Retina, Retinol, Retinal and the Natural History of Vitamin A as a Light Sensor

Abstract
:1. Sunlight and Vitamin A
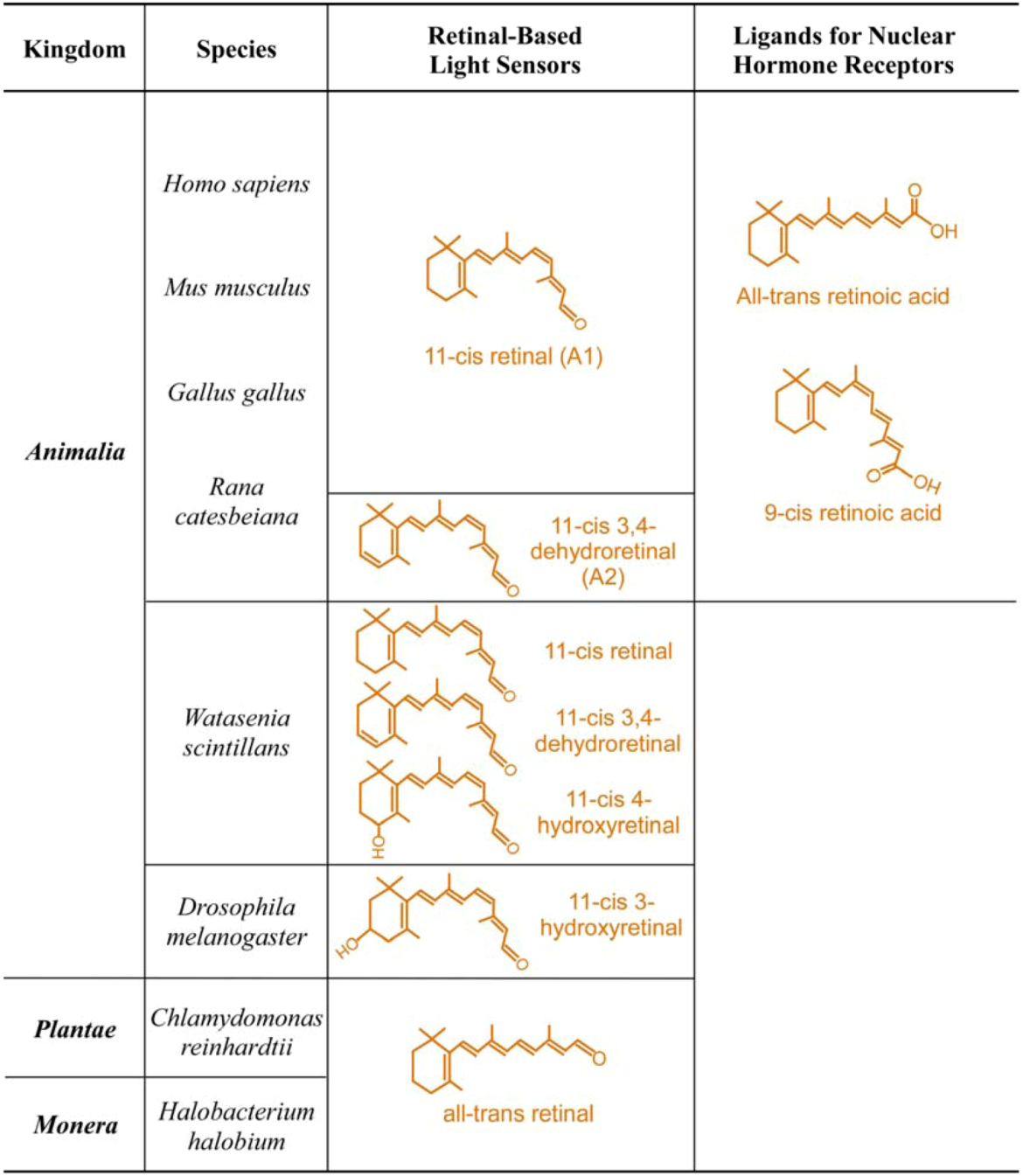
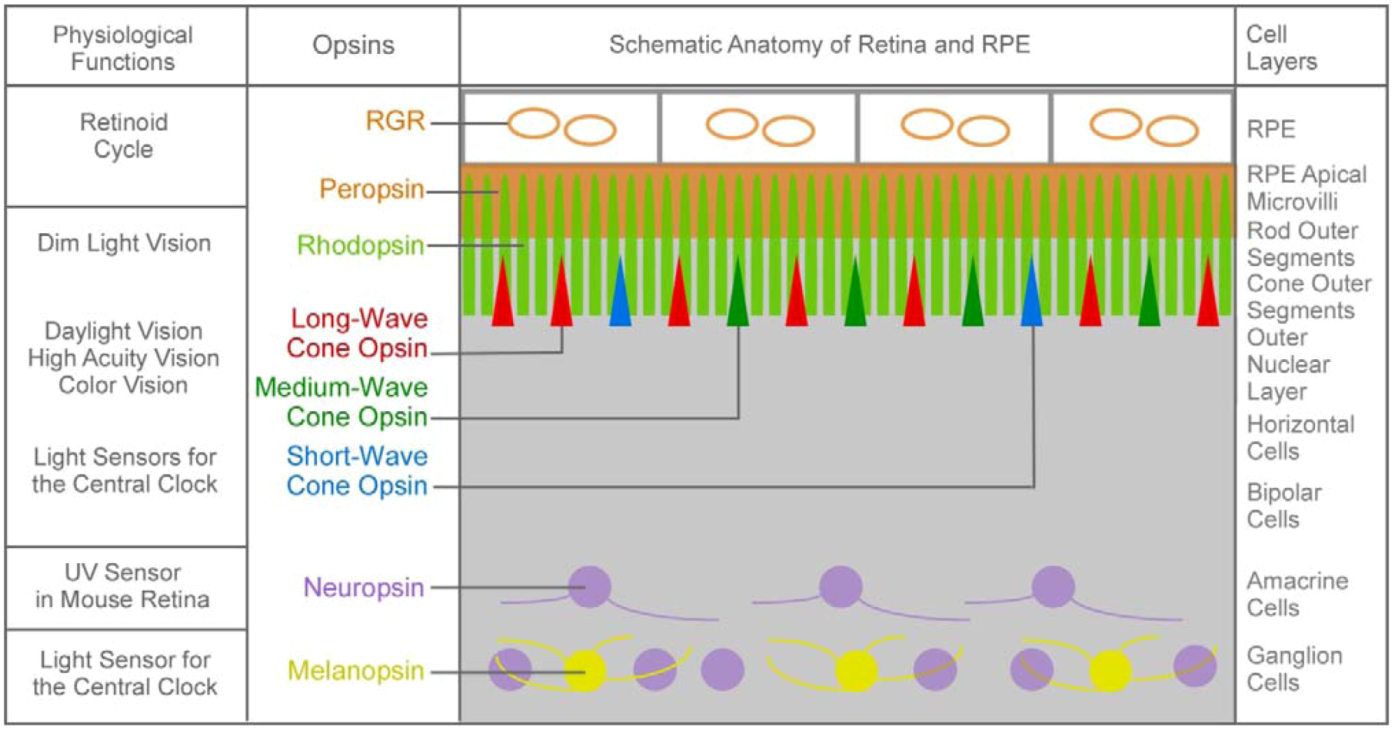
1.1. The First Major Switch in the Evolution of Vitamin A-Based Light Sensors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
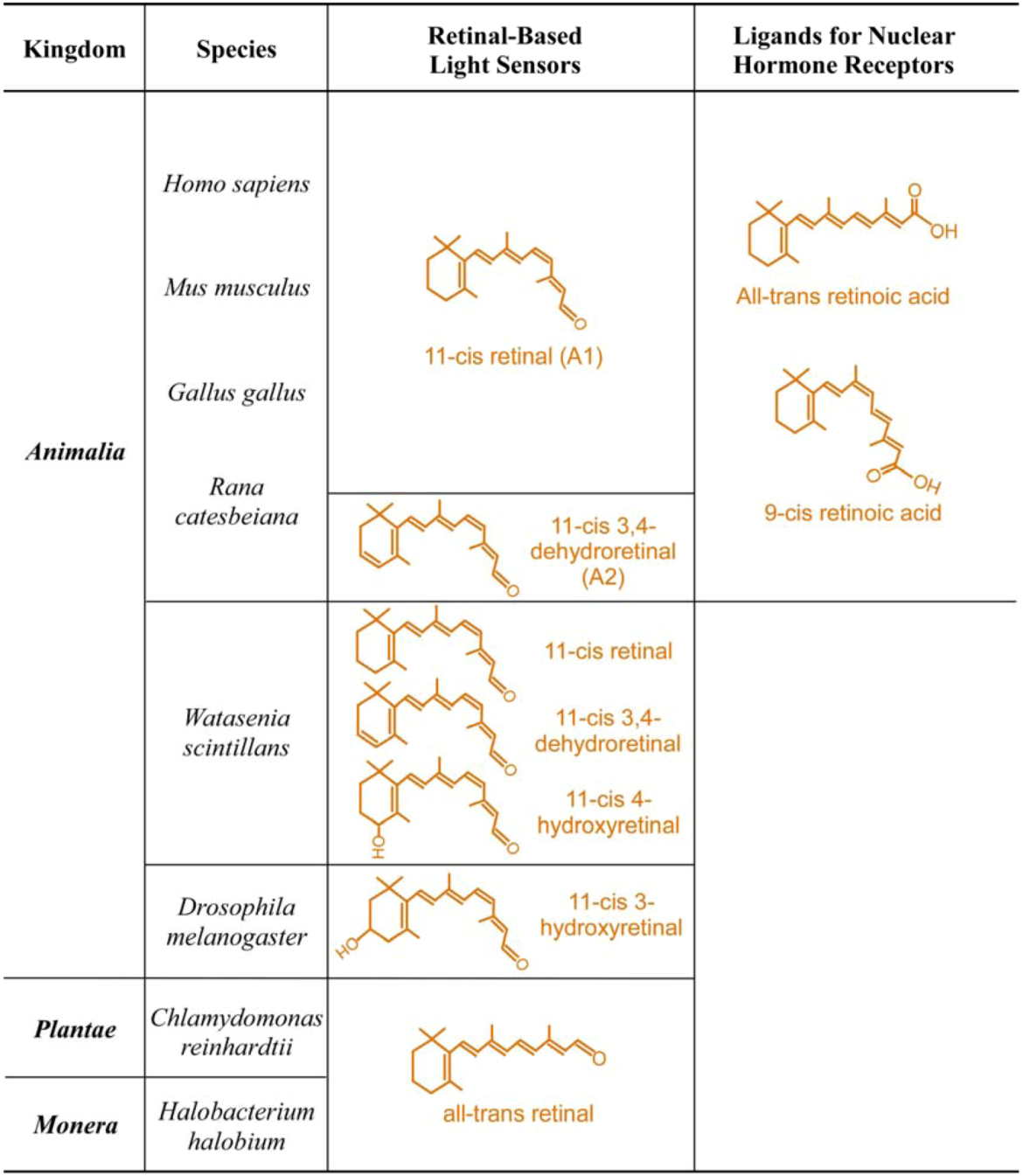
| Kingdom | Species | Photoreceptor Cell or Structure | Physiological Functions | Photoreceptor Proteins | Retinal Chomophore |
|---|---|---|---|---|---|
| Animalia | Homo sapiens Human | Cones | High luminescence vision and color vision + # | Long-wave cone pigment | 11-cis retinal |
| Medium-wave cone pigment | |||||
| Short-wave cone pigment | |||||
| Rod | Low luminescence vision + # | Rhodopsin | |||
| Light-sensitive ganglion cell | Light-sensing for the circadian clock and papillary reflex (#) | Melanopsin | |||
| Mus musculus Mouse | Cones | High luminescence vision and color vision + # | Medium-wave cone pigment UV cone pigment | 11-cis retinal | |
| Rod | Low luminescence vision + # | Rhodopsin | |||
| Light-sensitive ganglion cell | Light-sensing for the circadian clock and papillary reflex (#) | Melanopsin | |||
| Gallus gallus Chicken | Cones | High luminescence vision and color vision + # | Long-wave cone pigment Medium-wave cone pigment Short-wave cone pigment UV cone pigment | 11-cis retinal | |
| Rod | Low luminescence vision + # | Rhodopsin | |||
| Light-sensitive ganglion cell | Light-sensing for the circadian clock and papillary reflex (#) | Melanopsin | |||
| pinealocyte | Regulation of pineal circadian cycle | Pinopsin | |||
| Rana catesbeiana Frog | Rod and cones of adult frog | Vision on land and in water | Visual pigments | 11-cis retinal | |
| Photosensitive melanophore | Light-dependent melanosome migration | Melanopsin | |||
| Rod and cones of tadpole | Vision in water | Visual pigments | 11-cis-3,4-dehydroretinal | ||
| Watasenia scintillans Squid | Retinal photoreceptors | Vision in water | Visual pigments | 11-cis-3,4-dehydroretinal | |
| 11-cis-4-hydroxyretinal | |||||
| 11-cis retinal | |||||
