Selenium in Bone Health: Roles in Antioxidant Protection and Cell Proliferation

Abstract
:1. Introduction
2. Relationships of Se and Bone Metabolism
3. Mechanisms of Se in Supporting Bone Health
3.1. Se Metabolism
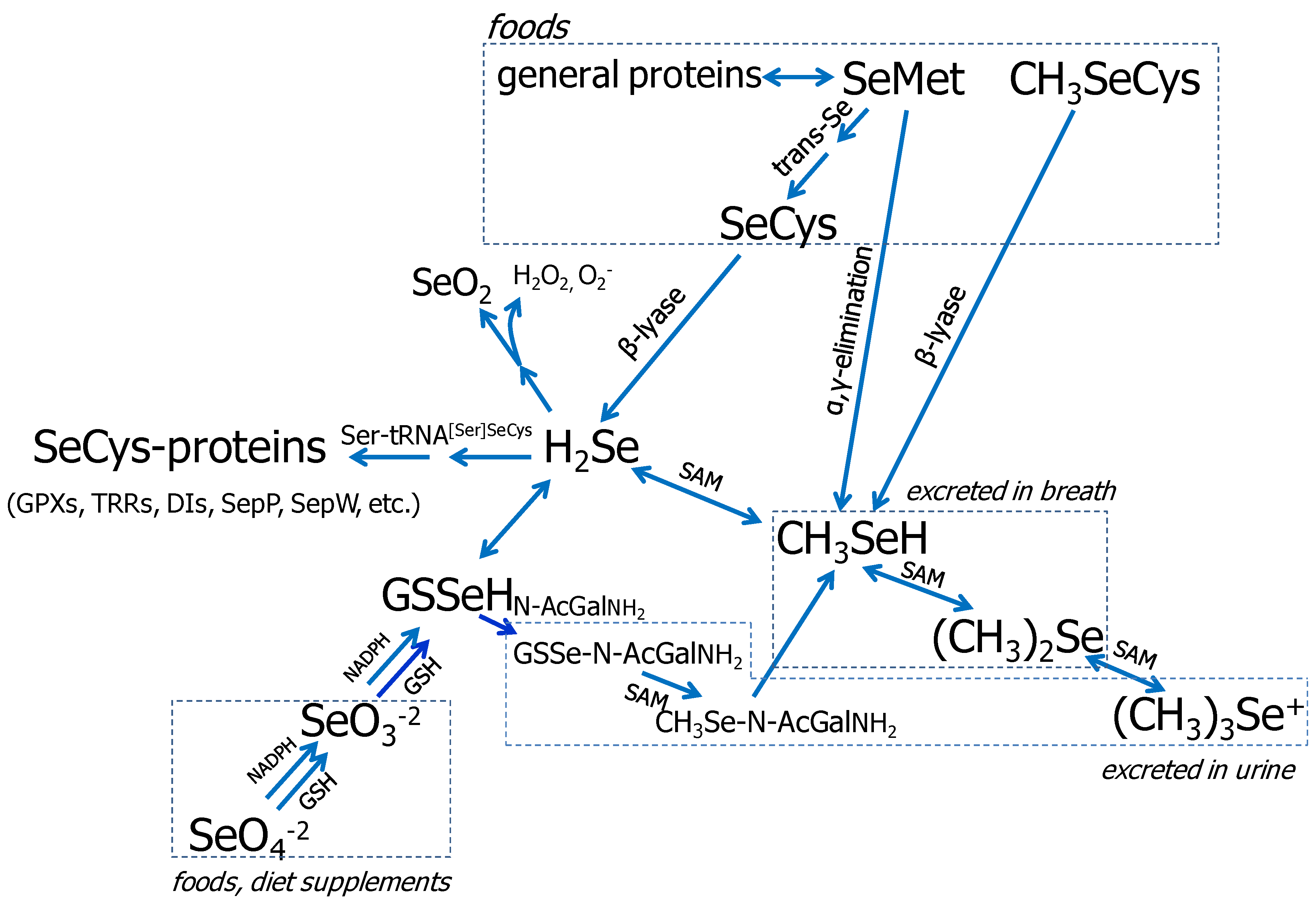
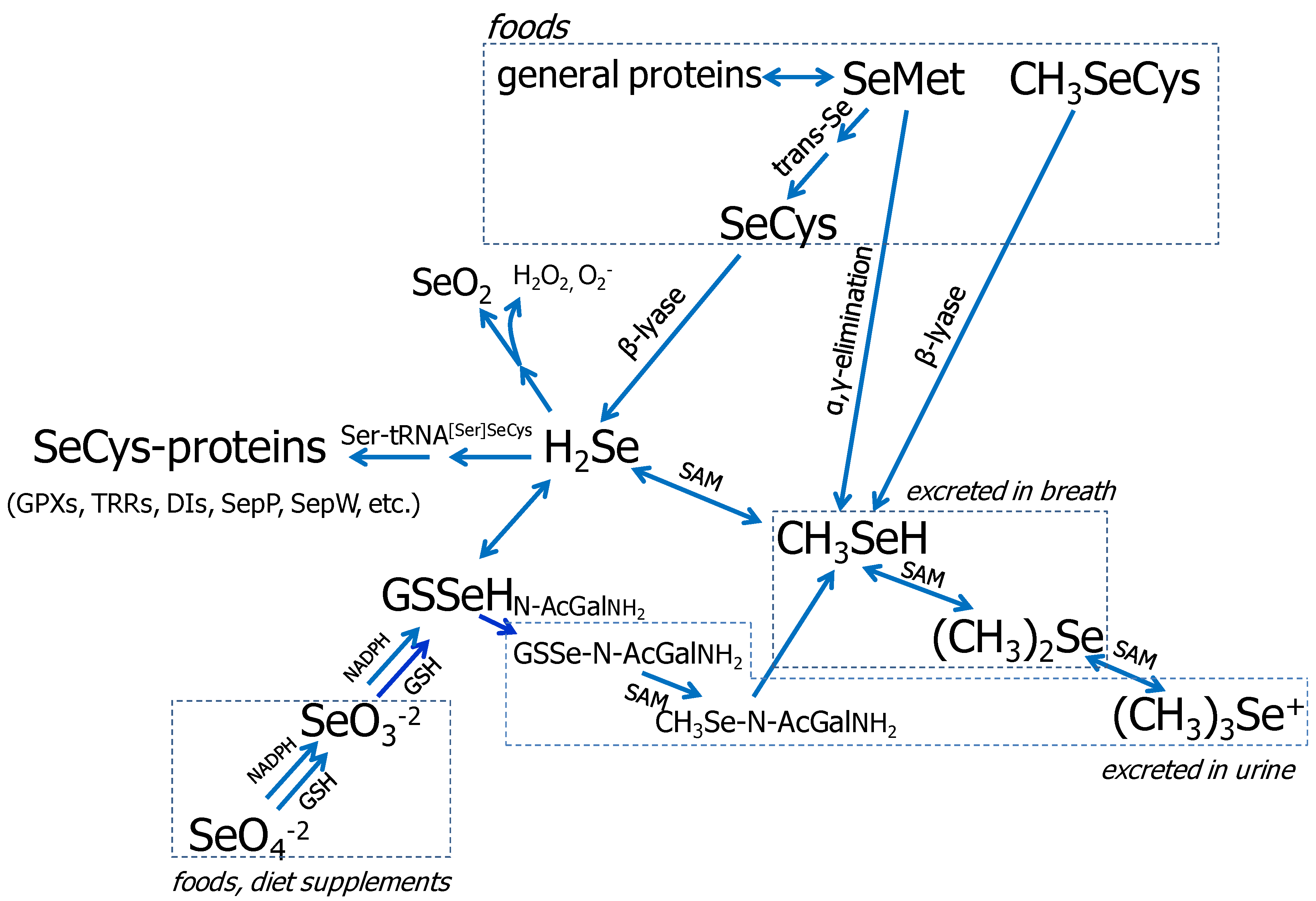{kind=link}
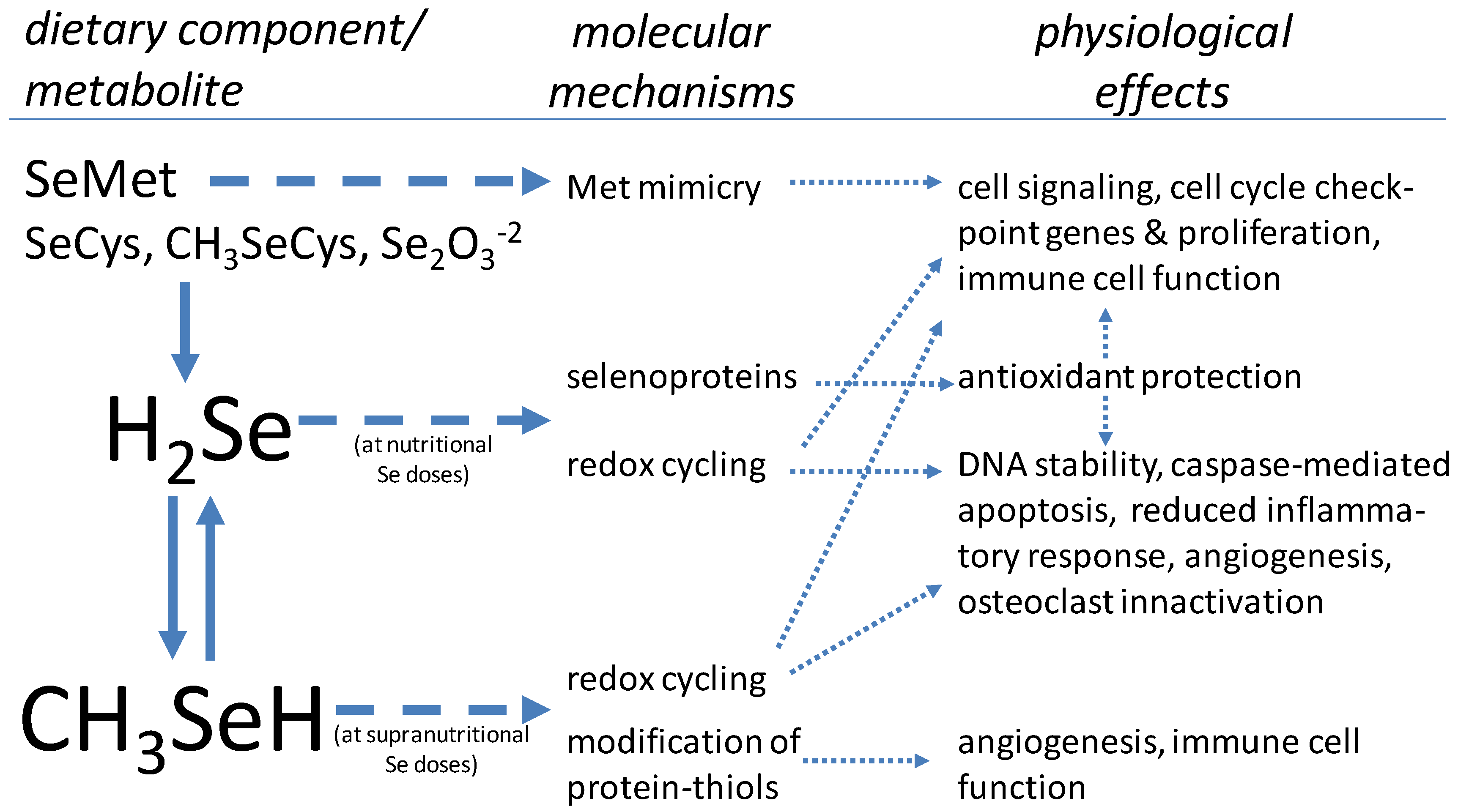
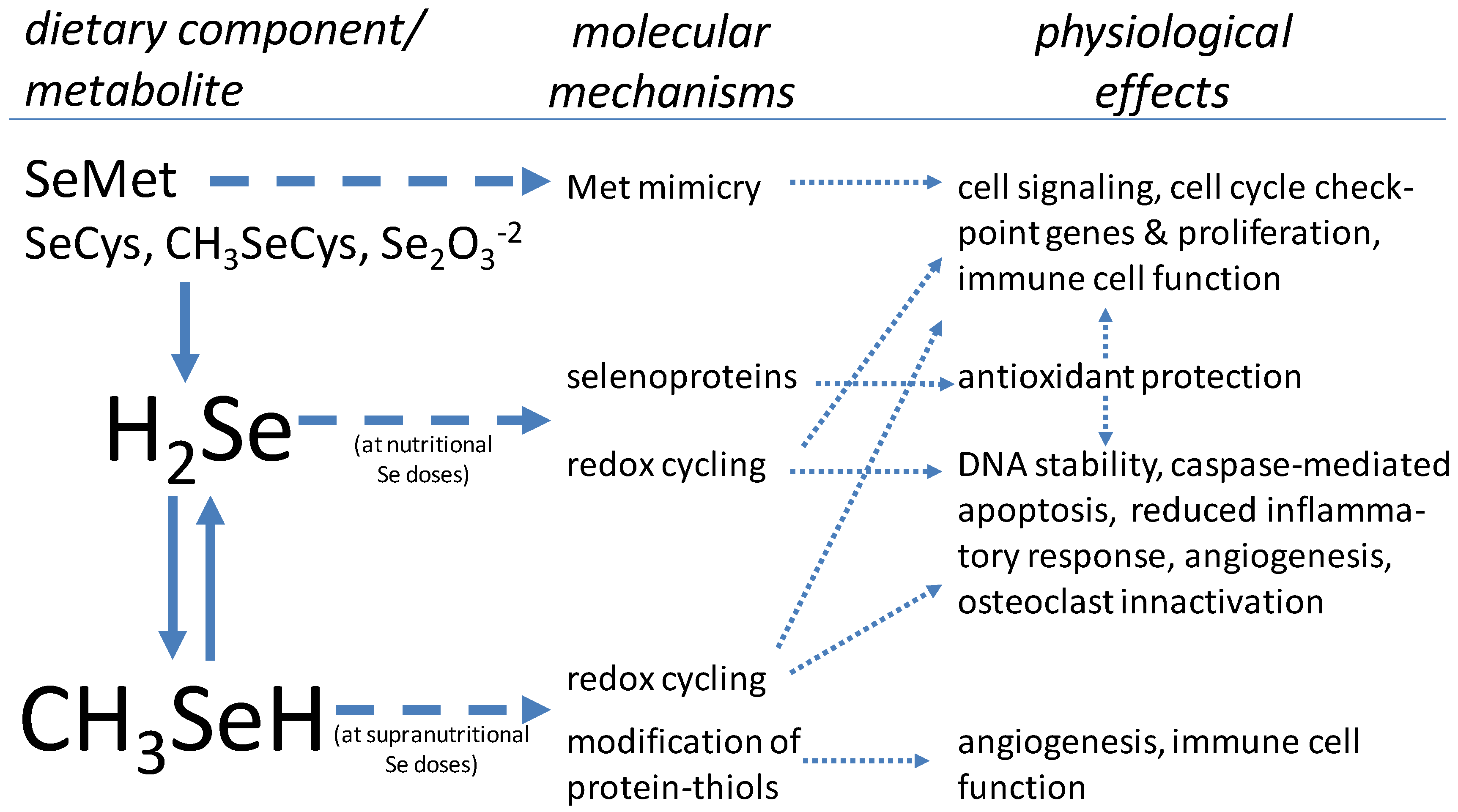{kind=link}
3.2. The Selenoproteins
3.3. Se in Protection from Oxidative Stress
3.4. Se in Cell Proliferation/Differentiation
3.5. Effects of High Doses of Se
4. Conclusions
Conflict of Interest
References
- Whanger, P.D. Selenium and its relationship to cancer: An update. Br. J. Nutr. 2004, 91, 11–28. [Google Scholar] [CrossRef]
- Rayman, M.P. Selenoproteins and human health: Insights from epidemiological data. Biochim. Biophys. Acta 2009, 1790, 1533–1540. [Google Scholar] [CrossRef] [Green Version]
- Davis, C.D.; Tsuji, P.A.; Milner, J.A. Selenoproteins and cancer prevention. Annu. Rev. Nutr. 2012, 32, 73–95. [Google Scholar] [CrossRef]
- Institute of Medicine, Food and Nutrition Board. Dietary Reference Intakes; National Academy Press: Washington, DC, USA, 2000. [Google Scholar]
- Johnson, C.C.; Fordyce, F.M.; Rayman, M.P. Symposium on ‘Geographical and geological influences on nutrition’: Factors controlling the distribution of selenium in the environment and their impact on health and nutrition. Proc. Nutr. Soc. 2010, 69, 119–132. [Google Scholar] [CrossRef]
- Moreno-Reyes, R.; Egrise, D.; Neve, J.; Pasteels, J.L.; Schoutens, A. Selenium deficiency-induced growth retardation is associated with an impaired bone metabolism and osteopenia. J. Bone Miner. Res. 2001, 16, 1556–1563. [Google Scholar]
- Cao, J.J.; Gregoire, B.R.; Zeng, H. Selenium deficiency decreases antioxidative capacity and is detrimental to bone microarchitecture in mice. J. Nutr. 2012, 142, 1526–1531. [Google Scholar] [CrossRef]
- Hoeg, A.; Gogakos, A.; Murphy, E.; Mueller, S.; Köhrle, J.; Reid, D.M.; Glüer, C.C.; Felsenberg, D.; Roux, C.; Eastell, R.; et al. Bone turnover and bone mineral density are independently related to selenium status in healthy euthyroid postmenopausal women. J. Clin. Endocrinol. Metab. 2012, 97, 4061–4070. [Google Scholar]
- Manolagas, S.C. From estrogen-centric to aging and oxidative stress: A revised perspective of the pathogenesis of osteoporosis. Endocr. Rev. 2010, 31, 266–300. [Google Scholar] [CrossRef]
- Zhang, J.; Munger, R.G.; West, N.A.; Cutler, D.R.; Wengreen, H.J.; Corcoran, C.D. Antioxidant intake and risk of osteoporotic hip fracture in Utah: An effect modified by smoking status. Am. J. Epidemiol. 2006, 163, 9–17. [Google Scholar]
