Mobilization of Stored Iron in Mammals: A Review

Abstract
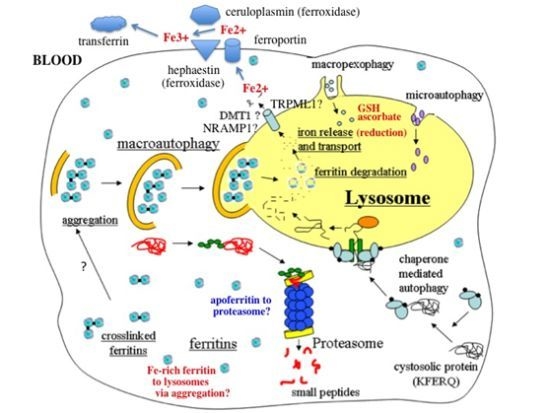
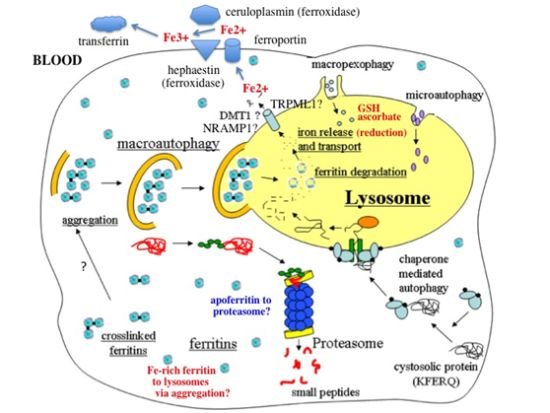
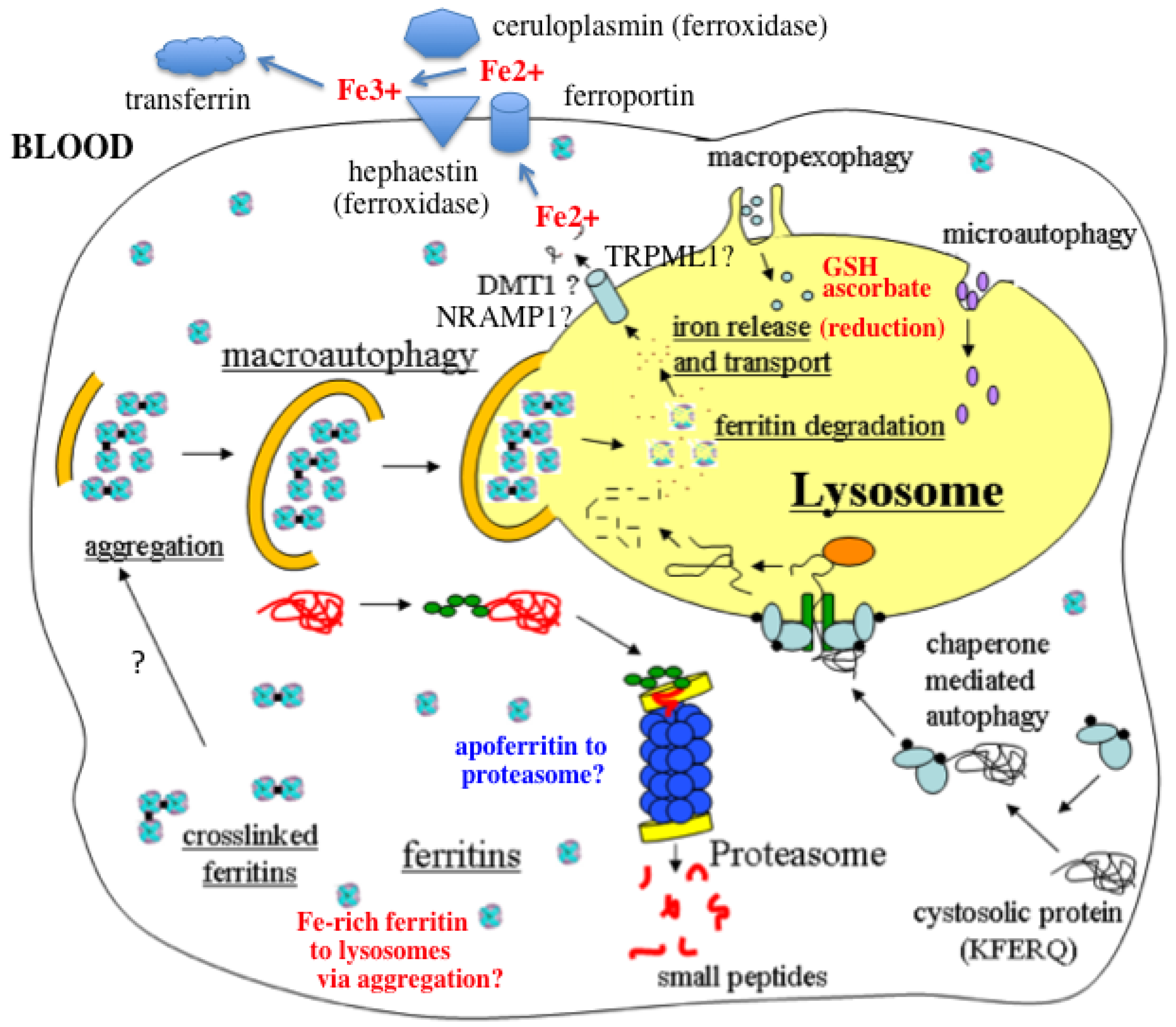
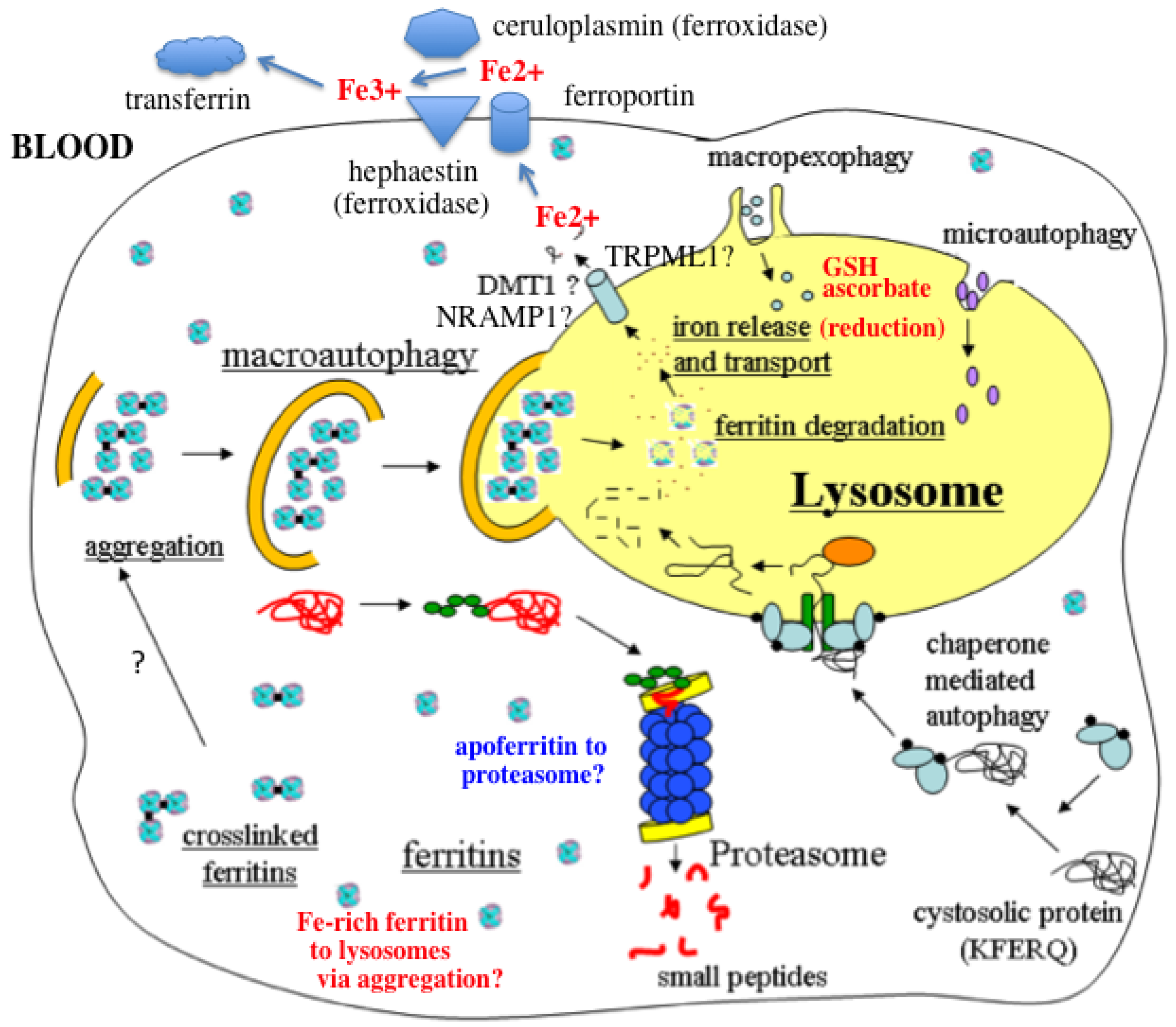
:
{kind=link}
{kind=link}
1. Introduction
2. Ferritin Structure and Formation for Iron Storage and Detoxification
3. Release of Stored Iron from Ferritin
3.1. Regulation of Ferritin Degradation
3.2. Mechanisms of Ferritin Degradation and Release of Its Iron
3.2.1. Lysosomal Ferritin Degradation
3.2.2. Proteasomal Degradation of Ferritin
3.3. Does Iron Exit Ferritin without the Protein First Being Degraded?
3.4. Solubilization of Ferritin Iron in Lysosomes
3.5. Transport and Distribution of Iron after Release from Ferritin in Lysosomes
4. Conclusions
- In response to a signal relating to iron deprivation (resulting in a drop in the level of the labile iron pool) and/or decreased oxygen tension, cells in various organs cause ferritin in the cytosol to be taken up by lysosomes via autophagy. The form of autophagy is unclear but both macro-autophagy and perhaps also ferritin aggregation might be involved. Iron-rich ferritin may be targeted preferentially. Possibly, iron-poor and apoferritin are degraded in the cytosol by the proteasome simultaneously, to minimize reincorporation of iron into ferritin.
