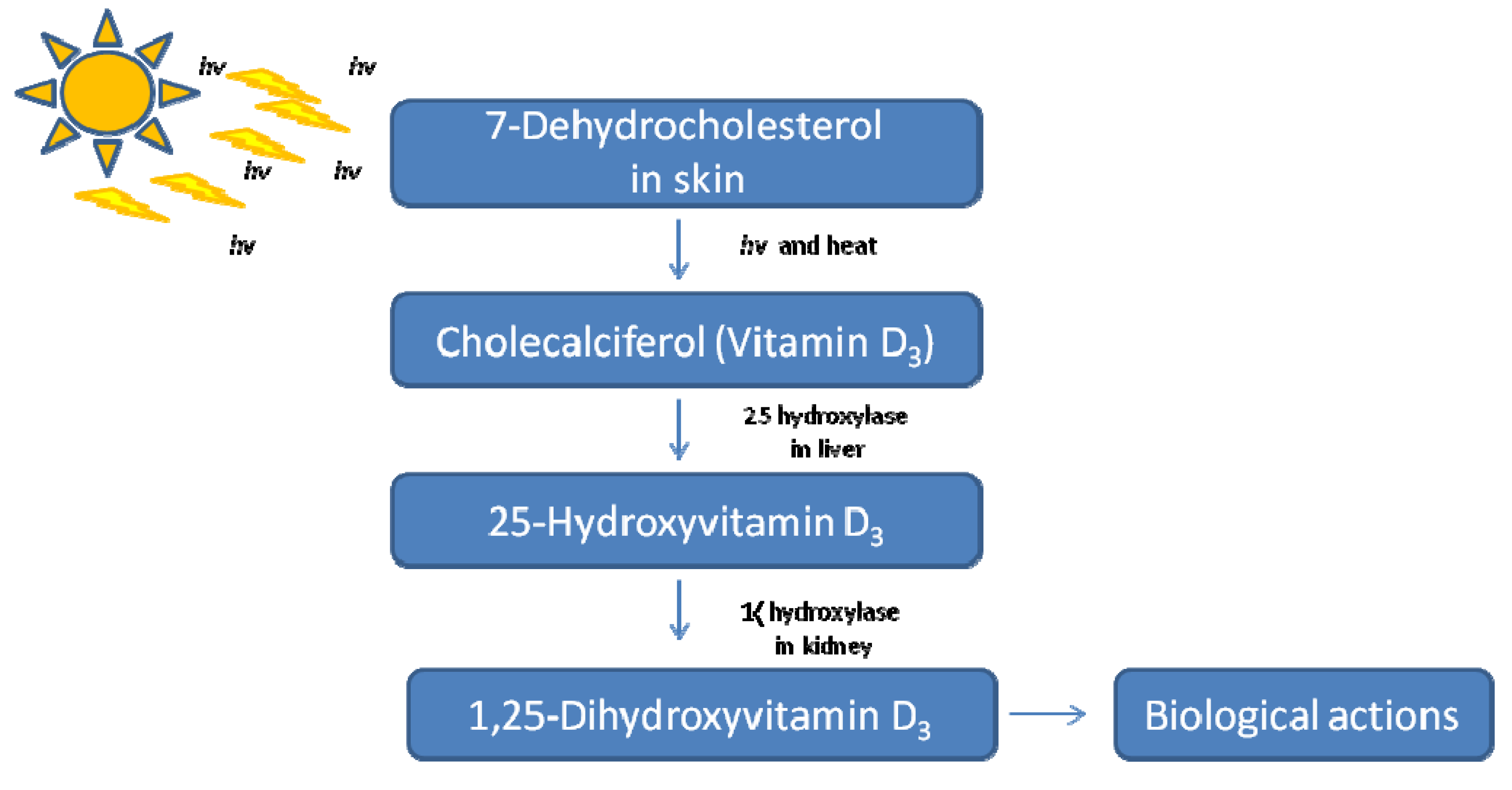
1.2. Vitamin D Genomic Action via the VDR
1.2.1. VDR Distribution, Dimerization and Function
1.2.2. VDR Structure and Role of Cofactors
1.2.3. Nongenomic Cytoplasmic Action
2. Vitamin D and Cancer
2.1. Vitamin D Mechanisms Regulating Cellular Proliferation and Growth
2.2. Cell Cycle and Apoptosis
2.3. Hypoxia, Oxidative Stress, HIF-1 and Angiogenesis
2.4. Interactions with Growth Factors that Mediate Transformation, Cell Adhesion and Metastasis
2.5. Autophagy
2.6. Lessons from Animal Models
- VDR-knockout mouse models of VDR (VDRKO or VDR−/−) vs. wild-type counterparts (VDRWT)
- Animals supplemented with vitamin D
- Animal models in which tumorigenesis is chemically induced before animals are supplemented with vitamin D or its analogues
- VDRKO models where tumors are implanted or transfected
- knockout animal or heterozygotes of other genes (e.g., APCmin/+ or nude mice) subsequently supplemented with vitamin D
2.7. Evidence for Vitamin D and Cancer
| Cancer | MR
(deaths/100,000/Year) * | US
[149,152] | Australia
[155] | China
[153,156] | France
[158] | Japan
[151] | Nordic countries
[162] | Spain
[157] |
|---|---|---|---|---|---|---|---|---|
| Lung | 69.4 | X | X | X | ||||
| Breast | 26.9 | X | X | X | X | X | X | |
| Colorectal | 24.5 | X | X | X | X | X | X | X |
| Prostate | 22.0 | X | X | X | ||||
| Colon | 20.1 | X | X | X | X | X | X | X |
| Pancreatic | 10.2 | X | X | X | X | X | ||
| Leukemia | 8.8 | X | X | X | X | |||
| Ovarian | 8.4 | X | X | X | ||||
| Gastric | 7.3 | X | X | X | X | X | X | |
| non-Hodgkin’s lymphoma (NHL) | 7.0 | X | X | X | ||||
| Bladder | 6.6 | X | X | X | X | |||
| Brain | 5.2 | X | ||||||
| Renal | 4.9 | X | X | |||||
| Esophageal | 4.8 | X | X | X | X | X | X | X |
| Rectal | 4.4 | X | X | X | X | X | ||
| Oral, pharyngeal | 4.0 | X | X | |||||
| Endometrial | 3.7 | X | X | X | ||||
| Cervical | 3.2 | X | X | |||||
| Gallbladder | 1.1 | X | X | X | X | |||
| Hodgkin’s lymphoma | 1.1 | X | X | |||||
| Thyroid | 0.4 | X | X | |||||
| Vulvar | 0.3 | X |
| Cancer | [171] | [154] | [172] | Others |
|---|---|---|---|---|
| Bladder | X * | X | X | [173] |
| Brain | ||||
| Breast | Case-control | |||
| Colon | X | Cohort | ||
| Colorectal | X | Cohort | ||
| Endometrial | [174] | |||
| Esophageal | X | |||
| Esophageal, squamous cell | X * | |||
| Gastric | X * | X * | ||
| Head and neck | X | [175] | ||
| Hepatoblastoma | [160] | |||
| Leukemia | X | |||
| Leukemia, acute lymphoblastic | [160] | |||
| Liver | X * | |||
| Lung | X * | X | ||
| Lung, adeno, squamous cell | X | |||
| NHL | X * | X | [160,176] | |
| Oral/pharyngeal | X | |||
| Ovarian | [177] | |||
| Pancreatic | X | X | X * | [178] |
| Pleura | X | |||
| Prostate | X * | X | ||
| Rectal | X * | Cohort | ||
| Renal | X * | X | X | [179] |
| Thyroid | X | [180] |
2.8. Clinical Trials
2.9. Cancer Survival
2.10. Hill’s Criteria for Causality
3. Recommendations/Conclusions
Abbreviations
| WSTF: | Williams syndrome transcription factor |
| WINAC: | WSTF including nucleosome assembly complex |
| MCF-7: | Michigan Cancer Foundation-7 human breast adenocarcinoma cell line |
| MART-10: | 19-nor-2α-(3-hydroxypropyl)-1α,25-Dihydroxyvitamin D3 |
| EB1089: | Seocalcitol |
| SW620: | Human colorectal adenocarcinoma cell line |
| PC/JW: | Human colorectal adenoma-derived epithelial cell line derived from adenomatous polyposis |
| HT29: | Human colorectal adenocarcinoma cell line HT29 |
| SW-480-ADH: | malignant colon cancer subline of human Dukes’ type B colorectal adenocarcinoma cell line SW-480 |
| LNCaP: | Human prostatic carcinoma cell line LNCaP |
| CL-1: | Human prostate cancer cell line derived from LNCaP |
| IGFBP3: | IGF-binding protein 3 |
| MCF10CA: | Human metastatic breast cancer cell line MCF10CA |
| HL60: | Human promyelocytic leukemia cells |
| mTOR: | mammalian target of rapamycin |
| CDK: | Cyclin-dependent kinase |
| APC: | Adenomatous polyposis coli |
| LPB-Tag: | Long Probasin Promoter-Large T Antigen |
Conflicts of Interest
References
- Severo, M.; Lopes, C.; Lucas, R.; Barros, H. Development of a tool for the assessment of calcium and vitamin D intakes in clinical settings. Osteoporos. Int. 2009, 20, 231–237. [Google Scholar]
- Calvo, M.S.; Babu, U.S.; Garthoff, L.H.; Woods, T.O.; Dreher, M.; Hill, G.; Nagaraja, S. Vitamin D2 from light-exposed edible mushrooms is safe, bioavailable and effectively supports bone growth in rats. Osteoporos. Int. 2013, 24, 197–207. [Google Scholar] [CrossRef]
- Hohman, E.E.; Martin, B.R.; Lachcik, P.J.; Gordon, D.T.; Fleet, J.C.; Weaver, C.M. Bioavailability and efficacy of vitamin D2 from UV-irradiated yeast in growing, vitamin D-deficient rats. J. Agric. Food Chem. 2011, 59, 2341–2346. [Google Scholar] [CrossRef]
- Heaney, R.P.; Armas, L.A.; French, C. All-Source basal vitamin D inputs are greater than previously thought and cutaneous inputs are smaller. J. Nutr. 2013, 143, 571–575. [Google Scholar] [CrossRef]
- Mellanby, E. An experimental investigation on rickets. Lancet 1919, 1, 407–412. [Google Scholar]
- McCollum, E.V.; Simmonds, N.; Becker, J.E.; Shipley, P.G. An experimental demonstration of the existence of a vitamin which promotes calcium deposition. J. Biol. Chem. 1922, 53, 293–298. [Google Scholar]
- Hess, A.F.; Unger, L.F. Cure of infantile rickets by sunlight. J. Am. Med. Assoc. 1921, 77, 39. [Google Scholar]
- Steenbock, H.; Black, A. A fat-soluble vitamins. XVII. The induction of growth-promoting and clcifying properties in a ration by exposure to ultraviolet light. J. Biol. Chem. 1924, 61, 405–422. [Google Scholar]
- Askew, F.A.; Bourdillon, B.R.; Bruce, H.M.; Jenkins, R.G.C. The distillation of vitamin D. Proc. R. Soc. 1931, B107, 76–90. [Google Scholar]
- Windaus, A.; Schenck, F.; von Werder, F. Uber das antirachitisch wirksame bestrahlungsproduct aus 7-dehydro-cholesterin. Hoppe-Seylers Z. Physiol. Chem. 1936, 241, 100–103. (in German). [Google Scholar]