- Saito, Y.; Yoshida, Y.; Akazawa, T.; Takahashi, K.; Niki, E. Cell death caused by selenium deficiency and protective effect of antioxidants. J. Biol. Chem. 2003, 278, 39428–39434. [Google Scholar]
- Köhrle, J.; Jakob, F.; Contempré, B.; Dumont, J.E. Selenium, the thyroid, and the endocrine system. Endocr. Rev. 2005, 26, 944–984. [Google Scholar] [CrossRef]
- Dreher, I.; Schütze, N.; Baur, A.; Hesse, K.; Schneider, D.; Köhrle, J.; Jakob, F. Selenoproteins are expressed in fetal human osteoblast-like cells. Biochem. Biophys. Res. Commun. 1998, 245, 101–107. [Google Scholar] [CrossRef]
- Jakob, F.; Becker, K.; Paar, E.; Ebert-Duemig, R.; Schütze, N. Expression and regulation of thioredoxin reductases and other selenoproteins in bone. Methods Enzymol. 2002, 347, 168–179. [Google Scholar] [CrossRef]
- Xu, Z.S.; Wang, X.Y.; Xiao, D.M.; Hu, L.F.; Lu, M.; Wu, Z.Y.; Bian, J.S. Hydrogen sulfide protects MC3T3-E1 osteoblastic cells against H2O2-induced oxidative damage-implications for the treatment of osteoporosis. Free Radic. Biol. Med. 2011, 50, 1314–1323. [Google Scholar] [CrossRef]
- Mody, N.; Parhami, F.; Sarafian, T.A.; Demer, L.L. Oxidative stress modulates osteoblastic differentiation of vascular and bone cells. Free Radic. Biol.Med. 2001, 31, 509–519. [Google Scholar] [CrossRef]
- Almeida, M.; Ambrogini, E.; Han, L.; Manolagas, S.C.; Jilka, R.L. Increased lipid oxidation causes oxidative stress, increased peroxisome proliferator-activated receptor-gamma expression, and diminished pro-osteogenic Wnt signaling in the skeleton. J. Biol. Chem. 2009, 284, 27438–27448. [Google Scholar]
- Ebert, R.; Ulmer, M.; Zeck, S.; Meissner-Weigl, J.; Schneider, D.; Stopper, H.; Schupp, N.; Kassem, M.; Jakob, F. Selenium supplementation restores the antioxidative capacity and prevents cell damage in bone marrow stromal cells in vitro. Stem Cells 2006, 24, 1226–1235. [Google Scholar] [CrossRef]
- Mlakar, S.J.; Osredkar, J.; Prezelj, J.; Marc, J. The antioxidant enzyme GPX1 gene polymorphisms are associated with low BMD and increased bone turnover markers. Dis. Markers 2010, 29, 71–80. [Google Scholar]
- Sun, J.; Sun, Q.; Lu, S. From selenoprotein to endochondral ossification: A novel mechanism with microRNAs potential in bone related diseases? Med. Hypotheses 2011, 77, 807–811. [Google Scholar] [CrossRef]
- Bos, S.D.; Kloppenburg, M.; Suchiman, E.; van Beelen, E.; Slagboom, P.E.; Meulenbelt, I. The role of plasma cytokine levels, CRP and Selenoprotein S gene variation in OA. Osteoarthritis Cartilage 2009, 17, 621–626. [Google Scholar] [CrossRef]