- In the lysosomes, the ferritin protein “shell” is degraded by cathepsins, exposing the nanocrystals of ferrihydrite inside, which dissolve in the lysosomal fluid upon reduction by glutathione, ascorbate, and other fluid constituents, the Fe2+ chelated by binding to them and to residual proteins present (including undegraded ferritin and perhaps also Hsp70 and metallothionein).
- Fe transporters in the lysosomal membrane, including DMT1, and depending upon the cell type also NRAMP1 and perhaps TRPML1, facilitate diffusion of Fe2+ into the cytosol, where it joins the labile iron pool, bound to potential chaperones that have and have not as yet been identified.
- Depending upon the cell type and perhaps also the signal, the new iron in the labile pool coming from ferritin via lysosomes is distributed to where it is needed within the cell (such as for Fe-S cluster or heme biosynthesis in the mitochondria) and/or exported into the extracellular fluid and blood via the plasma membrane transporter ferroportin and bind to the transport protein transferrin mediated by membrane-attached or soluble extracellular ferroxidases, like hephaestin and ceruloplasmin, respectively.
- Iron on transferrin then goes on mostly to develop reticulocytes in the bone marrow, but is also available to most other cells as needed, where it is taken up by receptor-mediated endocytosis of di- and monoferric-transferrin, and released from endosomes into the cytosol by DMT1 (and perhaps also other transmembrane transporters) after reduction of Fe3+ by Steap 3, other reductases, or endosomal ascorbate.