- McCollum, E.V. The paths to the discovery of vitamins A and D. J. Nutr. 1967, 91, 11–16. [Google Scholar]
- Wolf, G. The discovery of vitamin D: The contribution of adolf windaus. J. Nutr. 2004, 134, 1299–1302. [Google Scholar]
- Holick, M.F.; McNeill, S.C.; MacLaughlin, J.A.; Holick, S.A.; Clark, M.B.; Potts, J.T., Jr. Physiologic implications of the formation of previtamin D3 in skin. Trans. Assoc. Am. Phys. 1979, 92, 54–63. [Google Scholar]
- Holick, M.F.; Richtand, N.M.; McNeill, S.C.; Holick, S.A.; Frommer, J.E.; Henley, J.W.; Potts, J.T., Jr. Isolation and identification of previtamin D3 from the skin of rats exposed to ultraviolet irradiation. Biochemistry 1979, 18, 1003–1008. [Google Scholar] [CrossRef]
- Holick, S.A.; Holick, M.F.; MacLaughlin, J.A. Chemical synthesis of [1 beta-3H] 1 alpha, 25-dihydroxyvitamin D3 and [1 alpha-3H] 1 beta, 25-dihydroxyvitamin D2: Biological activity of 1 beta, 25-dihydroxyvitamin D3. Biochem. Biophys. Res. Commun. 1980, 97, 1031–1037. [Google Scholar] [CrossRef]
- Holick, M.F.; MacLaughlin, J.A.; Clark, M.B.; Holick, S.A.; Potts, J.T., Jr.; Anderson, R.R.; Blank, I.H.; Parrish, J.A.; Elias, P. Photosynthesis of previtamin D3 in human skin and the physiologic consequences. Science 1980, 210, 203–205. [Google Scholar]
- Zusman, I.; Hirsh, B.E.; Edelstein, S.; Ornoy, A. Transplacental effects of 1,25-dihydroxycholecalciferol and of 24,25-dihydroxycholecalciferol on the limb skeleton of fetuses and offspring rats. Acta Anat. (Basel) 1981, 111, 343–351. [Google Scholar] [CrossRef]
- Guo, Y.D.; Strugnell, S.; Back, D.W.; Jones, G. Transfected human liver cytochrome p-450 hydroxylates vitamin D analogs at different side-chain positions. Proc. Natl. Acad. Sci. USA 1993, 90, 8668–8672. [Google Scholar] [CrossRef]
- Cheng, J.B.; Levine, M.A.; Bell, N.H.; Mangelsdorf, D.J.; Russell, D.W. Genetic evidence that the human cyp2r1 enzyme is a key vitamin D 25-hydroxylase. Proc. Natl. Acad. Sci. USA 2004, 101, 7711–7715. [Google Scholar]
- Takeyama, K.; Kitanaka, S.; Sato, T.; Kobori, M.; Yanagisawa, J.; Kato, S. 25-Hydroxyvitamin D3 1alpha-hydroxylase and vitamin D synthesis. Science 1997, 277, 1827–1830. [Google Scholar] [CrossRef]
- Dilworth, F.J.; Scott, I.; Green, A.; Strugnell, S.; Guo, Y.D.; Roberts, E.A.; Kremer, R.; Calverley, M.J.; Makin, H.L.; Jones, G. Different mechanisms of hydroxylation site selection by liver and kidney cytochrome p450 species (cyp27 and cyp24) involved in vitamin D metabolism. J. Biol. Chem. 1995, 270, 16766–16774. [Google Scholar] [CrossRef]
- Zehnder, D.; Bland, R.; Williams, M.C.; McNinch, R.W.; Howie, A.J.; Stewart, P.M.; Hewison, M. Extrarenal expression of 25-hydroxyvitamin D(3)-1 alpha-hydroxylase. J. Clin. Endocrinol. Metab. 2001, 86, 888–894. [Google Scholar] [CrossRef]
- Baker, A.R.; McDonnell, D.P.; Hughes, M.; Crisp, T.M.; Mangelsdorf, D.J.; Haussler, M.R.; Pike, J.W.; Shine, J.; O’Malley, B.W. Cloning and expression of full-length cdna encoding human vitamin D receptor. Proc. Natl. Acad. Sci. USA 1988, 85, 3294–3298. [Google Scholar] [CrossRef]
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; Demay, M. Vitamin d and human health: Lessons from vitamin D receptor null mice. Endocr. Rev. 2008, 29, 726–776. [Google Scholar] [CrossRef]
- Bikle, D.D. Protective actions of vitamin D in UVB induced skin cancer. Photochem. Photobiol. Sci. 2012, 11, 1808–1816. [Google Scholar] [CrossRef]
- Rosen, C.J.; Adams, J.S.; Bikle, D.D.; Black, D.M.; Demay, M.B.; Manson, J.E.; Murad, M.H.; Kovacs, C.S. The nonskeletal effects of vitamin D: An endocrine society scientific statement. Endocr. Rev. 2012, 33, 456–492. [Google Scholar] [CrossRef]
- Mohr, S.B. A brief history of vitamin D and cancer prevention. Ann. Epidemiol. 2009, 19, 79–83. [Google Scholar] [CrossRef]
- Haussler, M.R.; Norman, A.W. Chromosomal receptor for a vitamin D metabolite. Proc. Natl. Acad. Sci. USA 1969, 62, 155–162. [Google Scholar] [CrossRef]
- McDonnell, D.P.; Mangelsdorf, D.J.; Pike, J.W.; Haussler, M.R.; O’Malley, B.W. Molecular cloning of complementary DNA encoding the avian receptor for vitamin D. Science 1987, 235, 1214–1217. [Google Scholar]
- Rochel, N.; Wurtz, J.M.; Mitschler, A.; Klaholz, B.; Moras, D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol. Cell 2000, 5, 173–179. [Google Scholar] [CrossRef]
- Lee, S.; Clark, S.A.; Gill, R.K.; Christakos, S. 1,25-Dihydroxyvitamin D3 and pancreatic beta-cell function: Vitamin D receptors, gene expression, and insulin secretion. Endocrinology 1994, 134, 1602–1610. [Google Scholar] [CrossRef]
- McGrath, J.J.; Feron, F.P.; Burne, T.H.; Mackay-Sim, A.; Eyles, D.W. Vitamin D3-implications for brain development. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 557–560. [Google Scholar]
- De Paula, F.J.; Dick-de-Paula, I.; Bornstein, S.; Rostama, B.; Le, P.; Lotinun, S.; Baron, R.; Rosen, C.J. VDR haploinsufficiency impacts body composition and skeletal acquisition in a gender-specific manner. Calcif. Tissue Int. 2011, 89, 179–191. [Google Scholar] [CrossRef]
- DeLuca, H.F. Overview of general physiologic features and functions of vitamin D. Am. J. Clin. Nutr. 2004, 80, 1689S–1696S. [Google Scholar]
- Zanatta, L.; Zamoner, A.; Zanatta, A.P.; Bouraima-Lelong, H.; Delalande, C.; Bois, C.; Carreau, S.; Silva, F.R. Nongenomic and genomic effects of 1alpha,25(OH)2 vitamin D3 in rat testis. Life Sci. 2011, 89, 515–523. [Google Scholar] [CrossRef]
- Wang, Y.; Zhu, J.; DeLuca, H.F. Where is the vitamin D receptor? Arch. Biochem. Biophys. 2012, 523, 123–133. [Google Scholar] [CrossRef]
- Haussler, M.R.; Whitfield, G.K.; Haussler, C.A.; Hsieh, J.C.; Thompson, P.D.; Selznick, S.H.; Dominguez, C.E.; Jurutka, P.W. The nuclear vitamin D receptor: Biological and molecular regulatory properties revealed. J. Bone Miner. Res. 1998, 13, 325–349. [Google Scholar] [CrossRef]