- Muthusami, S.; Ramachandran, I.; Muthusamy, B.; Vasudevan, G.; Prabhu, V.; Subramaniam, V.; Jagadeesan, A.; Narasimhan, S. Ovariectomy induces oxidative stress and impairs bone antioxidant system in adult rats. Clin. Chim. Acta 2005, 360, 81–86. [Google Scholar] [CrossRef]
- Ren, F.L.; Guo, X.; Zhang, R.J.; Wang, Sh.J.; Zuo, H.; Zhang, Z.T.; Geng, D.; Yu, Y.; Su, M. Effects of selenium and iodine deficiency on bone, cartilage growth plate and chondrocyte differentiation in two generations of rats. Osteoarthritis Cartilage 2007, 15, 1171–1177. [Google Scholar]
- Downey, C.M.; Horton, C.R.; Carlson, B.A.; Parsons, T.E.; Hatfield, D.L.; Hallgrímsson, B.; Jirik, F.R. Osteo-chondroprogenitor-specific deletion of the selenocysteine tRNA gene, Trsp, leads to chondronecrosis and abnormal skeletal development: A putative model for Kashin-Beck disease. PLoS Genet. 2009, 5, e1000616. [Google Scholar] [CrossRef]
- Allen, J.R.; Humphries, I.R.; Waters, D.L.; Roberts, D.C.; Lipson, A.H.; Howman-Giles, R.G.; Gaskin, K.J. Decreased bone mineral density in children with phenylketonuria. Am. J. Clin. Nutr. 1994, 59, 419–422. [Google Scholar]
- Zeman, J.; Bayer, M.; Stepán, J. Bone mineral density in patients with phenylketonuria. Acta Paediatr. 1999, 88, 1348–1351. [Google Scholar] [CrossRef]
- Wu, J.; Xu, G.L. Plasma selenium content, platelet glutathione peroxidase and superoxide dismutase activity of residents in Kashin-Beck disease affected area in China. J. Trace Elem. Electrolytes Health Dis. 1987, 1, 39–43. [Google Scholar]
- Chung, Y.W.; Kim, T.S.; Lee, S.Y.; Lee, S.H.; Choi, Y.; Kim, N.; Min, B.M.; Jeong, D.W.; Kim, I.Y. Selenite-induced apoptosis of osteoclasts mediated by the mitochondrial pathway. Toxicol. Lett. 2006, 160, 143–150. [Google Scholar] [CrossRef]
- Hiraoka, K.; Komiya, S.; Hamada, T.; Zenmyo, M.; Inoue, A. Osteosarcoma cell apoptosis induced by selenium. J. Orthop. Res. 2001, 19, 809–814. [Google Scholar] [CrossRef]
- Chen, Y.C.; Sosnoski, D.M.; Gandhi, U.H.; Novinger, L.J.; Prabhu, K.S.; Mastro, A.M. Selenium modifies the osteoblast inflammatory stress response to bone metastatic breast cancer. Carcinogenesis 2009, 30, 1941–1948. [Google Scholar] [CrossRef]
- Ip, C.; Birringer, M.; Block, E.; Kotrebai, M.; Tyson, J.F.; Uden, P.C.; Lisk, D.J. Chemical speciation influences comparative activity of selenium-enriched garlic and yeast in mammary cancer prevention. J. Agric. Food Chem. 2000, 48, 2062–2070. [Google Scholar] [CrossRef]