Acknowledgments
Conflicts of Interest
References
- Linder, M.C. Nutrition and Metabolism of Trace Elements. In Nutritional Biochemistry and Metabolism, 2nd ed.; Linder, M.C., Ed.; Elsevier/Appleton-Lange: New York, NY, USA, 1991; pp. 215–276. [Google Scholar]
- LeBlanc, S.; Barrick, M.D.; Arredondo, M. Heme carrier protein 1 transports heme and is involved in heme iron metabolism. Am. J. Physiol. Cell Physiol. 2012, 302, C1780–C1785. [Google Scholar] [CrossRef]
- Theil, E.C.; Chen, H.; Miranda, C.; Janser, H.; Elsenhans, B.; Nunez, M.T.; Pizarro, F.; Schumann, K. Absorption of iron from ferritin is independent of heme iron and ferrous salts in women and rat intestinal segments. J. Nutr. 2012, 142, 478–483. [Google Scholar] [CrossRef]
- Schaer, D.J.; Buehler, P.W.; Alayashi, A.; Belcher, J.D.; Vercellotti, G.M. Hemolysis and free hemoglobin revisited: Exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 2013, 121, 1276–1284. [Google Scholar] [CrossRef]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef]
- Muckenthaler, M.U.; Galy, B.; Hentze, M. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef]
- Clegg, G.A.; Fitton, J.E.; Harrison, P.M.; Treffry, A. Ferritin: Molecular structure and iron storage mechanisms. Prog. Biophys. Mol. Biol. 1980, 36, 56–86. [Google Scholar]
- Harrison, P.M.; Hoy, T.G.; Macara, I.G.; Hoare, R.J. Ferritin iron uptake and release. Structure-function relationships. Biochem. J. 1974, 143, 445–451. [Google Scholar]
- Linder, M.C.; Kakavandi, H.R.; Miller, P.; Nagel, G.N. Dissociation of ferritins. Arch. Biochem. Biophys. 1989, 269, 485–496. [Google Scholar] [CrossRef]
- Linder, M.C.; Goode, C.A.; Gonzalez, R.; Gottschling, C.; Gray, J.; Nagel, G.N. Heart tissue contains small and large aggregates of ferritin subunits. Arch. Biochem. Biophys. 1989, 273, 34–41. [Google Scholar] [CrossRef]
- Linder, M.C.; Nagel, G.N.; Roboz, M.; Hungerford, D., Jr. Heart cells contain a ferritin larger and more asymmetric than ferritins of other mammalian tissues. J. Biol. Chem. 1981, 256, 9104–9111. [Google Scholar]
- Vulimiri, L.; Linder, M.C.; Munro, H.N.; Catsimpoolas, N. Structural features of rat cardiac ferritins. Biochim. Biophys. Acta 1977, 491, 67–75. [Google Scholar] [CrossRef]
- Andrews, S.; Robinson, A.K.; Rodriguez-Quinones, F. Bacterial iron homeostasis. FEMS Microbiol. Rev. 2003, 27, 215–237. [Google Scholar] [CrossRef]
- Macedo, S.; Romao, C.V.; Mitchell, E.; Matias, P.M.; Liu, M.Y.; Xavier, A.V.; LeGall, J.; Teixeira, M.; Lindley, P.; Carrondo, M.A. The nature of the di-iron site in the bacterioferritin from Desulfovibrio desulfuricans. Nat. Struct. Biol. 2003, 10, 285–290. [Google Scholar] [CrossRef]
- Theil, E.C. Iron, ferritin, and nutrition. Annu. Rev. Nutr. 2004, 24, 327–343. [Google Scholar] [CrossRef]
- Campbell, C.H.; Ismail, R.; Linder, M.C. Ferritin mRNA is found on bound as well as free polyribosomes in rat heart. Arch. Biochem. Biophys. 1989, 273, 89–98. [Google Scholar] [CrossRef]
- Linder, M.C.; Madani, N.; Middleton, R.; Miremadi, A.; Cairo, G.; Tacchini, L.; Schiaffonati, L.; Rappocciolo, E. Ferritin synthesis on polyribosomes attached to the endoplasmic reticulum. J. Inorg. Biochem. 1992, 47, 229–240. [Google Scholar] [CrossRef]
- Tran, T.N.; Eubanks, S.; Zhou, C.Y.J.; Linder, M.C. Secretion of ferritin by rat hepatoma cells and its regulation by inflammatory cytokines and iron. Blood 1997, 90, 4979–4986. [Google Scholar] [Green Version]
- Levi, S.; Corsi, B.; Bosisio, M.; Invernizzi, R.; Volz, A.; Sanford, D.; Arosio, P.; Drysdale, J. A human mitochondrial ferritin encoded by an intronless gene. J. Biol. Chem. 2001, 276, 24437–24440. [Google Scholar] [Green Version]
- Richardson, D.R.; Lane, D.J.R.; Becker, E.M.; Huang, M.L.-H.; Whitnall, M.; Rahmanto, Y.S.; Sheftel, A.D.; Ponka, P. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc. Natl. Acad. Sci. USA 2010, 107, 10775–10782. [Google Scholar] [CrossRef]
- Theil, E.C.; Liu, X.S.; Tosha, T. Gated pores in the ferritin protein nanocage. Inorg. Chim. Acta 2008, 361, 868–874. [Google Scholar] [CrossRef]
- Liu, X.; Theil, E.C. Ferritins: Dynamic management of biological iron and oxygen chemistry. Acc. Chem. Res. 2005, 38, 167–175. [Google Scholar] [CrossRef]
- Vulimiri, L.; Catsimpoolas, N.; Griffith, A.L.; Linder, M.C.; Munro, H.N. Size and charge heterogeneity of rat tissue ferritins. Biochim. Biophys. Acta 1975, 412, 148–156. [Google Scholar] [CrossRef]