- Rachez, C.; Suldan, Z.; Ward, J.; Chang, C.P.; Burakov, D.; Erdjument-Bromage, H.; Tempst, P.; Freedman, L.P. A novel protein complex that interacts with the vitamin D3 receptor in a ligand-dependent manner and enhances VDR transactivation in a cell-free system. Genes Dev. 1998, 12, 1787–1800. [Google Scholar] [CrossRef]
- Christakos, S.; Dhawan, P.; Liu, Y.; Peng, X.; Porta, A. New insights into the mechanisms of vitamin D action. J. Cell. Biochem. 2003, 88, 695–705. [Google Scholar] [CrossRef]
- Deeb, K.K.; Trump, D.L.; Johnson, C.S. Vitamin D signalling pathways in cancer: Potential for anticancer therapeutics. Nat. Rev. Cancer 2007, 7, 684–700. [Google Scholar] [CrossRef]
- Teichert, A.; Arnold, L.A.; Otieno, S.; Oda, Y.; Augustinaite, I.; Geistlinger, T.R.; Kriwacki, R.W.; Guy, R.K.; Bikle, D.D. Quantification of the vitamin D receptor-coregulator interaction. Biochemistry 2009, 48, 1454–1461. [Google Scholar] [CrossRef]
- Gill, R.K.; Christakos, S. Identification of sequence elements in mouse calbindin-d28k gene that confer 1,25-dihydroxyvitamin D3- and butyrate-inducible responses. Proc. Natl. Acad. Sci. USA 1993, 90, 2984–2988. [Google Scholar] [CrossRef]
- Xie, Z.; Bikle, D.D. Cloning of the human phospholipase C-gamma1 promoter and identification of a DR6-type vitamin D-responsive element. J. Biol. Chem. 1997, 272, 6573–6577. [Google Scholar] [CrossRef]
- Kurokawa, R.; Yu, V.C.; Naar, A.; Kyakumoto, S.; Han, Z.; Silverman, S.; Rosenfeld, M.G.; Glass, C.K. Differential orientations of the DNA-binding domain and carboxy-terminal dimerization interface regulate binding site selection by nuclear receptor heterodimers. Genes Dev. 1993, 7, 1423–1435. [Google Scholar] [CrossRef]
- Crofts, L.A.; Hancock, M.S.; Morrison, N.A.; Eisman, J.A. Multiple promoters direct the tissue-specific expression of novel n-terminal variant human vitamin D receptor gene transcripts. Proc. Natl. Acad. Sci. USA 1998, 95, 10529–10534. [Google Scholar] [CrossRef]
- Sunn, K.L.; Cock, T.A.; Crofts, L.A.; Eisman, J.A.; Gardiner, E.M. Novel n-terminal variant of human VDR. Mol. Endocrinol. 2001, 15, 1599–1609. [Google Scholar] [CrossRef]
- Hsieh, J.C.; Shimizu, Y.; Minoshima, S.; Shimizu, N.; Haussler, C.A.; Jurutka, P.W.; Haussler, M.R. Novel nuclear localization signal between the two DNA-binding zinc fingers in the human vitamin D receptor. J. Cell. Biochem. 1998, 70, 94–109. [Google Scholar] [CrossRef]
- Michigami, T.; Suga, A.; Yamazaki, M.; Shimizu, C.; Cai, G.; Okada, S.; Ozono, K. Identification of amino acid sequence in the hinge region of human vitamin D receptor that transfers a cytosolic protein to the nucleus. J. Biol. Chem. 1999, 274, 33531–33538. [Google Scholar]
- Kato, S.; Fujiki, R.; Kitagawa, H. Vitamin D receptor (VDR) promoter targeting through a novel chromatin remodeling complex. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 173–178. [Google Scholar] [CrossRef]
- Fujiki, R.; Kim, M.S.; Sasaki, Y.; Yoshimura, K.; Kitagawa, H.; Kato, S. Ligand-Induced transrepression by VDR through association of WSTF with acetylated histones. EMBO J. 2005, 24, 3881–3894. [Google Scholar] [CrossRef]
- Kato, S.; Fujiki, R.; Kim, M.S.; Kitagawa, H. Ligand-Induced transrepressive function of VDR requires a chromatin remodeling complex, WINAC. J. Steroid Biochem. Mol. Biol. 2007, 103, 372–380. [Google Scholar] [CrossRef]
- Oda, Y.; Sihlbom, C.; Chalkley, R.J.; Huang, L.; Rachez, C.; Chang, C.P.; Burlingame, A.L.; Freedman, L.P.; Bikle, D.D. Two distinct coactivators, DRIP/mediator and SRC/p160, are differentially involved in VDR transactivation during keratinocyte differentiation. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 273–276. [Google Scholar] [CrossRef]
- Oda, Y.; Chalkley, R.J.; Burlingame, A.L.; Bikle, D.D. The transcriptional coactivator drip/mediator complex is involved in vitamin D receptor function and regulates keratinocyte proliferation and differentiation. J. Investig. Dermatol. 2010, 130, 2377–2388. [Google Scholar] [CrossRef]
- Garcia-Bassets, I.; Kwon, Y.S.; Telese, F.; Prefontaine, G.G.; Hutt, K.R.; Cheng, C.S.; Ju, B.G.; Ohgi, K.A.; Wang, J.; Escoubet-Lozach, L.; et al. Histone methylation-dependent mechanisms impose ligand dependency for gene activation by nuclear receptors. Cell 2007, 128, 505–518. [Google Scholar] [CrossRef]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef]
- Rachez, C.; Lemon, B.D.; Suldan, Z.; Bromleigh, V.; Gamble, M.; Naar, A.M.; Erdjument-Bromage, H.; Tempst, P.; Freedman, L.P. Ligand-dependent transcription activation by nuclear receptors requires the DRIP complex. Nature 1999, 398, 824–828. [Google Scholar] [CrossRef]
- Rachez, C.; Gamble, M.; Chang, C.P.; Atkins, G.B.; Lazar, M.A.; Freedman, L.P. The DRIP complex and SRC-1/p160 coactivators share similar nuclear receptor binding determinants but constitute functionally distinct complexes. Mol. Cell Biol. 2000, 20, 2718–2726. [Google Scholar] [CrossRef]
- Segal, J. Action of the thyroid hormone at the level of the plasma membrane. Endocr. Res. 1989, 15, 619–649. [Google Scholar] [CrossRef]
- Morley, P.; Whitfield, J.F.; Vanderhyden, B.C.; Tsang, B.K.; Schwartz, J.L. A new, nongenomic estrogen action: The rapid release of intracellular calcium. Endocrinology 1992, 131, 1305–1312. [Google Scholar] [CrossRef]
- Nemere, I.; Safford, S.E.; Rohe, B.; DeSouza, M.M.; Farach-Carson, M.C. Identification and characterization of 1,25D3-membrane-associated rapid response, steroid (1,25d3-MARRS) binding protein. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 281–285. [Google Scholar] [CrossRef]
- Caffrey, J.M.; Farach-Carson, M.C. Vitamin D3 metabolites modulate dihydropyridine-sensitive calcium currents in clonal rat osteosarcoma cells. J. Biol. Chem. 1989, 264, 20265–20274. [Google Scholar]
- Baran, D.T.; Sorensen, A.M.; Honeyman, T.W.; Ray, R.; Holick, M.F. 1 alpha,25-dihydroxyvitamin D3-induced increments in hepatocyte cytosolic calcium and lysophosphatidylinositol: Inhibition by pertussis toxin and 1 beta,25-dihydroxyvitamin D3. J. Bone Miner. Res. 1990, 5, 517–524. [Google Scholar] [CrossRef]