- Schrauzer, G.N. Nutritional selenium supplements: Product types, quality, and safety. J. Am. Coll. Nutr. 2001, 20, 1–4. [Google Scholar]
- Ganther, H.E. Selenium metabolism, selenoproteins and mechanisms of cancer prevention: Complexities with thioredoxin reductase. Carcinogenesis 1999, 20, 1657–1666. [Google Scholar] [CrossRef]
- Ip, C.; Ganther, H. Efficacy of trimethylselenonium versus selenite in cancer chemoprevention and its modulation by arsenite. Carcinogenesis 1988, 9, 1481–1484. [Google Scholar] [CrossRef]
- Gladyshev, V.N.; Hatfield, D.L. Selenocysteine-containing proteins in mammals. J. Biomed. Sci. 1999, 6, 151–160. [Google Scholar] [CrossRef]
- Sunde, R.A. Selenium. In Present Knowledge in Nutrition, 9th; Bowman, B.A., Russell, R.M., Eds.; ILSI Press Inc: Washington, DC, USA, 2006; pp. 480–497. [Google Scholar]
- Kobayashi, Y.; Ogra, Y.; Ishiwata, K.; Takayama, H.; Aimi, N.; Suzuki, K.T. Selenosugars are key and urinary metabolites for selenium excretion within the required to low-toxic range. Proc. Natl. Acad. Sci. USA 2002, 99, 15932–15936. [Google Scholar]
- Rayman, M.P. Selenium in cancer prevention: A review of the evidence and mechanism of action. Proc. Nutr. Soc. 2005, 64, 527–542. [Google Scholar] [CrossRef]
- Combs, G.F., Jr. Status of selenium in prostate cancer prevention. Br. J. Cancer 2004, 91, 195–199. [Google Scholar]
- Ip, C.; Ganther, H.E. Activity of methylated forms of selenium in cancer prevention. Cancer Res. 1990, 50, 1206–1211. [Google Scholar]
- Zeng, H. Selenium as an essential micronutrient: Roles in cell cycle and apoptosis. Molecules 2009, 14, 1263–1278. [Google Scholar] [CrossRef]
- Kaushal, N.; Bansal, M.P. Dietary selenium variation-induced oxidative stress modulates CDC2/cyclin B1 expression and apoptosis of germ cells in mice testis. J. Nutr. Biochem. 2007, 18, 553–564. [Google Scholar] [CrossRef]
- Udagawa, N.; Takahashi, N.; Akatsu, T.; Tanaka, H.; Sasaki, T.; Nishihara, T.; Koga, T.; Martin, T.J.; Suda, T. Origin of osteoclasts: Mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc. Natl. Acad. Sci. USA 1990, 87, 7260–7264. [Google Scholar]
- Christensen, M.J.; Nartey, E.T.; Hada, A.L.; Legg, R.L.; Barzee, B.R. High selenium reduces NF-kappaB-regulated gene expression in uninduced human prostate cancer cells. Nutr. Cancer 2007, 58, 197–204. [Google Scholar] [CrossRef]
- Duntas, L.H. Selenium and inflammation: Underlying anti-inflammatory mechanisms. Horm. Metab. Res. 2009, 41, 443–437. [Google Scholar]