- Vulimiri, L.; Linder, M.C.; Munro, H.N. Sex difference in the distribution and iron responsiveness of rat cardiac and skeletal muscle ferritins. Biochim. Biophys. Acta 1977, 497, 280–287. [Google Scholar] [CrossRef]
- Linder, M.C.; Moor, J.R.; Munro, H.N.; Morris, H.P. Structural differences in ferritins from normal and malignant rat tissues. Biochim. Biophys. Acta 1975, 386, 409–421. [Google Scholar] [CrossRef]
- Linder, M.C.; Wright, K.; Madara, J. Concentration, structure and iron-saturation of ferritins from normal human lung and lung tumors with graded histopathology. Enzyme 1982, 27, 189–198. [Google Scholar]
- Linder, M.C.; Munro, H.N. Metabolic and chemical features of ferritin, a series of iron inducible tissue proteins. Am. J. Pathol. 1973, 72, 263–282. [Google Scholar]
- Liu, X.; Jin, W.; Theil, E.C. Opening protein pores with chaotropes enhances Fe reduction and chelation of Fe from the ferritin biomineral. Proc. Natl. Acad. Sci. USA 2003, 100, 3653–3658. [Google Scholar] [CrossRef]
- May, C.A.; Grady, J.K.; Laue, T.M.; Poli, M.; Arosio, P.; Chasteen, N.D. The sedimentation properties of ferritins. New insights and analysis of methods of nanoparticle preparation. Biochim. Biophys. Acta 2010, 1800, 858–870. [Google Scholar] [CrossRef]
- Munro, H.N.; Linder, M.C. Ferritin: Structure, biosynthesis, and role in iron metabolism. Physiol. Rev. 1978, 58, 317–396. [Google Scholar]
- Theil, E.C. Ferritin protein nanocages use ion channels, catalytic sites, and nucleation channels to manage iron/oxygen chemistry. Curr. Opin. Chem. Biol. 2011, 15, 304–311. [Google Scholar] [CrossRef]
- Toussaint, L.; Bertrand, L.; Hue, L.; Crichton, R.R.; Declercq, J.P. High resolution E-ray structures of human apoferritin H chains mutants correlates with their activity and metal binding sites. J. Mol. Biol. 2007, 365, 440–452. [Google Scholar] [CrossRef]
- Philpott, C.C. Coming into view: Eukaryotic iron chaperones and intracellular iron delivery. J. Biol. Chem. 2012, 2878, 13518–13523. [Google Scholar] [CrossRef]
- Shi, H.; Bencze, K.Z.; Stemmler, T.L.; Philpott, C.C. A cytosolic iron chaperone that delivers iron to ferritin. Science 2008, 320, 1207–1210. [Google Scholar] [CrossRef]
- Nam, H.; Wang, C.Y.; Zhang, L.; Zhang, W.; Hojyo, S.; Fukada, T.; Knutson, M.D. ZIP14 and DMT1 in the liver, pancreas, and heart are differentially regulated by iron deficiency and overload: Implications for tissue iron uptake in iron-related disorders. Haematologica 2013, 98, 1049–1057. [Google Scholar] [CrossRef]
- Kahlon, O.; Cabantchik, Z.I. The labile iron pool: Characterization, measurement, and participation in cellular processes (1). Free Radic. Biol. Med. 2002, 33, 1037–1046. [Google Scholar] [CrossRef]
- Ward, D.M.; Kaplan, J. Ferroportin-mediated iron transport: Expression and regulation. Biochim. Biophys. Acta 2012, 1823, 1426–1433. [Google Scholar] [CrossRef]
- Lane, D.J.R.; Chikhani, S.; Richardson, V.; Richardson, D.R. Transferrin iron uptake is stimulated by ascorbate via an intracellular reductive mechanism. Biochem. Biophys. Acta 2013, 1833, 1527–1541. [Google Scholar] [CrossRef]
- Arosio, P.; Ingrassia, R.; Cavadini, P. Ferritins: A family of molecules for iron storage, antioxidation and more. Biochim. Biophys. Acta 2009, 1790, 589–599. [Google Scholar] [CrossRef]
- Torti, F.M.; Torti, S.V. Regulation of ferritin genes and protein. Blood 2002, 99, 3505–3516. [Google Scholar] [CrossRef]
- Tsuji, Y. JunD activates transcription of human ferritin H gene through an antioxidant defense element during oxidative stress. Oncogene 2005, 24, 7567–7578. [Google Scholar] [CrossRef]
- Truty, J.; Malpe, R.; Linder, M.C. Iron prevents ferritin turnover in hepatic cells. J. Biol. Chem. 2001, 276, 48775–48780. [Google Scholar] [CrossRef]
- Kidane, T.Z.; Sauble, E.; Linder, M.C. Release of iron from ferritin requires lysosomal activity. Am. J. Physiol. Cell Physiol. 2006, 291, C445–C455. [Google Scholar] [CrossRef]
- Linder, M.C.; Zerounian, N.A.; Moriya, M.; Malpe, R. Iron and copper homeostasis and intestinal absorption, using the Caco2 cell model system. BioMetals 2003, 16, 145–160. [Google Scholar] [CrossRef]
- Moriya, M.; Linder, M.C. Vesicular transport and apotransferrin in intestinal iron absorption, as shown in the Caco2 cell model. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G301–G309. [Google Scholar] [CrossRef]
- Dailey, H.A.; Meissner, P.N. Erythroid heme biosynthesis and its disorders. Cold Spring Harb. Perspect. Med. 2013, 3. [Google Scholar] [CrossRef]