| Drosophila melanogaster Fly | R1 to R7 photoreceptors | Vision | Visual pigments | 11-cis-3-hydroxyretinal | |
| Plantae | Chlamydomonas reinhardtii Green algea | Eye spot | Phototactic response | Chlamyopsin | All-trans retinal |
| Photophobic response | |||||
| Monera | Halobacterium halobium Bacteria | Halobacterium halobium | Light-driven chloride pump | Halorhodopsin | All-trans retinal |
| Light-driven proton pump | Bacteriorhodopsin | ||||
| Phototactic response | Sensory rhodopsin I | ||||
| Photophobic response | Sensory rhodopsin II |
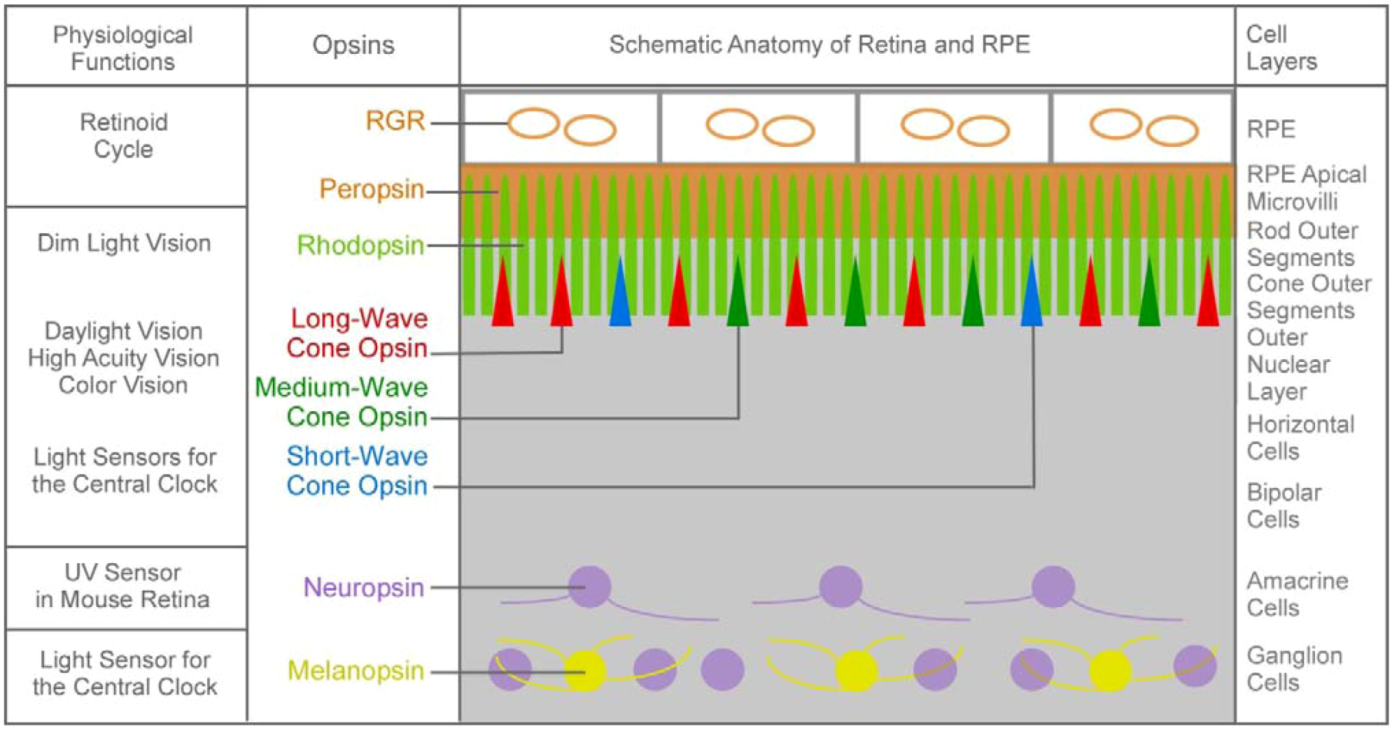
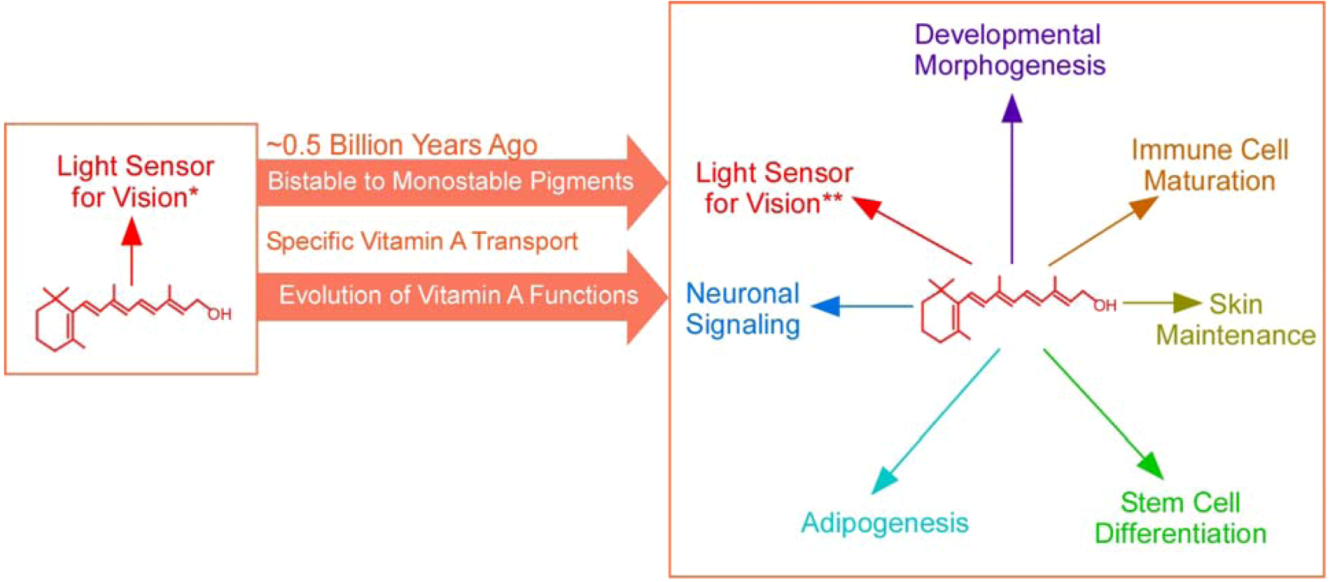
1.2. The Second Major Switch in the Evolution of Vitamin A-Based Light Sensors
| Kingdom | Monera | Plantae | Animalia | |||||
|---|---|---|---|---|---|---|---|---|
| Species | Halobacterium halobium | Chlamydomonas reinhardtii | Drosophila melanogaster | Watasenia scintillans | Rana catesbeiana | Gallus gallus | Mus musculus | Homo sapiens |
| Light sensing | Vitamin A-based light sensors for vision or the equivalent of vision | |||||||
| Opsins | Light-driven pumps or light-gated ion channels | All visual pigments in the animal kingdom are G-protein coupled receptors | ||||||
| Chromophore | All-trans retinal | 11-cis retinal | ||||||
| Light-induced isomerization | All-trans to 13-cis | 11-cis to all-trans | ||||||
| Photolability | Bistable pigments | Monostable pigments for vision | ||||||
| Regeneration after photobleaching | Light-dependent | Enzymatic | ||||||
| Vitamin A functions | Vitamin A’s only function is light absorption | Vitamin A has diverse biological functions (e.g., regulating cell growth and differentiation in development and in adult) | ||||||
| Toxicity of free retinoid | Relatively low | High | ||||||
| Vitamin A transport | No known mechanism dedicated to long-range vitamin A transport | The emergence of the RBP/STRA6 system for sustained, specific, efficient and controlled delivery | ||||||
| Advantages | Bistable pigment | Monostable pigment |
|---|---|---|
| Disadvantages | ||
| Chromophore Release | Chromophore is not released after photoisomerization | Chromophore is released after every photoisomerization event |