- Méplan, C.; Hesketh, J. The influence of selenium and selenoprotein gene variants on colorectal cancer risk. Mutagenesis 2012, 27, 177–186. [Google Scholar]
- Hawkes, W.C.; Alkan, Z. Regulation of redox signaling by selenoproteins. Biol. Trace Elem. Res. 2010, 134, 235–251. [Google Scholar] [CrossRef]
- Steinbrenner, H.; Sies, H. Protection against reactive oxygen species by selenoproteins. Biochim. Biophys. Acta 2009, 1790, 1478–1485. [Google Scholar] [CrossRef]
- Day, R.M.; Suzuki, Y.J. Cell proliferation, reactive oxygen and cellular glutathione. Dose Response 2006, 3, 425–442. [Google Scholar]
- Stone, J.R.; Yang, S. Hydrogen peroxide: A signaling messenger. Antioxid. Redox Signal. 2006, 8, 243–270. [Google Scholar] [CrossRef]
- Vardatsikos, G.; Sahu, A.; Srivastava, A.K. The insulin-like growth factor family: Molecular mechanisms, redox regulation, and clinical implications. Antioxid. Redox Signal. 2009, 11, 1165–1190. [Google Scholar] [CrossRef]
- Purushothaman, D.; Sarin, A. Cytokine-dependent regulation of NADPH oxidase activity and the consequences for activated T cell homeostasis. J. Exp. Med. 2009, 206, 1515–1523. [Google Scholar] [CrossRef]
- Lee, J.; Giordano, S.; Zhang, J. Autophagy, mitochondria and oxidative stress: Cross-talk and redox signalling. Biochem. J. 2012, 441, 523–540. [Google Scholar]
- McKeehan, W.L.; Hamilton, W.G.; Ham, R.G. Selenium is an essential trace nutrient for growth of WI-38 diploid human fibroblasts. Proc. Natl. Acad. Sci. USA 1976, 73, 2023–2027. [Google Scholar]
- Guilbert, L.J.; Iscove, N.N. Partial replacement of serum by selenite, transferrin, albumin and lecithin in haemopoietic cell cultures. Nature 1976, 263, 594–595. [Google Scholar] [CrossRef]
- Zeng, H. Selenite and selenomethionine promote HL-60 cell cycle progression. J. Nutr. 2002, 132, 674–679. [Google Scholar]
- Hu, B.; Mitra, J.; van den, H.S.; Enders, G.H. S and G2 phase roles for Cdk2 revealed by inducible expression of a dominant-negative mutant in human cells. Mol. Cell. Biol. 2001, 21, 2755–2766. [Google Scholar]
- Gille, H.; Downward, J. Multiple ras effector pathways contribute to G(1) cell cycle progression. J. Biol. Chem. 1999, 274, 22033–22040. [Google Scholar] [CrossRef]
- Irmak, M.B.; Ince, G.; Ozturk, M.; Cetin-Atalay, R. Acquired tolerance of hepatocellular carcinoma cells to selenium deficiency: A selective survival mechanism? Cancer Res. 2003, 63, 6707–6715. [Google Scholar]
- Zeng, H.; Botnen, J.H. Selenium is critical for cancer-signaling gene expression but not cell proliferation in human colon Caco-2 cells. Biofactors 2007, 31, 155–164. [Google Scholar] [CrossRef]