- Bridges, K.R.; Hoffman, K.E. The effects of ascorbic acid on the intracellular metabolism of iron and ferritin. J. Biol. Chem. 1986, 261, 14273–14277. [Google Scholar]
- Asano, T.; Komatsu, M.; Yamaguchi-Iwai, Y.; Ishikawa, F.; Mizushima, N.; Iwai, K. Distinct mechanisms of ferritin delivery to lysosomes in iron-depleted and iron-replete cells. Mol. Cell. Biol. 2011, 31, 2040–2052. [Google Scholar] [CrossRef]
- Faa, G.; Terlizzo, M.; Gerosa, C.; Congiu, T.; Angelucci, E. Patterns of iron distribution in liver cells in β-thalassemia studied in X-ray microanalysis. Haematologica 2002, 87, 479–484. [Google Scholar]
- Garner, G.; Roberg, K.; Brunk, U.T. Endogenous ferritin protects cells with iron-laden lysosomes against oxidative stress. Free Radic. Res. 1998, 29, 103–114. [Google Scholar] [CrossRef]
- Houghton, P.B.; James, E.M.; Williams, W.J.; Henderson, W.J. The fine structure of dense lysosomes isolated from rat spleen. Beitr. Pathol. 1976, 157, 244–250. [Google Scholar] [CrossRef]
- Persson, H.L.; Nilsson, K.J.; Brunk, U.T. Novel cellular defenses against iron and oxidation: Ferritin and autophagocytosis preserve lysosomal stability in airway epithelium. Redox Rep. 2001, 6, 57–63. [Google Scholar] [CrossRef]
- Kurz, T.; Eaton, J.W.; Brunk, U.T. The role of lysosomes in iron metabolism and recycling. Int. J. Biochem. Cell Biol. 2011, 43, 1686–1697. [Google Scholar] [CrossRef]
- Droga-Mazovec, G.; Bojic, L.; Petelin, A.; Ivanova, S.; Romih, R.; Repnik, U.; Salvesen, G.S.; Stoka, V.; Turk, V.; Turk, B. Cysteine cathepsins trigger caspase-dependent cell death through cleavage of bid and antiapoptotic Bcl-2 homologues. J. Biol. Chem. 2008, 283, 19140–19150. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Terman, A.; Kurz, T.; Navratil, M.; Arriaga, E.; Brunk, U. Mitochondrial turnover and aging of long-lived postmitotic cells: The mitochondrial-lysosomal axis theory of aging. Antioxid. Redox Signal. 2010, 12, 503–535. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Cuervo, A.M.; Dunn, W.A., Jr.; Levine, B.; van der Klei, I.; Seglen, P.O. How shall I eat thee? Autophagy 2007, 3, 413–416. [Google Scholar]
- Park, C.; Cuervo, A.M. Selective autophagy: Talking with the UPS. Cell. Biochem. Biophys. 2013, 67, 3–13. [Google Scholar] [CrossRef]
- Mehlhase, J.; Sandig, G.; Pantopoulos, K.; Grune, T. Oxidation-induced ferritin turnover in microglial cells: Role of proteasome. Free Radic. Biol. Med. 2005, 38, 276–285. [Google Scholar] [CrossRef]
- Bridges, K.R. Ascorbic acid inhibits lysosomal autophagy of ferritin. J. Biol. Chem. 1987, 262, 14773–14778. [Google Scholar]
- Hoffman, K.E.; Yanelli, K.; Bridges, K.R. Ascorbic acid and iron metabolism: Alterations in lysosomal function. Am. J. Clin. Nutr. 1991, 54, 1188S–1192S. [Google Scholar]
- Roberts, S.; Bomford, A. Ferritin iron kinetics and protein turnover in K562 cells. J. Biol. Chem. 1988, 263, 19181–19187. [Google Scholar]
- Vaisman, B.; Fibach, E.; Konijn, A.M. Utilization of intracellular ferritin iron for hemoglobin synthesis in developing human erythroid precursors. Blood 1997, 90, 831–838. [Google Scholar] [Green Version]
- Radisky, D.C.; Kaplan, J. Iron in cytosolic ferritin can be recycled through lysosomal degradation in human fibroblasts. Biochem. J. 1998, 336, 201–205. [Google Scholar] [Green Version]
- Ollinger, K.; Roberg, K. Nutrient deprivation of cultured rat hepatocytes increases the desferrioxamine-available iron pool and augments the sensitivity to hydrogen peroxide. J. Biol. Chem. 1997, 272, 23707–23711. [Google Scholar] [CrossRef]
- Larson, J.A.; Howie, H.L.; So, M. Neisseria meningitidis accelerates ferritin degradation in host epithelial cells to yield an essential iron source. Mol. Microbiol. 2004, 53, 807–820. [Google Scholar] [CrossRef]
- Kwok, J.C.; Richardson, D.R. Examination of the mechanisms involved in doxorubicin-mediated iron accumulation in ferritin: Studies using metabolic inhibitors, protein synthesis inhibitors, and lysosomotropic agents. Mol. Pharmacol. 2004, 65, 81–195. [Google Scholar]
- Zhang, Y.; Mikhael, M.; Xu, D.; Li, Y.; Soe-Lin, S.; Ning, B.; Li, W.; Nie, G.; Zhao, Y.; Ponka, P. Lysosomal proteolysis is the primary degradation pathway for cytosolic ferritin and cytosolic ferritin degradation is necessary for iron exit. Antioxid. Redox Signal. 2010, 13, 999–1009. [Google Scholar] [CrossRef]