| Regeneration Mechanism’s Complexity | The pigment can regenerate itself using light | Depends on multiple enzymatic steps and two cell types to regenerate every released chromophore molecule |
| Consumption of Cellular Energy | Does not depend on cellular energy to regenerate after bleaching and is much more energy efficient | Depends on the cellular energy of two cell types to regenerate every released chromophore molecule |
| The need of New Vitamin A-Based Chromophore | Vitamin A-based chromophore is only needed during the initial production of the bistable pigment | Constant recycling of retinoid between two cell types during daytime leads to inevitable loss of the chromophore and demands new supply |
| Sensitivity to Vitamin A Deficiency | Relatively low | High (the eye is the human organ most sensitive to vitamin A deficiency) |
| Long-Term Toxicity | No toxic retinal is released after light bleaching of the pigment | Toxic retinal is released after every photoisomerization event; free retinal can lead to toxic A2E formation |
| Frequency of the (Enzymatic) Visual Cycle | Infrequent (A visual cycle is used to recycle chromophore released from degraded opsins) | Highly frequent (A visual cycle is used after every photoisomerization event to regenerate bleached pigment) |
| “Wasteful” Regeneration | Little or no wasteful regeneration that consumes cellular energy | Constant regeneration of bleached rhodospin in bright daylight when the rod is completely saturated is highly wasteful |
| Regeneration in the Dark | Depends on light to regenerate; can regenerate in the dark only during the initial formation of the pigment | Due to its ability to be regenerated in complete darkness, it is more sensitive for nighttime vision |
| Consequence of Photon Absorption | Activation or regeneration | Activation only |
| Encoding Wavelength Information of Light | Each pigment has two kinds of spectral sensitivity (for bleaching and regeneration) | Each pigment has a distinct spectral sensitivity and is perhaps more precise in encoding wavelength information for color vision |