- Williams, M.S.; Henkart, P.A. Role of reactive oxygen intermediates in TCR-induced death of T cell blasts and hybridomas. J. Immunol. 1996, 157, 2395–2402. [Google Scholar]
- Bhaskaram, P. Micronutrient malnutrition, infection, and immunity: An overview. Nutr. Rev. 2002, 60, S40–S45. [Google Scholar] [CrossRef]
- Arthur, J.R.; McKenzie, R.C.; Beckett, G.J. Selenium in the immune system. J. Nutr. 2003, 133, 1457–1459. [Google Scholar]
- Shrimali, R.K.; Irons, R.D.; Carlson, B.A.; Sano, Y.; Gladyshev, V.N.; Park, J.M.; Hatfield, D.L. Selenoproteins mediate T cell immunity through an antioxidant mechanism. J. Biol. Chem. 2008, 283, 20181–20185. [Google Scholar]
- Schütze, N.; Bachthaler, M.; Lechner, A.; Köhrle, J.; Jakob, F. Identification by differential display PCR of the selenoprotein thioredoxin reductase as a 1 alpha,25(OH)2-vitamin D3-responsive gene in human osteoblasts—Regulation by selenite. Biofactors 1998, 7, 299–310. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. The RANKL/RANK/OPG pathway. Curr. Osteoporos. Rep. 2007, 5, 98–104. [Google Scholar] [CrossRef]
- Teitelbaum, S.L.; Ross, F.P. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 2003, 4, 638–649. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, Y.; Ries, W.; Key, L. Expression of Nox4 in osteoclasts. J. Cell Biochem. 2004, 92, 238–248. [Google Scholar] [CrossRef]
- Steinbeck, M.J.; Kim, J.K.; Trudeau, M.J.; Hauschka, P.V.; Karnovsky, M.J. Involvement of hydrogen peroxide in the differentiation of clonal HD-11EM cells into osteoclast-like cells. J. Cell Physiol. 1998, 176, 574–587. [Google Scholar]
- Moon, H.J.; Ko, W.K.; Han, S.W.; Kim, D.S.; Hwang, Y.S.; Park, H.K.; Kwon, I.K. Antioxidants, like coenzyme Q10, selenite, and curcumin, inhibited osteoclast differentiation by suppressing reactive oxygen species generation. Biochem. Biophys. Res. Commun. 2012, 418, 247–253. [Google Scholar] [CrossRef]
- Lean, J.M.; Jagger, C.J.; Kirstein, B.; Fuller, K.; Chambers, T.J. Hydrogen peroxide is essential for estrogen-deficiency bone loss and osteoclast formation. Endocrinology 2005, 146, 728–735. [Google Scholar]
- Sheweita, S.A.; Khoshhal, K.I. Calcium metabolism and oxidative stress in bone fractures: Role of antioxidants. Curr. Drug Metab. 2007, 8, 519–525. [Google Scholar] [CrossRef]
- Bai, X.C.; Lu, D.; Bai, J.; Zheng, H.; Ke, Z.Y.; Li, X.M.; Luo, S.Q. Oxidative stress inhibits osteoblastic differentiation of bone cells by ERK and NF-kappaB. Biochem. Biophys. Res. Commun. 2004, 314, 197–207. [Google Scholar]