- Kurz, T.; Gustafsson, B.; Brunk, U.T. Cell sensitivity to oxidative stress is influenced by ferritin autophagy. Free Radic. Biol. Med. 2011, 50, 1647–1658. [Google Scholar] [CrossRef]
- Sulzer, D.; Mosharov, E.; Talloczy, Z.; Zucca, F.A.; Simon, J.D.; Zecca, L. Neuronal pigmented autophagic vacuoles: Lipofuscin, neuromelanin, and ceroid as macroautophagic responses during aging and disease. J. Neurochem. 2008, 106, 24–36. [Google Scholar] [CrossRef]
- Ghosh, M.; Carlsson, F.; Laskar, A.; Yuan, X.-M.; Li, W. Lysosomal membtane permeabilization causes oxidative stress and ferritin induction in macrophages. FEBS Lett. 2011, 585, 623–629. [Google Scholar] [CrossRef]
- Kurz, T.; Terman, A.; Gustafsson, B.; Brunk, U.T. Lysosomes in iron metabolism, ageing and apoptosis. Histochem. Cell Biol. 2008, 129, 389–406. [Google Scholar] [CrossRef]
- Andrews, G.K. Regulation of metallothionein gene expression by oxidative stress and metal ions. Biochem. Pharmacol. 2000, 59, 95–104. [Google Scholar] [CrossRef]
- Ruttkay-Nedecky, B.; Nejdl, L.; Gumulec, J.; Zitka, O.; Masarik, M.; Eckschlager, T.; Stiborova, M.; Adam, V.; Kizek, R. The role of metallothionein in oxidative stress. Int. J. Mol. Sci. 2013, 14, 6044–6066. [Google Scholar] [CrossRef]
- Doulias, P.T.; Kotoglou, P.; Tenopoulou, M.; Keramisanou, D.; Tzavaras, T.; Brunk, U.T.; Galaris, D.; Angelidis, C. Involvement of heat shock protein-70 in the mechanism of hydrogen peroxide-induced DNA damage: The role of lysosomes and iron. Free Radic. Biol. Med. 2007, 42, 567–577. [Google Scholar] [CrossRef]
- Kurz, T.; Brunk, U.T. Autophagy of HSP70 and chelation of lysosomal iron in a non-redox active form. Autophagy 2009, 5, 93–95. [Google Scholar] [CrossRef]
- Baird, S.K.; Kurz, T.; Brunk, U.T. Metallothionein protects against oxidative stress-induced lysosomal destabilization. Biochem. J. 2006, 394, 275–283. [Google Scholar] [CrossRef]
- Garner, G.; Li, W.; Roberg, K.; Brunk, U.T. On the cytoprotective role of ferritin in macrophages and its ability to enhance lysosomal stability. Free Radic. Res. 1997, 27, 487–500. [Google Scholar] [CrossRef]
- Epsztejn, S.; Glickstein, H.; Picard, V.; Slotki, I.N.; Breuer, W.; Beaumont, C.; Cabantchik, Z.I. H-Ferritin subunit overexpression in erythroid cells reduces the oxidative stress response and induces multidrug resistance properties. Blood 1999, 94, 3593–3603. [Google Scholar]
- Orino, K.; Tsuji, Y.; Torti, F.M.; Torti, S.V. Adenovirus E1A blocks oxidant-dependent ferritin induction and sensitizes cells to pro-oxidant cytotoxicity. FEBS Lett. 1999, 462, 334–338. [Google Scholar]
- Li, W.; Yang, Q.; Mao, Z. Chaperone-mediated autophagy: Machinery, regulation and biological consequences. Cell. Mol. Life Sci. 2011, 68, 749–763. [Google Scholar] [CrossRef]
- Cuervo, A.M. Autophagy: Many paths to the same end. Mol. Cell. Biochem. 2004, 263, 55–72. [Google Scholar] [CrossRef]
- Mizushima, N. Methods for monitoring autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2491–2502. [Google Scholar] [CrossRef]
- Fuqua, B.K.; Vulpe, C.D.; Anderson, G.J. Intestinal iron absorption. J. Trace Elem. Med. Biol. 2012, 26, 115–119. [Google Scholar] [CrossRef]
- Kettern, N.; Dreiseidler, M.; Tawo, R.; Hohfeld, J. Chaperone-assisted degradation: Multiple paths to destruction. Biol. Chem. 2010, 391, 481–489. [Google Scholar]
- Kaushik, S.; Cuervo, A.M. Autophagy as a cell-repair mechanism: Activation of chaperone-mediated autophagy during oxidative stress. Mol. Aspects Med. 2006, 27, 444–454. [Google Scholar] [CrossRef]
- Massey, A.; Kiffin, R.; Cuervo, A.M. Pathophysiology of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2420–2434. [Google Scholar] [CrossRef]
- Gamerdinger, M.; Carra, A.; Behl, C. Emerging roles of molecular chaperones and co-chaperones in sselective autophagy: Focus on BAG proteins. J. Mol. Med. 2011, 89, 1175–1182. [Google Scholar] [CrossRef]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Didik, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef]
- Grune, T.; Jung, T.; Merker, K.; Davies, K.J. Decreased proteolysis caused by protein aggregates, inclusion bodies, plaques, lipofusion, ceroid, and “aggresomes” during oxidative stress, aging, and disease. Int. J. Biochem. Cell Biol. 2004, 36, 2519–2530. [Google Scholar] [CrossRef]
- De Domenico, I.; Ward, D.M.; Kaplan, J. Specific iron chelators determine the route of ferritin degradation. Blood 2009, 114, 4546–4551. [Google Scholar] [CrossRef]