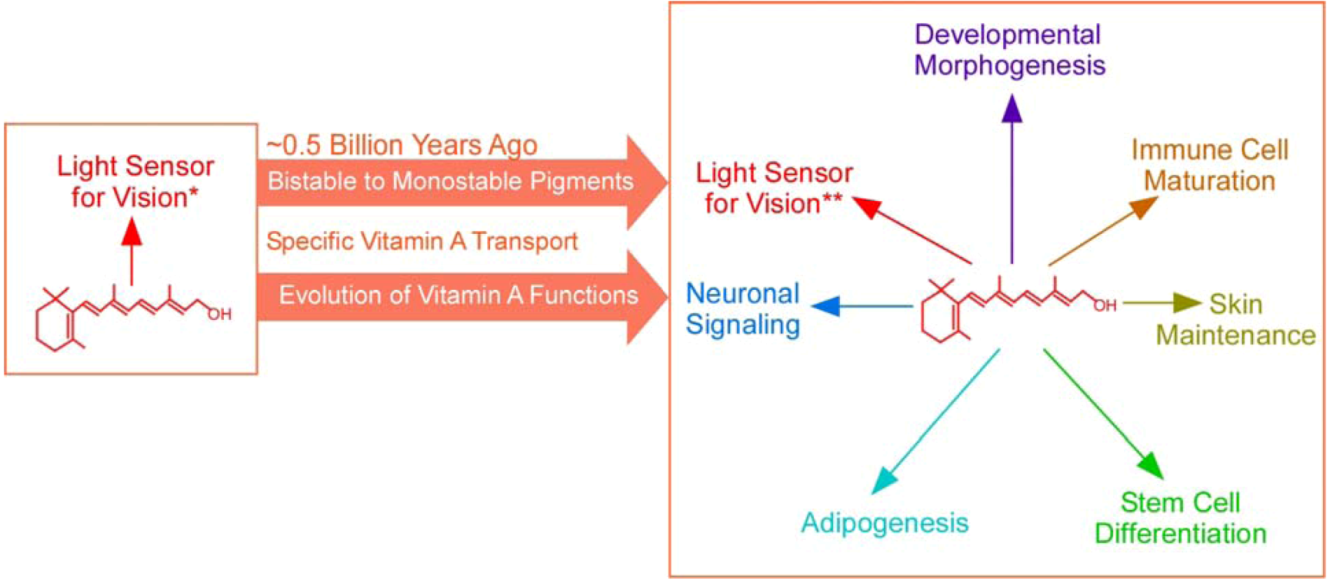
2. Broadening of the Biological Functions of Vitamin A
2.1. Expanding Biological Functions of Vitamin A
2.2. Retinoid Toxicity Associated with the Evolution of Vitamin A Functions
| Appropriate Amount |  Vitamin A Derivatives | Excessive Amount | Evolutionary Origin of Toxicity | ||
| Known Biochemical Basis of Functions | Examples of Biological Functions | Example of Toxicity | Biochemical Basis of Toxicity | ||
| One the least toxic retinoids; stored by binding to retinol binding proteins | Vitamin A storage and transport | Retinol (Vitamin A alcohol) | Pathological symptoms associated with hypervitaminosis A | Excessive vitamin A intake overwhelms and bypasses dedicated and specific delivery pathway to cause toxicity | Expanding biological roles of vitamin A |
| One the least toxic retinoids; stored as a lipid | Vitamin A storage and transport | Retinyl Ester (Vitamin A ester) | Excessive retinyl ester in the blood is toxic | Excessive retinyl esters can be converted to biologically active retinoids to cause toxicity | Expanding biological roles of vitamin A |
| The chromophore for opsins, the photoreceptor proteins for vision and the biological clock | Light absorption for vision and for regulating the biological clock | Retinal (Vitamin A aldehyde) | Excessive accumulation of retinal in retina causes photoreceptor degeneration | Random protein modification through Schiff-base formation; mediates photo-oxidative damage | Choice of monostable pigments that constantly release free retinal in daylight |
| Activates nuclear hormone receptors; regulates protein translation | Regulating the growth and differentiation from embryogenesis to adulthood; regulating learning and memory | Retinoic Acid (Vitamin A acid) | Systemic random diffusion of retinoic acid is toxic to many adult organs; also a potent teratogen | The most toxic retinoid due to its activity in activating or suppressing gene expression | Expanding biological roles of vitamin A |