- Liu, H.; Bian, W.; Liu, S.; Huang, K. Selenium protects bone marrow stromal cells against hydrogen peroxide-induced inhibition of osteoblastic differentiation by suppressing oxidative stress and ERK signaling pathway. Biol. Trace Elem. Res. 2012, 150, 441–450. [Google Scholar] [CrossRef]
- Zeng, H.; Briske-Anderson, M.; Wu, M.; Moyer, M.P. Methylselenol, a selenium metabolite, plays common and different role in cancerous colon HCT116 cell and noncancerous NCM460 colon cell proliferation. Nutr. Cancer 2012, 64, 128–135. [Google Scholar] [CrossRef]
- Wang, Z.; Jiang, C.; Lu, J. Induction of caspase-mediated apoptosis and cell-cycle G1 arrest by selenium metabolite methylselenol. Mol. Carcinog. 2002, 34, 113–120. [Google Scholar] [CrossRef]
- Zeng, H.; Briske-Anderson, M.; Idso, J.P.; Hunt, C.D. The selenium metabolite methylselenol inhibits the migration and invasion potential of HT1080 tumor cells. J. Nutr. 2006, 136, 1528–1532. [Google Scholar]
- Sinha, R.; Said, T.K.; Medina, D. Organic and inorganic selenium compounds inhibit mouse mammary cell growth in vitro by different cellular pathways. Cancer Lett. 1996, 107, 277–284. [Google Scholar] [CrossRef]
- Kaeck, M.; Lu, J.; Strange, R.; Ip, C.; Ganther, H.E.; Thompson, H.J. Differential induction of growth arrest inducible genes by selenium compounds. Biochem. Pharmacol. 1997, 53, 921–926. [Google Scholar]
- Zeng, H.; Davis, C.D. Down-regulation of proliferating cell nuclear antigen gene expression occurs during cell cycle arrest induced by human fecal water in colonic HT-29 cells. J. Nutr. 2003, 133, 2682–2687. [Google Scholar]
- Jiang, C.; Wang, Z.; Ganther, H.; Lu, J. Distinct effects of methylseleninic acid versus selenite on apoptosis, cell cycle, and protein kinase pathways in DU145 human prostate cancer cells. Mol. Cancer Ther. 2002, 1, 1059–1066. [Google Scholar]
- Kim, A.; Oh, J.H.; Park, J.M.; Chung, A.S. Methylselenol generated from selenomethionine by methioninase downregulates integrin expression and induces caspase-mediated apoptosis of B16F10 melanoma cells. J. Cell Physiol. 2007, 212, 386–400. [Google Scholar] [CrossRef]
- Miki, K.; Xu, M.; Gupta, A.; Ba, Y.; Tan, Y.; Al-Refaie, W.; Bouvet, M.; Makuuchi, M.; Moossa, A.R.; Hoffman, R.M. Methioninase cancer gene therapy with selenomethionine as suicide prodrug substrate. Cancer Res. 2001, 61, 6805–6810. [Google Scholar]
- Zeng, H.; Wu, M.; Botnen, J.H. Methylselenol, a selenium metabolite, induces cell cycle arrest in G1 phase and apoptosis via the extracellular-regulated kinase 1/2 pathway and other cancer signaling genes. J. Nutr. 2009, 139, 1613–1618. [Google Scholar] [CrossRef]