- Seglen, P.O.; Gordon, P.B. 3-Methyl adenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef]
- Wu, Y.-T.; Tan, W.-L.; Shui, G.; Bauvy, C.; Huang, Q.; Wenk, M.R.; Ong, C.-N.; Codogno, P.; Shen, H.-M. Dual role of 3methyladenine in modulation of autophagy via different temporal patterns of inhibition on Class I and III phosphoinositide 3-kinase. J. Biol. Chem. 2010, 285, 10850–10861. [Google Scholar] [CrossRef]
- Williams, A.; Jahreiss, L.; Sarkar, S.; Saiki, S.; Menzies, F.M.; Ravikumar, B.; Rubinsztein, D.C. Aggregate-prone proteins are cleared from the cytosol by autophagy: Therapeutic implications. Curr. Top. Dev. Biol. 2006, 76, 89–101. [Google Scholar] [CrossRef]
- Harrison, P.M.; Gregory, D.W. Evidence for the existence of stable “aggregates” in horse ferritin and apoferritin. J. Mol. Biol. 1965, 14, 626–629. [Google Scholar] [CrossRef]
- Lee, S.S.C.; Richter, G.W. The monomers and oligomers of ferritin and apoferritin: Association and dissociation. Biochemistry 1967, 15, 65–70. [Google Scholar]
- Williams, M.A.; Harrison, P.M. Electron microscopic and chemical studies of oligomers of ferritin. Biochem. J. 1968, 110, 265–280. [Google Scholar]
- Welch, K.D.; Reilly, C.A.; Aust, S.D. The role of cysteine residues in the oxidation of ferritin. Free Radic. Biol. Med. 2001, 33, 399–408. [Google Scholar] [CrossRef]
- Welch, K.D.; van Eden, M.E.; Aust, S.D. Modification of ferritin during iron loading. Free Radic. Biol. Med. 2001, 31, 999–1006. [Google Scholar] [CrossRef]
- Hasan, M.R.; Morishima, D.; Tomita, K.; Katsuki, M.; Kotani, S. Identification of a 250 kDa putative microtubule-associated protein as bovine ferritin: Evidence for ferritin-microtubule interaction. FEBS J. 2005, 272, 822–831. [Google Scholar] [CrossRef]
- Hasan, M.R.; Koikawa, S.; Kotani, S.; Miyamoto, S.; Nakagawa, H. Ferritin forms dynamic oligomers to associate with microtubules in vivo: Implication for the role of microtubules in iron metabolism. Exp. Cell Res. 2006, 312, 1950–1960. [Google Scholar] [CrossRef]
- Rechsteiner, M.; Rogers, S.W. PEST sequences and regulation by proteolysis. Trends Biochem. Sci. 1996, 21, 267–271. [Google Scholar]
- Adams, J. The proteasome: Structure, function, and role in the cell. Cancer Treat. Rev. 2003, 29, S3–S9. [Google Scholar] [CrossRef]
- Shringarpure, R.; Grune, T.; Mehlhase, J.; Davies, K.J.A. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. J. Biol. Chem. 2003, 278, 311–318. [Google Scholar] [CrossRef]
- Rock, K.L.; Gramm, C.; Rothstein, L.; Clark, K.; Stein, R.; Dick, L.; Hwang, D.; Goldberg, A.L. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 1994, 78, 761–771. [Google Scholar] [CrossRef]
- Rudeck, M.; Volk, T.; Sitte, N.; Grune, T. Ferritin oxidation in vitro: Implication of iron release and degradation by the 20S proteasome. IUBMB Life 2000, 49, 451–456. [Google Scholar] [CrossRef]
- De Domenico, I.; Vaughn, M.B.; Li, L.; Bagley, D.; Musci, G.; Ward, D.M.; Kaplan, J. Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. EMBO J. 2006, 25, 5396–5404. [Google Scholar] [CrossRef]
- Lloyd, J.B.; Cable, H.; Rice-Evans, C. Evidence that desferrioxamine cannot enter cells by passive diffusion. Biochem. Pharmacol. 1991, 41, 1361–1363. [Google Scholar] [CrossRef]
- Tosha, T.; Behera, R.K.; Ng, H.L.; Bhattahali, O.; Alber, T.; Theil, E. Ferritin protein nanocage ion channels: Gating by N-terminal extensions. J. Biol. Chem. 2012, 287, 13016–13025. [Google Scholar]
- Jin, W.; Takagi, H.; Pancorbo, B.; Theil, E.C. “Opening” the ferritin pore for iron release by mutation of conserved amino acids at interhelix and loop sites. Biochemistry 2001, 40, 7525–7532. [Google Scholar] [CrossRef]
- Takagi, H.; Shi, D.; Ha, Y.; Allewell, N.M.; Theil, E.C. Localized unfolding at the junction of three ferritin subunits. A mechanism for iron release? J. Biol. Chem. 273, 1998, 18685–18688. [Google Scholar]
- Liu, X.S.; Patterson, L.D.; Miller, M.J.; Theil, E.C. Peptides selected for the protein nanocage pores change the rate of iron recovery from the ferritin mineral. J. Biol. Chem. 2007, 282, 31821–31825. [Google Scholar]
- Nguyen, T.; Linder, M.C. California State University: Fullerton, CA, USA, Unpublished work. 2010.
- Morgan, J.; La, A.; Nguyen, T.; Sauble, E.; Gonzalez, A.; Nguyen, A.; Linder, M.C. Release of Iron from Cellular Iron Stores in Lysosomes: Potential Involvement of Divalent Metal Transporter 1 (DMT1) and Common Metabolites. In Proceedings of the Metals at EB2012, San Diego, CA, USA, 21–25 April 2012.