 | | A2E (Retinal Derivative) | The toxic fluorophore that accumulates in the RPE of Stargard disease patients and in aging human eyes | Photo-oxidative damage; Inhibits lysosomal enzymes and retinoid isomerase; activates the complement system | Choice of monostable pigments that constantly release free retinal in daylight |
3. The Emergence of a Specific and Stable Vitamin A Transport Mechanism that Coincided with Major Changes in Vitamin A Functions
| RBP-Bound Retinol in Blood | Retinyl Ester in Blood | |
|---|---|---|
| Tissue Origin | Primarily the liver | Primarily the small intestine |
| Source of Vitamin A | Vitamin A stored in the liver, the primary organ for vitamin A storage | Dietary vitamin A immediately after absorption by the small intestine |
| Ability to Mobilize Liver-Stored Vitamin A | Yes | No |
| Dependence on Immediate Diatary Intake | No | Yes |
| Regulation of its Concentration in the Blood | Yes | No |
| As a Source of Vitamin A During the Absence of Food | Yes | No |
| As a Source of Vitamin A in the Absence of Vitamin A in Food | Yes | No |
| Nature of the Carrier Protein(s) in the Blood | The only known natural ligand of RBP is retinol | Retinyl esters are carried by lipoproteins such as chylomicron remnants, which contain many kinds of lipids |
| Cellular Uptake Specificity | Cellular retinol uptake by the RBP receptor is not associated with cellular uptake of many other kinds of lipids | Cellular retinyl ester uptake is associated with cellular uptake of many other kinds of lipids |
| Regulatory Mechanism of Vitamin A Uptake | Unknown | Unknown |
| As a Cause of Vitamin A Toxicity in Human | No (Healthy people maintain micromolar concentrations in the blood) | Yes (An increase above 10% in retinyl esters in the blood is a sign of vitamin A overload in human) |
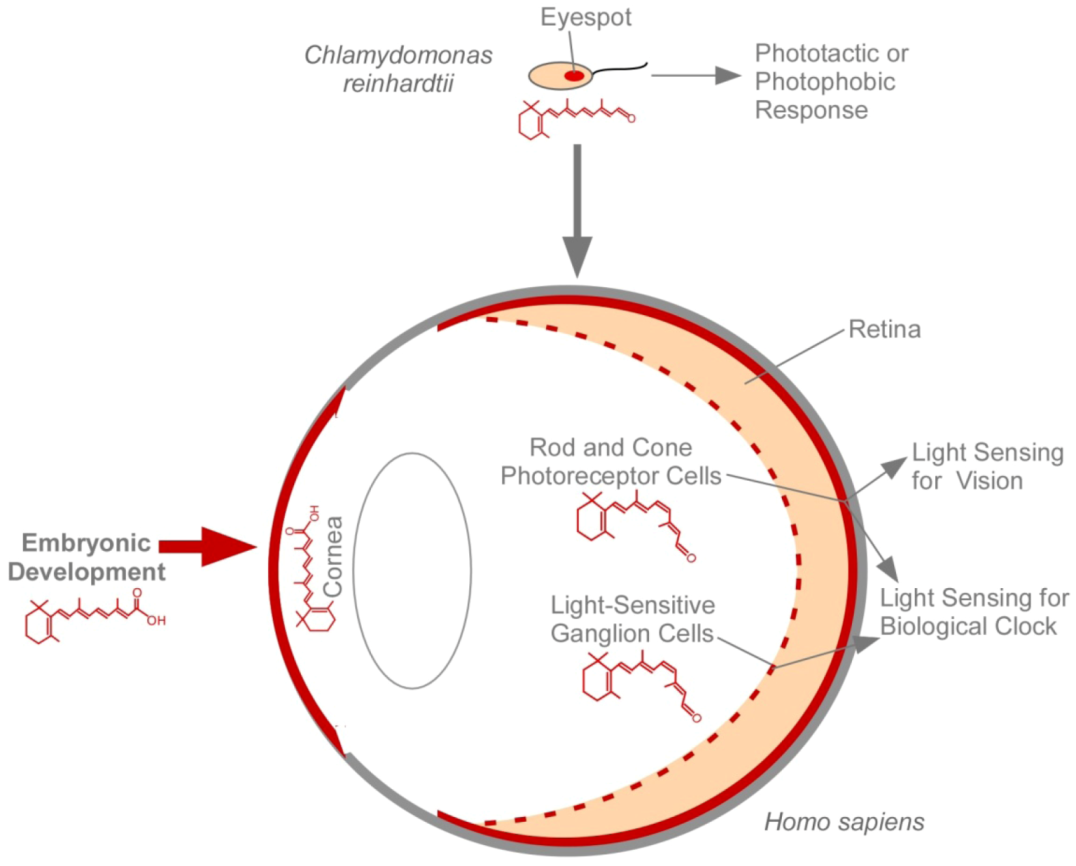
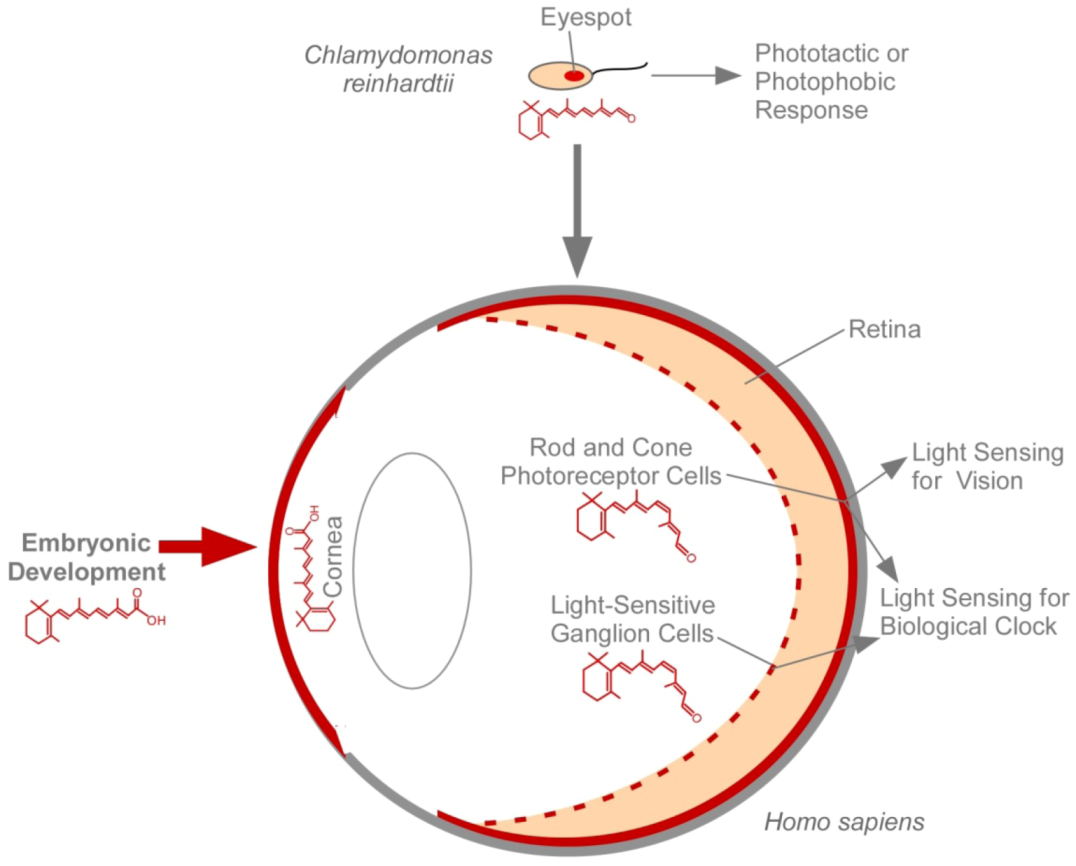
4. The Eye and Vitamin A
| The Most Sensitive Organ in Mouse | The Most Sensitive Organ in Human | The Most Severe Systemic Phenotype | |
|---|---|---|---|
| Vitamin A deficiency | The Eye | The Eye | Embryonic Lethality |
| Loss of RBP | The Eye | The Eye | Embryonic Lethality |
| Loss of STRA6 | The Eye | The Eye | Embryonic Lethality |
5. Conclusion
Acknowledgment
References