- Wali, R.K.; Baum, C.L.; Sitrin, M.D.; Brasitus, T.A. 1,25(OH)2 vitamin D3 stimulates membrane phosphoinositide turnover, activates protein kinase C, and increases cytosolic calcium in rat colonic epithelium. J. Clin. Investig. 1990, 85, 1296–1303. [Google Scholar] [CrossRef]
- Morelli, S.; de Boland, A.R.; Boland, R.L. Generation of inositol phosphates, diacylglycerol and calcium fluxes in myoblasts treated with 1,25-dihydroxyvitamin D3. Biochem. J. 1993, 289, 675–679. [Google Scholar]
- Baran, D.T.; Sorensen, A.M. Rapid actions of 1 alpha-25-dihydroxyvitamin D3 physiologic role. Proc. Soc. Exp. Biol. Med. 1994, 207, 175–179. [Google Scholar] [CrossRef]
- Sitrin, M.D.; Bissonnette, M.; Bolt, M.J.; Wali, R.; Khare, S.; Scaglione-Sewell, B.; Skarosi, S.; Brasitus, T.A. Rapid effects of 1,25(OH)2 vitamin D3 on signal transduction systems in colonic cells. Steroids 1999, 64, 137–142. [Google Scholar] [CrossRef]
- Norman, A.W.; Ishizuka, S.; Okamura, W.H. Ligands for the vitamin D endocrine system: Different shapes function as agonists and antagonists for genomic and rapid response receptors or as a ligand for the plasma vitamin D binding protein. J. Steroid Biochem. Mol. Biol. 2001, 76, 49–59. [Google Scholar] [CrossRef]
- Nemere, I.; Garbi, N.; Hammerling, G.; Hintze, K.J. Role of the 1,25D3-MARRS receptor in the 1,25(OH)2D3-stimulated uptake of calcium and phosphate in intestinal cells. Steroids 2012, 77, 897–902. [Google Scholar] [CrossRef]
- Shi, H.; Norman, A.W.; Okamura, W.H.; Sen, A.; Zemel, M.B. 1alpha,25-Dihydroxyvitamin D3 modulates human adipocyte metabolism via nongenomic action. FASEB J. 2001, 15, 2751–2753. [Google Scholar]
- Richard, C.L.; Farach-Carson, M.C.; Rohe, B.; Nemere, I.; Meckling, K.A. Involvement of 1,25D3-MARRS (membrane associated, rapid response steroid-binding), a novel vitamin D receptor, in growth inhibition of breast cancer cells. Exp. Cell Res. 2010, 316, 695–703. [Google Scholar] [CrossRef]
- Jensen, S.S.; Madsen, M.W.; Lukas, J.; Binderup, L.; Bartek, J. Inhibitory effects of 1alpha,25-dihydroxyvitamin D(3) on the G(1)-S phase-controlling machinery. Mol. Endocrinol. 2001, 15, 1370–1380. [Google Scholar] [CrossRef]
- Salehi-Tabar, R.; Nguyen-Yamamoto, L.; Tavera-Mendoza, L.E.; Quail, T.; Dimitrov, V.; An, B.S.; Glass, L.; Goltzman, D.; White, J.H. Vitamin D receptor as a master regulator of the c-MYC/MXD1 network. Proc. Natl. Acad. Sci. USA 2012, 109, 18827–18832. [Google Scholar] [CrossRef]
- Meyer, M.B.; Goetsch, P.D.; Pike, J.W. VDR/RXR and TCF4/β-catenin cistromes in colonic cells of colorectal tumor origin: Impact on c-FOS and c-MYC gene expression. Mol. Endocrinol. 2012, 26, 37–51. [Google Scholar] [CrossRef]
- Washington, M.N.; Kim, J.S.; Weigel, N.L. 1α,25-dihydroxyvitamin D3 inhibits C4–2 prostate cancer cell growth via a retinoblastoma protein (Rb)-independent G1 arrest. Prostat 2011, 71, 98–110. [Google Scholar] [CrossRef]
- Li, P.; Li, C.; Zhao, X.; Zhang, X.; Nicosia, S.V.; Bai, W. p27(Kip1) stabilization and G(1) arrest by 1,25-dihydroxyvitamin D(3) in ovarian cancer cells mediated through down-regulation of cyclin E/cyclin-dependent kinase 2 and Skp1-Cullin-F-box protein/Skp2 ubiquitin ligase. J. Biol. Chem. 2004, 279, 25260–25267. [Google Scholar]
- Akutsu, N.; Lin, R.; Bastien, Y.; Bestawros, A.; Enepekides, D.J.; Black, M.J.; White, J.H. Regulation of gene expression by 1alpha,25-dihydroxyvitamin D3 and its analog EB1089 under growth-inhibitory conditions in squamous carcinoma cells. Mol. Endocrinol. 2001, 15, 1127–1139. [Google Scholar] [CrossRef]
- Chiang, K.C.; Yeh, C.N.; Chen, S.C.; Shen, S.C.; Hsu, J.T.; Yeh, T.S.; Pang, J.H.; Su, L.J.; Takano, M.; Kittaka, A.; et al. MART-10, a new generation of vitamin D analog, is more potent than 1alpha,25-dihydroxyvitamin D(3) in inhibiting cell proliferation and inducing apoptosis in ER+ MCF-7 breast cancer cells. Evid. Based Complement. Alternat. Med. 2012, 2012, 310872. [Google Scholar]
- Baudet, C.; Chevalier, G.; Chassevent, A.; Canova, C.; Filmon, R.; Larra, F.; Brachet, P.; Wion, D. 1,25-Dihydroxyvitamin D3 induces programmed cell death in a rat glioma cell line. J. Neurosci. Res. 1996, 46, 540–550. [Google Scholar] [CrossRef]
- Bao, B.Y.; Hu, Y.C.; Ting, H.J.; Lee, Y.F. Androgen signaling is required for the vitamin D-mediated growth inhibition in human prostate cancer cells. Oncogene 2004, 23, 3350–3360. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell. Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Losso, J.N.; Bawadi, H.A. Hypoxia inducible factor pathways as targets for functional foods. J. Agric. Food Chem. 2005, 53, 3751–3768. [Google Scholar] [CrossRef]
- Peehl, D.M.; Shinghal, R.; Nonn, L.; Seto, E.; Krishnan, A.V.; Brooks, J.D.; Feldman, D. Molecular activity of 1,25-dihydroxyvitamin D3 in primary cultures of human prostatic epithelial cells revealed by cDNA microarray analysis. J. Steroid Biochem. Mol. Biol. 2004, 92, 131–141. [Google Scholar] [CrossRef]
- Bao, B.Y.; Ting, H.J.; Hsu, J.W.; Lee, Y.F. Protective role of 1 alpha, 25-dihydroxyvitamin D3 against oxidative stress in nonmalignant human prostate epithelial cells. Int. J. Cancer 2008, 122, 2699–2706. [Google Scholar] [CrossRef]
- Fleet, J.C.; DeSmet, M.; Johnson, R.; Li, Y. Vitamin D and cancer: A review of molecular mechanisms. Biochem. J. 2012, 441, 61–76. [Google Scholar] [CrossRef]
- Ben-Shoshan, M.; Amir, S.; Dang, D.T.; Dang, L.H.; Weisman, Y.; Mabjeesh, N.J. 1alpha,25-Dihydroxyvitamin D3 (calcitriol) inhibits hypoxia-inducible factor-1/vascular endothelial growth factor pathway in human cancer cells. Mol. Cancer Ther. 2007, 6, 1433–1439. [Google Scholar] [CrossRef]
- Chung, I.; Han, G.; Seshadri, M.; Gillard, B.M.; Yu, W.D.; Foster, B.A.; Trump, D.L.; Johnson, C.S. Role of vitamin D receptor in the antiproliferative effects of calcitriol in tumor-derived endothelial cells and tumor angiogenesis in vivo. Cancer Res. 2009, 69, 967–975. [Google Scholar] [CrossRef]