- Kiremidjian-Schumacher, L.; Roy, M. Effect of selenium on the immunocompetence of patients with head and neck cancer and on adoptive immunotherapy of early and established lesions. Biofactors 2001, 14, 161–168. [Google Scholar] [CrossRef]
- Hoffmann, F.W.; Hashimoto, A.C.; Shafer, L.A.; Dow, S.; Berry, M.J.; Hoffmann, P.R. Dietary selenium modulates activation and differentiation of CD4+ T cells in mice through a mechanism involving cellular free thiols. J. Nutr. 2010, 140, 1155–1161. [Google Scholar] [CrossRef]
- Zachara, B.A.; Pawluk, H.; Bloch-Boguslawska, E.; Sliwka, K.M.; Korenkiewicz, J.; Skok, Z.; Ryć, K. Tissue level, distribution, and total body selenium content in healthy and diseased humans in Poland. Arch. Environ. Health 2001, 56, 461–466. [Google Scholar] [CrossRef]
- Raisbeck, M.F. Selenosis. Vet. Clin. North Am. Food Anim. Pract. 2000, 16, 465–480. [Google Scholar]
- Pedrera-Zamorano, J.D.; Calderon-García, J.F.; Roncero-Martin, R.; Mañas-Nuñez, P.; Moran, J.M.; Lavado-Garcia, J.M. The protective effect of calcium on bone mass in postmenopausal women with high selenium intake. J. Nutr. Health Aging 2012, 16, 743–748. [Google Scholar] [CrossRef]
- Yazar, M.; Sarban, S.; Kocyigit, A.; Isikan, U.E. Synovial fluid and plasma selenium, copper, zinc, and iron concentrations in patients with rheumatoid arthritis and osteoarthritis. Biol. Trace Elem. Res. 2005, 106, 123–132. [Google Scholar] [CrossRef]
- Canter, P.H.; Wider, B.; Ernst, E. The antioxidant vitamins A, C, E and selenium in the treatment of arthritis: A systematic review of randomized clinical trials. Rheumatology 2007, 46, 1223–1233. [Google Scholar]
- Turan, B.; Can, B.; Delilbasi, E. Selenium combined with vitamin E and vitamin C restores structural alterations of bones in heparin-induced osteoporosis. Clin. Rheumatol. 2003, 22, 432–436. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zeng, H.; Cao, J.J.; Combs, G.F., Jr. Selenium in Bone Health: Roles in Antioxidant Protection and Cell Proliferation. Nutrients 2013, 5, 97-110. https://doi.org/10.3390/nu5010097
Zeng H, Cao JJ, Combs GF Jr. Selenium in Bone Health: Roles in Antioxidant Protection and Cell Proliferation. Nutrients. 2013; 5(1):97-110. https://doi.org/10.3390/nu5010097
Chicago/Turabian StyleZeng, Huawei, Jay J. Cao, and Gerald F. Combs, Jr. 2013. "Selenium in Bone Health: Roles in Antioxidant Protection and Cell Proliferation" Nutrients 5, no. 1: 97-110. https://doi.org/10.3390/nu5010097