- Brine, D.T.; Iqbal, M. Diffuse and global solar spectral irradiance under cloudless skies. Sol. Energy 1983, 30, 447–453. [Google Scholar] [CrossRef]
- Lean, J. Evolution of the sun’s spectral irradiance since the maunder minimum. Geophys. Res. Lett. 2000, 27, 2425–2428. [Google Scholar] [CrossRef]
- Nathans, J. The evolution and physiology of human color vision: Insights from molecular genetic studies of visual pigments. Neuron 1999, 24, 299–312. [Google Scholar] [CrossRef]
- Spudich, J.L.; Yang, C.S.; Jung, K.H.; Spudich, E.N. Retinylidene proteins: Structures and functions from archaea to humans. Annu. Rev. Cell Dev. Biol. 2000, 16, 365–392. [Google Scholar] [CrossRef]
- Nathans, J.; Piantanida, T.P.; Eddy, R.L.; Shows, T.B.; Hogness, D.S. Molecular genetics of inherited variation in human color vision. Science 1986, 232, 203–210. [Google Scholar]
- Nathans, J.; Thomas, D.; Hogness, D.S. Molecular genetics of human color vision: The genes encoding blue, green, and red pigments. Science 1986, 232, 193–202. [Google Scholar]
- Nathans, J. Rhodopsin: Structure, function, and genetics. Biochemistry 1992, 31, 4923–4931. [Google Scholar] [CrossRef]
- Khorana, H.G. Rhodopsin, photoreceptor of the rod cell. An emerging pattern for structure and function. J. Biol. Chem. 1992, 267, 1–4. [Google Scholar]
- Hubbell, W.L.; Altenbach, C.; Hubbell, C.M.; Khorana, H.G. Rhodopsin structure, dynamics, and activation: A perspective from crystallography, site-directed spin labeling, sulfhydryl reactivity, and disulfide cross-linking. Adv. Protein Chem. 2003, 63, 243–290. [Google Scholar] [CrossRef]
- Palczewski, K. G protein-coupled receptor rhodopsin. Annu. Rev. Biochem. 2006, 75, 743–767. [Google Scholar] [CrossRef]
- Provencio, I.; Rodriguez, I.R.; Jiang, G.; Hayes, W.P.; Moreira, E.F.; Rollag, M.D. A novel human opsin in the inner retina. J. Neurosci. 2000, 20, 600–605. [Google Scholar]
- Hattar, S.; Liao, H.W.; Takao, M.; Berson, D.M.; Yau, K.W. Melanopsin-containing retinal ganglion cells: Architecture, projections, and intrinsic photosensitivity. Science 2002, 295, 1065–1070. [Google Scholar] [CrossRef]