- Dormoy, V.; Beraud, C.; Lindner, V.; Coquard, C.; Barthelmebs, M.; Brasse, D.; Jacqmin, D.; Lang, H.; Massfelder, T. Vitamin D3 triggers antitumor activity through targeting hedgehog signaling in human renal cell carcinoma. Carcinogenesis 2012, 33, 2084–2093. [Google Scholar] [CrossRef]
- Yu, H.; Rohan, T. Role of the insulin-like growth factor family in cancer development and progression. J. Natl. Cancer Inst. 2000, 92, 1472–1489. [Google Scholar] [CrossRef]
- Figueroa, J.A.; de Raad, S.; Tadlock, L.; Speights, V.O.; Rinehart, J.J. Differential expression of insulin-like growth factor binding proteins in high versus low gleason score prostate cancer. J. Urol. 1998, 159, 1379–1383. [Google Scholar] [CrossRef]
- Kriebitzsch, C.; Verlinden, L.; Eelen, G.; Tan, B.K.; van Camp, M.; Bouillon, R.; Verstuyf, A. The impact of 1,25(OH)2D3 and its structural analogs on gene expression in cancer cells—A microarray approach. Anticancer Res. 2009, 29, 3471–3483. [Google Scholar]
- Krishnan, A.V.; Shinghal, R.; Raghavachari, N.; Brooks, J.D.; Peehl, D.M.; Feldman, D. Analysis of vitamin D-regulated gene expression in LNCaP human prostate cancer cells using cDNA microarrays. Prostate 2004, 59, 243–251. [Google Scholar] [CrossRef]
- Lee, H.J.; Liu, H.; Goodman, C.; Ji, Y.; Maehr, H.; Uskokovic, M.; Notterman, D.; Reiss, M.; Suh, N. Gene expression profiling changes induced by a novel gemini vitamin D derivative during the progression of breast cancer. Biochem. Pharmacol. 2006, 72, 332–343. [Google Scholar] [CrossRef]
- Swami, S.; Raghavachari, N.; Muller, U.R.; Bao, Y.P.; Feldman, D. Vitamin D growth inhibition of breast cancer cells: Gene expression patterns assessed by cDNA microarray. Breast Cancer Res. Treat. 2003, 80, 49–62. [Google Scholar] [CrossRef]
- Chen, A.; Davis, B.H.; Sitrin, M.D.; Brasitus, T.A.; Bissonnette, M. Transforming growth factor-beta 1 signaling contributes to caco-2 cell growth inhibition induced by 1,25(OH)(2)D(3). Am. J. Physiol. Gastrointest Liver Physiol. 2002, 283, G864–G874. [Google Scholar]
- Tong, W.M.; Hofer, H.; Ellinger, A.; Peterlik, M.; Cross, H.S. Mechanism of antimitogenic action of vitamin D in human colon carcinoma cells: Relevance for suppression of epidermal growth factor-stimulated cell growth. Oncol. Res. 1999, 11, 77–84. [Google Scholar]
- Yanagisawa, J.; Yanagi, Y.; Masuhiro, Y.; Suzawa, M.; Watanabe, M.; Kashiwagi, K.; Toriyabe, T.; Kawabata, M.; Miyazono, K.; Kato, S. Convergence of transforming growth factor-beta and vitamin D signaling pathways on SMAD transcriptional coactivators. Science 1999, 283, 1317–1321. [Google Scholar] [CrossRef]
- Schlange, T.; Matsuda, Y.; Lienhard, S.; Huber, A.; Hynes, N.E. Autocrine WNT signaling contributes to breast cancer cell proliferation via the canonical WNT pathway and EGFR transactivation. Breast Cancer Res. 2007, 9, R63. [Google Scholar] [CrossRef]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. Beta-Catenin-Driven cancers require a yap1 transcriptional complex for survival and tumorigenesis. Cell 2012, 151, 1457–1473. [Google Scholar] [CrossRef]
- Behrens, J. Everything you would like to know about wnt signaling. Sci. Signal. 2013, 6, pe17. [Google Scholar] [CrossRef]
- Vadlakonda, L.; Pasupuleti, M.; Pallu, R. Role of pi3k-akt-mtor and wnt signaling pathways in transition of g1-s phase of cell cycle in cancer cells. Front. Oncol. 2013, 3, 85. [Google Scholar]
- Brabletz, T.; Jung, A.; Dag, S.; Hlubek, F.; Kirchner, T. Beta-Catenin regulates the expression of the matrix metalloproteinase-7 in human colorectal cancer. Am. J. Pathol. 1999, 155, 1033–1038. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H.E. Wnt genes. Cell 1992, 69, 1073–1087. [Google Scholar] [CrossRef]
- Nusse, R. The wnt gene family in tumorigenesis and in normal development. J. Steroid Biochem. Mol. Biol. 1992, 43, 9–12. [Google Scholar] [CrossRef]
- Nusse, R. Wnt signaling and stem cell control. Cell Res. 2008, 18, 523–527. [Google Scholar] [CrossRef]
- Jamora, C.; DasGupta, R.; Kocieniewski, P.; Fuchs, E. Links between signal transduction, transcription and adhesion in epithelial bud development. Nature 2003, 422, 317–322. [Google Scholar] [CrossRef]
- Kovalenko, P.L.; Zhang, Z.; Cui, M.; Clinton, S.K.; Fleet, J.C. 1,25 dihydroxyvitamin d-mediated orchestration of anticancer, transcript-level effects in the immortalized, non-transformed prostate epithelial cell line, rwpe1. BMC Genomics 2010, 11, 26. [Google Scholar] [CrossRef]
- Palmer, H.G.; Gonzalez-Sancho, J.M.; Espada, J.; Berciano, M.T.; Puig, I.; Baulida, J.; Quintanilla, M.; Cano, A.; de Herreros, A.G.; Lafarga, M.; et al. Vitamin d(3) promotes the differentiation of colon carcinoma cells by the induction of e-cadherin and the inhibition of beta-catenin signaling. J. Cell Biol. 2001, 154, 369–387. [Google Scholar] [CrossRef]
- Pendas-Franco, N.; Aguilera, O.; Pereira, F.; Gonzalez-Sancho, J.M.; Munoz, A. Vitamin d and wnt/beta-catenin pathway in colon cancer: Role and regulation of dickkopf genes. Anticancer Res. 2008, 28, 2613–2623. [Google Scholar]
- Beildeck, M.E.; Islam, M.; Shah, S.; Welsh, J.; Byers, S.W. Control of tcf-4 expression by vdr and vitamin d in the mouse mammary gland and colorectal cancer cell lines. PLoS One 2009, 4, e7872. [Google Scholar]
- Rohan, J.N.; Weigel, N.L. 1alpha,25-dihydroxyvitamin d3 reduces c-myc expression, inhibiting proliferation and causing g1 accumulation in c4–2 prostate cancer cells. Endocrinology 2009, 150, 2046–2054. [Google Scholar] [CrossRef]
- Rawson, J.B.; Sun, Z.; Dicks, E.; Daftary, D.; Parfrey, P.S.; Green, R.C.; Gallinger, S.; McLaughlin, J.R.; Wang, P.P.; Knight, J.A.; et al. Vitamin d intake is negatively associated with promoter methylation of the wnt antagonist gene dkk1 in a large group of colorectal cancer patients. Nutr. Cancer 2012, 64, 919–928. [Google Scholar] [CrossRef]
- Aguilera, O.; Pena, C.; Garcia, J.M.; Larriba, M.J.; Ordonez-Moran, P.; Navarro, D.; Barbachano, A.; Lopez de Silanes, I.; Ballestar, E.; Fraga, M.F.; et al. The wnt antagonist dickkopf-1 gene is induced by 1alpha,25-dihydroxyvitamin d3 associated to the differentiation of human colon cancer cells. Carcinogenesis 2007, 28, 1877–1884. [Google Scholar] [CrossRef]