- Harris, D.C.; Aisen, P. Facilitation of Fe(II) autoxidation by Fe(III) complexing agents. Biochim. Biophys. Acta 1973, 329, 156–158. [Google Scholar] [CrossRef]
- Gray, L.A.; Kidane, T.Z.; Nguyen, A.; Akagi, S.; Petrasek, K.; Chu, Y.-L.; Nguyen, T.; Cabrera, A.; Kantardjieff, K.; Mason, A.Z.; et al. Copper proteins and ferroxidases in human plasma in that of wild type and ceruloplasmin knockout mice. Biochem. J. 2009, 419, 237–245. [Google Scholar] [CrossRef]
- Chen, H.; Attieh, Z.K.; Syed, B.A.; Kuo, Y.M.; Stevens, V.; Fuqua, B.K.; Anderson, H.S.; Naylor, C.E.; Evans, R.W.; Gambling, L.; et al. Identification of zyklopen, a new member of the vertebrate multicopper ferroxidase family, and characterization in rodents and human cells. J. Nutr. 2010, 140, 1728–1735. [Google Scholar] [CrossRef]
- Sauble, E.N. Mechanisms of Ferritin Degradation in Lysosomes. Master’s Thesis, California State University, June 2007. [Google Scholar]
- Gruenheid, S.; Pinner, E.; Desjardins, M.; Gros, P. Natural resistance to infection with intracellular pathogens: The Nramp1 protein is recruited to the membrane of the phagosome. J. Exp. Med. 1997, 185, 717–730. [Google Scholar] [CrossRef]
- Forbes, J.R.; Gros, P. Iron, manganese, and cobalt transport by Nramp1 (Slc11a1) and Nramp2 (Slc11a2) expressed at the plasma membrane. Blood 2003, 102, 1884–1892. [Google Scholar] [CrossRef]
- Soe-Lin, S.; Apte, S.S.; Andriopoulos, B., Jr.; Andrews, M.C.; Schranzhofer, M.; Kahawita, T.; Garcia-Santos, D.; Ponka, P. Nramp1 promotes efficient macrophage recycling of iron following erythrophagocytosis in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 5960–5965. [Google Scholar] [CrossRef]
- Soe-Lin, S.; Apte, S.S.; Mikhael, M.R.; Kayembe, L.K.; Nie, G.; Ponka, P. Both Nramp1 and DMT1 are necessary for efficient macrophage iron recycling. Exp. Hematol. 2010, 38, 609–617. [Google Scholar] [CrossRef]
- Sun, M.; Goldin, E.; Stahl, S.; Falardeau, J.L.; Kennedy, J.C.; Acierno, J.S., Jr.; Bove, C.; Kaneski, C.R.; Nagle, J.; Bromley, M.C.; et al. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum. Mol. Genet. 2000, 9, 2471–2478. [Google Scholar] [CrossRef]
- Venkatachalam, K.; Hofmann, I.; Monteil, C. Lysosomal localization of TRPML3 depends on TRPML2 and the mucolipidosis-associated protein TRPML1. J. Biol. Chem. 2006, 281, 17517–17527. [Google Scholar] [CrossRef]
- Dong, X.P.; Cheng, X.; Mills, E.; Delling, M.; Wang, F.; Kurz, T.; Xu, H. The type IV mucolipidosis-associated protein TRPML1 is an endolysosomal iron release channel. Nature 2008, 455, 992–996. [Google Scholar] [CrossRef]
- Eichelsdoerfer, J.L.; Evans, J.A.; Slaugenhaupt, S.A.; Cuajungco, M.P. Zinc dyshomeostasis is linked with the loss of mucolipidosis IV-associated TRPML1 ion channel. J. Biol. Chem. 2010, 285, 34304–34308. [Google Scholar]
- Wang, C.Y.; Jenkitkasemwong, S.; Duarte, S.; Sparkman, B.K.; Mackenzie, B.; Knutson, M.D. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J. Biol. Chem. 2012, 287, 34032–34043. [Google Scholar]
- Mikhael, M.; Sheftel, A.D.; Ponka, P. Ferritin does not donate its iron for haem synthesis in macrophages. Biochem. J. 2010, 429, 463–471. [Google Scholar] [CrossRef]
- Devireddy, L.R.; Hart, D.O.; Goetz, D.H.; Green, M.R. A mammalian siderophore synthesized by an enzyme with a bacterial homolog involved in enterobactin production. Cell 2010, 141, 1006–1017. [Google Scholar] [CrossRef]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef]
- Zhang, A.; Zhang, F.; An, P.; Guo, X.; Shen, Y.; Tao, Y.; Wu, Q.; Zhang, Y.; Yu, Y.; Ning, B.; et al. Ferroportin 1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood 2011, 118, 1912–1922. [Google Scholar] [CrossRef]
- Alvarez-Hernandez, X.; Smith, M.; Glass, J. The effect of apotransferrin on iron release from Caco2 cells, an intestinal epithelial cell line. Blood 1998, 91, 3974–3979. [Google Scholar]
- Ma, Y.; Specian, R.D.; Yeh, K.Y.; Yeh, M.; Rodriguez-Paris, J.; Glass, J. The transcytosis of divalent metal transporter 1 and apo-transferrin during iron uptake in intestinal epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G965–G974. [Google Scholar]
- Nunez, M.T.; Nunez-Millacura, C.; Beltran, M.; Tapia, V.; Alvarez-Hernandez, X. Apotransferrin and holotransferrin undergo different endocytic cycles in intestinal epithelia (Caco2) cells. J. Biol. Chem. 1997, 272, 19425–19428. [Google Scholar]
- Nemeth, E.; Ganz, T. Regulation of iron metabolism by hepcidin. Annu. Rev. Nutr. 2007, 26, 323–342. [Google Scholar] [CrossRef]
- Sow, F.B.; Florence, W.C.; Satoskar, A.R.; Schlesinger, L.S.; Zwilling, B.S.; Lafuse, W.P. Expression and localization of hepcidin in macrophages: A role in host defense against tuberculosis. J. Leukoc. Biol. 2007, 82, 934–945. [Google Scholar] [CrossRef]
- Delaby, C.; Rondeau, C.; Pouzet, C.; Willemetz, A.; Pilard, N.; Canonne-Hergaux, F. Subcellular localization of iron and heme metabolism related proteins at early stages of erythropoiesis. PLoS One 2012, 7, e42199. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Linder, M.C. Mobilization of Stored Iron in Mammals: A Review. Nutrients 2013, 5, 4022-4050. https://doi.org/10.3390/nu5104022
Linder MC. Mobilization of Stored Iron in Mammals: A Review. Nutrients. 2013; 5(10):4022-4050. https://doi.org/10.3390/nu5104022
Chicago/Turabian StyleLinder, Maria C. 2013. "Mobilization of Stored Iron in Mammals: A Review" Nutrients 5, no. 10: 4022-4050. https://doi.org/10.3390/nu5104022
APA StyleLinder, M. C. (2013). Mobilization of Stored Iron in Mammals: A Review. Nutrients, 5(10), 4022-4050. https://doi.org/10.3390/nu5104022