- Panda, S.; Provencio, I.; Tu, D.C.; Pires, S.S.; Rollag, M.D.; Castrucci, A.M.; Pletcher, M.T.; Sato, T.K.; Wiltshire, T.; Andahazy, M.; et al. Melanopsin is required for non-image-forming photic responses in blind mice. Science 2003, 301, 525–527. [Google Scholar] [CrossRef]
- Dacey, D.M.; Liao, H.W.; Peterson, B.B.; Robinson, F.R.; Smith, V.C.; Pokorny, J.; Yau, K.W.; Gamlin, P.D. Melanopsin-expressing ganglion cells in primate retina signal colour and irradiance and project to the lgn. Nature 2005, 433, 749–754. [Google Scholar] [CrossRef]
- Melyan, Z.; Tarttelin, E.E.; Bellingham, J.; Lucas, R.J.; Hankins, M.W. Addition of human melanopsin renders mammalian cells photoresponsive. Nature 2005, 433, 741–745. [Google Scholar] [CrossRef]
- Do, M.T.; Kang, S.H.; Xue, T.; Zhong, H.; Liao, H.W.; Bergles, D.E.; Yau, K.W. Photon capture and signalling by melanopsin retinal ganglion cells. Nature 2009, 457, 281–287. [Google Scholar]
- Guler, A.D.; Ecker, J.L.; Lall, G.S.; Haq, S.; Altimus, C.M.; Liao, H.W.; Barnard, A.R.; Cahill, H.; Badea, T.C.; Zhao, H.; et al. Melanopsin cells are the principal conduits for rod-cone input to non-image-forming vision. Nature 2008, 453, 102–105. [Google Scholar]
- Sun, H.; Gilbert, D.J.; Copeland, N.G.; Jenkins, N.A.; Nathans, J. Peropsin, a novel visual pigment-like protein located in the apical microvilli of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 1997, 94, 9893–9898. [Google Scholar]
- Shen, D.; Jiang, M.; Hao, W.; Tao, L.; Salazar, M.; Fong, H.K. A human opsin-related gene that encodes a retinaldehyde-binding protein. Biochemistry 1994, 33, 13117–13125. [Google Scholar] [CrossRef]
- Morimura, H.; Saindelle-Ribeaudeau, F.; Berson, E.L.; Dryja, T.P. Mutations in RGR, encoding a light-sensitive opsin homologue, in patients with retinitis pigmentosa. Nat. Genet. 1999, 23, 393–394. [Google Scholar] [CrossRef]
- Wenzel, A.; Oberhauser, V.; Pugh, E.N., Jr.; Lamb, T.D.; Grimm, C.; Samardzija, M.; Fahl, E.; Seeliger, M.W.; Reme, C.E.; von Lintig, J. The retinal G protein-coupled receptor (RGR) enhances isomerohydrolase activity independent of light. J. Biol. Chem. 2005, 280, 29874–29884. [Google Scholar]