- Villaggio, B.; Soldano, S.; Cutolo, M. 1,25-Dihydroxyvitamin D3 downregulates aromatase expression and inflammatory cytokines in human macrophages. Clin. Exp. Rheumatol. 2012, 30, 934–938. [Google Scholar]
- Kaler, P.; Augenlicht, L.; Klampfer, L. Macrophage-derived il-1beta stimulates wnt signaling and growth of colon cancer cells: A crosstalk interrupted by vitamin d3. Oncogene 2009, 28, 3892–3902. [Google Scholar] [CrossRef]
- Bao, B.Y.; Yao, J.; Lee, Y.F. 1alpha,25-Dihydroxyvitamin d3 suppresses interleukin-8-mediated prostate cancer cell angiogenesis. Carcinogenesis 2006, 27, 1883–1893. [Google Scholar] [CrossRef]
- Tse, A.K.; Zhu, G.Y.; Wan, C.K.; Shen, X.L.; Yu, Z.L.; Fong, W.F. 1alpha,25-Dihydroxyvitamin d3 inhibits transcriptional potential of nuclear factor kappa b in breast cancer cells. Mol. Immunol. 2010, 47, 1728–1738. [Google Scholar] [CrossRef]
- Hoyer-Hansen, M.; Jaattela, M. Autophagy: An emerging target for cancer therapy. Autophagy 2008, 4, 574–580. [Google Scholar]
- Bristol, M.L.; Di, X.; Beckman, M.J.; Wilson, E.N.; Henderson, S.C.; Maiti, A.; Fan, Z.; Gewirtz, D.A. Dual functions of autophagy in the response of breast tumor cells to radiation: Cytoprotective autophagy with radiation alone and cytotoxic autophagy in radiosensitization by vitamin d 3. Autophagy 2012, 8, 739–753. [Google Scholar] [CrossRef]
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381. [Google Scholar] [CrossRef]
- Karantza-Wadsworth, V.; Patel, S.; Kravchuk, O.; Chen, G.; Mathew, R.; Jin, S.; White, E. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev. 2007, 21, 1621–1635. [Google Scholar] [CrossRef]
- Wang, J. Beclin 1 bridges autophagy, apoptosis and differentiation. Autophagy 2008, 4, 947–948. [Google Scholar]
- Wang, J.; Lian, H.; Zhao, Y.; Kauss, M.A.; Spindel, S. Vitamin d3 induces autophagy of human myeloid leukemia cells. J. Biol. Chem. 2008, 283, 25596–25605. [Google Scholar]
- Lozy, F.; Karantza, V. Autophagy and cancer cell metabolism. Semin. Cell Dev. Biol. 2012, 23, 395–401. [Google Scholar] [CrossRef]
- Kato, S.; Takeyama, K.; Kitanaka, S.; Murayama, A.; Sekine, K.; Yoshizawa, T. In vivo function of VDR in gene expression-VDR knock-out mice. J. Steroid Biochem. Mol. Biol. 1999, 69, 247–251. [Google Scholar] [CrossRef]
- Li, Y.C.; Pirro, A.E.; Amling, M.; Delling, G.; Baron, R.; Bronson, R.; Demay, M.B. Targeted ablation of the vitamin d receptor: An animal model of vitamin d-dependent rickets type ii with alopecia. Proc. Natl. Acad. Sci. USA 1997, 94, 9831–9835. [Google Scholar] [CrossRef]
- Nakagawa, K.; Kawaura, A.; Kato, S.; Takeda, E.; Okano, T. Metastatic growth of lung cancer cells is extremely reduced in vitamin d receptor knockout mice. J. Steroid Biochem. Mol. Biol. 2004, 89–90, 545–547. [Google Scholar] [CrossRef]
- Kallay, E.; Pietschmann, P.; Toyokuni, S.; Bajna, E.; Hahn, P.; Mazzucco, K.; Bieglmayer, C.; Kato, S.; Cross, H.S. Characterization of a vitamin d receptor knockout mouse as a model of colorectal hyperproliferation and DNA damage. Carcinogenesis 2001, 22, 1429–1435. [Google Scholar] [CrossRef]
- Kallay, E.; Bareis, P.; Bajna, E.; Kriwanek, S.; Bonner, E.; Toyokuni, S.; Cross, H.S. Vitamin d receptor activity and prevention of colonic hyperproliferation and oxidative stress. Food Chem. Toxicol. 2002, 40, 1191–1196. [Google Scholar] [CrossRef]
- Zinser, G.; Packman, K.; Welsh, J. Vitamin d(3) receptor ablation alters mammary gland morphogenesis. Development 2002, 129, 3067–3076. [Google Scholar]
- Mehta, R.G.; Moriarty, R.M.; Mehta, R.R.; Penmasta, R.; Lazzaro, G.; Constantinou, A.; Guo, L. Prevention of preneoplastic mammary lesion development by a novel vitamin d analogue, 1alpha-hydroxyvitamin d5. J. Natl. Cancer Inst. 1997, 89, 212–218. [Google Scholar] [CrossRef]
- Mehta, R.; Hawthorne, M.; Uselding, L.; Albinescu, D.; Moriarty, R.; Christov, K. Prevention of N-methyl-N-nitrosourea-induced mammary carcinogenesis in rats by 1alpha-hydroxyvitamin d(5). J. Natl. Cancer Inst. 2000, 92, 1836–1840. [Google Scholar] [CrossRef]
- Jacobson, E.A.; James, K.A.; Newmark, H.L.; Carroll, K.K. Effects of dietary fat, calcium, and vitamin d on growth and mammary tumorigenesis induced by 7,12-dimethylbenz(a)anthracene in female sprague-dawley rats. Cancer Res. 1989, 49, 6300–6303. [Google Scholar]
- VanWeelden, K.; Flanagan, L.; Binderup, L.; Tenniswood, M.; Welsh, J. Apoptotic regression of MCF-7 xenografts in nude mice treated with the vitamin d3 analog, Eb1089. Endocrinology 1998, 139, 2102–2110. [Google Scholar] [CrossRef]
- Welsh, J. Vitamin d and breast cancer: Insights from animal models. Am. J. Clin. Nutr. 2004, 80, 1721S–1724S. [Google Scholar]
- Zinser, G.M.; Welsh, J. Accelerated mammary gland development during pregnancy and delayed postlactational involution in vitamin d3 receptor null mice. Mol. Endocrinol. 2004, 18, 2208–2223. [Google Scholar] [CrossRef]
- Welsh, J. Cellular and molecular effects of vitamin d on carcinogenesis. Arch. Biochem. Biophys. 2012, 523, 107–114. [Google Scholar] [CrossRef]
- Xu, H.; McCann, M.; Zhang, Z.; Posner, G.H.; Bingham, V.; El-Tanani, M.; Campbell, F.C. Vitamin d receptor modulates the neoplastic phenotype through antagonistic growth regulatory signals. Mol. Carcinog. 2009, 48, 758–772. [Google Scholar] [CrossRef]
- Xu, H.; Posner, G.H.; Stevenson, M.; Campbell, F.C. Apc(min) modulation of vitamin d secosteroid growth control. Carcinogenesis 2010, 31, 1434–1441. [Google Scholar] [CrossRef]
- Xie, Z.; Komuves, L.; Yu, Q.C.; Elalieh, H.; Ng, D.C.; Leary, C.; Chang, S.; Crumrine, D.; Yoshizawa, T.; Kato, S.; et al. Lack of the vitamin d receptor is associated with reduced epidermal differentiation and hair follicle growth. J. Investig. Dermatol. 2002, 118, 11–16. [Google Scholar] [CrossRef]
- Zinser, G.M.; Sundberg, J.P.; Welsh, J. Vitamin d(3) receptor ablation sensitizes skin to chemically induced tumorigenesis. Carcinogenesis 2002, 23, 2103–2109. [Google Scholar] [CrossRef]
- Zinser, G.M.; Tribble, E.; Valrance, M.; Urben, C.M.; Knutson, J.C.; Mazess, R.B.; Strugnell, S.A.; Welsh, J. 1,24(s)-dihydroxyvitamin d2, an endogenous vitamin d2 metabolite, inhibits growth of breast cancer cells and tumors. Anticancer Res. 2005, 25, 235–241. [Google Scholar]
- Mordan-McCombs, S.; Brown, T.; Wang, W.L.; Gaupel, A.C.; Welsh, J.; Tenniswood, M. Tumor progression in the lpb-tag transgenic model of prostate cancer is altered by vitamin d receptor and serum testosterone status. J. Steroid Biochem. Mol. Biol. 2010, 121, 368–371. [Google Scholar] [CrossRef]
- Garland, C.F.; Garland, F.C. Do sunlight and vitamin d reduce the likelihood of colon cancer? Int. J. Epidemiol. 1980, 9, 227–231. [Google Scholar] [CrossRef]
- Garland, F.C.; Garland, C.F.; Gorham, E.D.; Young, J.F. Geographic variation in breast cancer mortality in the united states: A hypothesis involving exposure to solar radiation. Prev. Med. 1990, 19, 614–622. [Google Scholar] [CrossRef]
- Lefkowitz, E.S.; Garland, C.F. Sunlight, vitamin d, and ovarian cancer mortality rates in US women. Int. J. Epidemiol. 1994, 23, 1133–1136. [Google Scholar] [CrossRef]
- Grant, W.B. Ecological studies of the UVB-vitamin d-cancer hypothesis. Anticancer Res. 2012, 32, 223–236. [Google Scholar]
- Grant, W.B. Update on evidence that support a role of solar ultraviolet-b irradiance in reducing cancer risk. Anti-Cancer Agents Med. Chem. 2013, 13, 140–146. [Google Scholar] [CrossRef]
- Grant, W.B.; Garland, C.F. The association of solar ultraviolet b (UVB) with reducing risk of cancer: Multifactorial ecologic analysis of geographic variation in age-adjusted cancer mortality rates. Anticancer Res. 2006, 26, 2687–2699. [Google Scholar]
- Grant, W.B. An estimate of premature cancer mortality in the U.S. Due to inadequate doses of solar ultraviolet-b radiation. Cancer 2002, 94, 1867–1875. [Google Scholar] [CrossRef]
- Mizoue, T. Ecological study of solar radiation and cancer mortality in japan. Health Phys. 2004, 87, 532–538. [Google Scholar] [CrossRef]
- Boscoe, F.P.; Schymura, M.J. Solar ultraviolet-b exposure and cancer incidence and mortality in the united states, 1993–2002. BMC Cancer 2006, 6, 264. [Google Scholar] [CrossRef]
- Chen, W.; Clements, M.; Rahman, B.; Zhang, S.; Qiao, Y.; Armstrong, B.K. Relationship between cancer mortality/incidence and ambient ultraviolet b irradiance in china. Cancer Causes Control. 2010, 21, 1701–1709. [Google Scholar] [CrossRef]
- Lin, S.W.; Wheeler, D.C.; Park, Y.; Cahoon, E.K.; Hollenbeck, A.R.; Freedman, D.M.; Abnet, C.C. Prospective study of ultraviolet radiation exposure and risk of cancer in the united states. Int. J. Cancer 2012, 131, E1015–E1023. [Google Scholar] [CrossRef]
- Astbury, A. Non Uniformity in Cancer Mortality in the USA and Australia Appears to Share a Common Origin; TRIUMF: Vancouver, BC, USA, 2005. [Google Scholar]
- Grant, W.B. Does solar ultraviolet irradiation affect cancer mortality rates in china? Asian Pac. J. Cancer Prev. 2007, 8, 236–242. [Google Scholar]
- Grant, W.B. An ecologic study of cancer mortality rates in spain with respect to indices of solar UVB irradiance and smoking. Int. J. Cancer 2007, 120, 1123–1128. [Google Scholar] [CrossRef]
- Grant, W.B. An ecological study of cancer incidence and mortality rates in france with respect to latitude, an index for vitamin d production. Deramato-Endocrinology 2010, 2, 62–67. [Google Scholar] [CrossRef]
- Grant, W.B. An ecological study of cancer mortality rates in california, 1950–64, with respect to solar UVB and smoking indices. Dermato-Endocrinology 2012, 4, 176–182. [Google Scholar] [CrossRef]
- Lombardi, C.; Heck, J.E.; Cockburn, M.; Ritz, B. Solar UV radiation and cancer in young children. Cancer Epidemiol. Biomark. Prev. 2013, 22, 1118–1128. [Google Scholar] [CrossRef]
- Fioletov, V.E.; McArthur, L.J.; Mathews, T.W.; Marrett, L. Estimated ultraviolet exposure levels for a sufficient vitamin d status in north america. J. Photochem. Photobiol. B 2010, 100, 57–66. [Google Scholar] [CrossRef]
- Grant, W.B. Role of solar UVB irradiance and smoking in cancer as inferred from cancer incidence rates by occupation in nordic countries. Dermato-Endocrinology 2012, 4, 203–211. [Google Scholar] [CrossRef]
- Brittingham, A.; de la Cruz, G.P. Ancestry 2000. Census 2000 Brief ck2br-35; U.S. Dept. of Commerce: Washington, DC, USA, 2004. [Google Scholar]
- Grant, W.B. A multicountry ecological study of risk-modifying factors for prostate cancer: Apolipoprotein e epsilon4 as a risk factor and cereals as a risk reduction factor. Anticancer Res. 2010, 30, 189–199. [Google Scholar]
- Lehrer, S. Possible relationship of the apolipoprotein e (apoe) epsilon4 allele to prostate cancer. Br. J. Cancer 1998, 78, 1398. [Google Scholar] [CrossRef]
- Grant, W.B. Relation between prediagnostic serum 25-hydroxyvitamin d level and incidence of breast, colorectal, and other cancers. J. Photochem. Photobiol. B 2010, 101, 130–136. [Google Scholar] [CrossRef]
- Devesa, S.S.; Grauman, D.J.; Blot, W.J.; Pennello, G.A.; Hoover, R.N.; Fraumeni, J.F.J. Atlas of Cancer Mortality in the United States, 1950–1994. In NIH Publication No. 99–4564; National Institute of Health: Rockville, MD, USA, 1999. [Google Scholar]
- Grant, W.B. An ecological study of cancer mortality rates in the united states with respect to solar ultraviolet-b doses, smoking, alcohol consumption and urban/rural residence. Dermato-Endocrinology 2010, 2, 68–76. [Google Scholar] [CrossRef]
- Grant, W.B. Effect of interval between serum draw and follow-up period on relative risk of cancer incidence with respect to 25-hydroxyvitamin d level: Implications for meta-analyses and setting vitamin d guidelines. Dermato-Endocrinology 2011, 3, 199–204. [Google Scholar]
- Grant, W.B. Effect of follow-up time on the relation between prediagnostic serum 25-hydroxyvitamin d and all-cause mortality rate. Dermato-Endocrinology 2012, 4, 198–202. [Google Scholar] [CrossRef]
- Giovannucci, E.; Liu, Y.; Rimm, E.B.; Hollis, B.W.; Fuchs, C.S.; Stampfer, M.J.; Willett, W.C. Prospective study of predictors of vitamin d status and cancer incidence and mortality in men. J. Natl. Cancer Inst. 2006, 98, 451–459. [Google Scholar] [CrossRef]
- Afzal, S.; Bojesen, S.E.; Nordestgaard, B.G. Low plasma 25-hydroxyvitamin d and risk of tobacco-related cancer. Clin. Chem. 2013, 59, 771–780. [Google Scholar] [CrossRef]
- Mondul, A.M.; Weinstein, S.J.; Mannisto, S.; Snyder, K.; Horst, R.L.; Virtamo, J.; Albanes, D. Serum vitamin d and risk of bladder cancer. Cancer Res. 2010, 70, 9218–9223. [Google Scholar]
- Epstein, E.; Lindqvist, P.G.; Geppert, B.; Olsson, H. A population-based cohort study on sun habits and endometrial cancer. Br. J. Cancer 2009, 101, 537–540. [Google Scholar] [CrossRef]
- Orell-Kotikangas, H.; Schwab, U.; Osterlund, P.; Saarilahti, K.; Makitie, O.; Makitie, A.A. High prevalence of vitamin d insufficiency in patients with head and neck cancer at diagnosis. Head Neck 2012, 34, 1450–1455. [Google Scholar] [CrossRef]
- Kricker, A.; Armstrong, B.K.; Hughes, A.M.; Goumas, C.; Smedby, K.E.; Zheng, T.; Spinelli, J.J.; de Sanjose, S.; Hartge, P.; Melbye, M.; et al. Personal sun exposure and risk of non hodgkin lymphoma: A pooled analysis from the interlymph consortium. Int. J. Cancer 2008, 122, 144–154. [Google Scholar] [CrossRef]
- Toriola, A.T.; Surcel, H.M.; Calypse, A.; Grankvist, K.; Luostarinen, T.; Lukanova, A.; Pukkala, E.; Lehtinen, M. Independent and joint effects of serum 25-hydroxyvitamin d and calcium on ovarian cancer risk: A prospective nested case-control study. Eur. J. Cancer 2010, 46, 2799–2805. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Ng, K.; Bao, Y.; Kraft, P.; Stampfer, M.J.; Michaud, D.S.; Ma, J.; Buring, J.E.; Sesso, H.D.; Lee, I.M.; et al. Plasma 25-hydroxyvitamin d and risk of pancreatic cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 82–91. [Google Scholar] [CrossRef]
- Joh, H.K.; Giovannucci, E.L.; Bertrand, K.A.; Lim, S.; Cho, E. Predicted plasma 25-hydroxyvitamin d and risk of renal cell cancer. J. Natl. Cancer Inst. 2013, 105, 726–732. [Google Scholar] [CrossRef]
- Roskies, M.; Dolev, Y.; Caglar, D.; Hier, M.P.; Mlynarek, A.; Majdan, A.; Payne, R.J. Vitamin d deficiency as a potentially modifiable risk factor for thyroid cancer. J. Otolaryngol. Head Neck Surg. 2012, 41, 160–163. [Google Scholar]
- Shimada, T.; Urakawa, I.; Isakova, T.; Yamazaki, Y.; Epstein, M.; Wesseling-Perry, K.; Wolf, M.; Salusky, I.B.; Juppner, H. Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. J. Clin. Endocrinol. Metab. 2010, 95, 578–585. [Google Scholar] [CrossRef]
- Oh, E.Y.; Ansell, C.; Nawaz, H.; Yang, C.H.; Wood, P.A.; Hrushesky, W.J. Global breast cancer seasonality. Breast Cancer Res. Treat. 2010, 123, 233–243. [Google Scholar] [CrossRef]
- Lappe, J.M.; Travers-Gustafson, D.; Davies, K.M.; Recker, R.R.; Heaney, R.P. Vitamin d and calcium supplementation reduces cancer risk: Results of a randomized trial. Am. J. Clin. Nutr. 2007, 85, 1586–1591. [Google Scholar]
- Peterlik, M.; Grant, W.B.; Cross, H.S. Calcium, vitamin d and cancer. Anticancer Res. 2009, 29, 3687–3698. [Google Scholar]
- Bolland, M.J.; Grey, A.; Gamble, G.D.; Reid, I.R. Calcium and vitamin d supplements and health outcomes: A reanalysis of the women’s health initiative (whi) limited-access data set. Am. J. Clin. Nutr. 2011, 94, 1144–1149. [Google Scholar] [CrossRef]
- Garland, C.F.; French, C.B.; Baggerly, L.L.; Heaney, R.P. Vitamin d supplement doses and serum 25-hydroxyvitamin d in the range associated with cancer prevention. Anticancer Res. 2011, 31, 607–611. [Google Scholar]
- Lappe, J.M.; Heaney, R.P. Why randomized controlled trials of calcium and vitamin d sometimes fail. Dermato-Endocrinology 2012, 4, 95–100. [Google Scholar] [CrossRef]
- Ng, K.; Meyerhardt, J.A.; Wu, K.; Feskanich, D.; Hollis, B.W.; Giovannucci, E.L.; Fuchs, C.S. Circulating 25-hydroxyvitamin d levels and survival in patients with colorectal cancer. J. Clin. Oncol. 2008, 26, 2984–2991. [Google Scholar] [CrossRef]
- Grant, W.B.; Peiris, A.N. Differences in vitamin d status may account for unexplained disparities in cancer survival rates between african and white americans. Dermato-Endocrinology 2012, 4, 85–94. [Google Scholar] [CrossRef]
- Ginde, A.A.; Liu, M.C.; Camargo, C.A., Jr. Demographic differences and trends of vitamin d insufficiency in the us population, 1988–2004. Arch. Intern. Med. 2009, 169, 626–632. [Google Scholar] [CrossRef]
- Hill, A.B. The environment and disease: Association or causation? Proc. R. Soc. Med. 1965, 58, 295–300. [Google Scholar]
- Grant, W.B. How strong is the evidence that solar ultraviolet b and vitamin d reduce the risk of cancer? An examination using hill’s criteria for causality. Dermato-Endocrinology 2009, 1, 17–24. [Google Scholar] [CrossRef]
- Mohr, S.B.; Gorham, E.D.; Alcaraz, J.E.; Kane, C.I.; Macera, C.A.; Parsons, J.K.; Wingard, D.L.; Garland, C.F. Does the evidence for an inverse relationship between serum vitamin d status and breast cancer risk satisfy the hill criteria? Dermato-Endocrinology 2012, 4, 152–157. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moukayed, M.; Grant, W.B. Molecular Link between Vitamin D and Cancer Prevention. Nutrients 2013, 5, 3993-4021. https://doi.org/10.3390/nu5103993
Moukayed M, Grant WB. Molecular Link between Vitamin D and Cancer Prevention. Nutrients. 2013; 5(10):3993-4021. https://doi.org/10.3390/nu5103993
Chicago/Turabian StyleMoukayed, Meis, and William B. Grant. 2013. "Molecular Link between Vitamin D and Cancer Prevention" Nutrients 5, no. 10: 3993-4021. https://doi.org/10.3390/nu5103993
APA StyleMoukayed, M., & Grant, W. B. (2013). Molecular Link between Vitamin D and Cancer Prevention. Nutrients, 5(10), 3993-4021. https://doi.org/10.3390/nu5103993