- Radu, R.A.; Hu, J.; Peng, J.; Bok, D.; Mata, N.L.; Travis, G.H. Retinal pigment epithelium-retinal G protein receptor-opsin mediates light-dependent translocation of all-trans-retinyl esters for synthesis of visual chromophore in retinal pigment epithelial cells. J. Biol. Chem. 2008, 283, 19730–19738. [Google Scholar] [CrossRef]
- Applebury, M.L.; Antoch, M.P.; Baxter, L.C.; Chun, L.L.; Falk, J.D.; Farhangfar, F.; Kage, K.; Krzystolik, M.G.; Lyass, L.A.; Robbins, J.T. The murine cone photoreceptor: A single cone type expresses both s and m opsins with retinal spatial patterning. Neuron 2000, 27, 513–523. [Google Scholar] [CrossRef]
- Tarttelin, E.E.; Bellingham, J.; Hankins, M.W.; Foster, R.G.; Lucas, R.J. Neuropsin (Opn5): A novel opsin identified in mammalian neural tissue. FEBS Lett. 2003, 554, 410–416. [Google Scholar] [CrossRef]
- Kojima, D.; Mori, S.; Torii, M.; Wada, A.; Morishita, R.; Fukada, Y. UV-sensitive photoreceptor protein OPN5 in humans and mice. PLoS One 2011, 6, e26388. [Google Scholar]
- Yokoyama, S.; Zhang, H.; Radlwimmer, F.B.; Blow, N.S. Adaptive evolution of color vision of the Comoran coelacanth (Latimeria chalumnae). Proc. Natl. Acad. Sci. USA 1999, 96, 6279–6284. [Google Scholar] [CrossRef]
- Owens, G.L.; Windsor, D.J.; Mui, J.; Taylor, J.S. A fish eye out of water: Ten visual opsins in the four-eyed fish, anableps anableps. PLoS One 2009, 4, e5970. [Google Scholar] [CrossRef]
- Merbs, S.L.; Nathans, J. Role of hydroxyl-bearing amino acids in differentially tuning the absorption spectra of the human red and green cone pigments. Photochem. Photobiol. 1993, 58, 706–710. [Google Scholar] [CrossRef]
- Sun, H.; Macke, J.P.; Nathans, J. Mechanisms of spectral tuning in the mouse green cone pigment. Proc. Natl. Acad. Sci. USA 1997, 94, 8860–8865. [Google Scholar] [CrossRef]
- Fasick, J.I.; Lee, N.; Oprian, D.D. Spectral tuning in the human blue cone pigment. Biochemistry 1999, 38, 11593–11596. [Google Scholar] [CrossRef]
- Kochendoerfer, G.G.; Lin, S.W.; Sakmar, T.P.; Mathies, R.A. How color visual pigments are tuned. Trends Biochem. Sci. 1999, 24, 300–305. [Google Scholar] [CrossRef]
- Lin, S.W.; Sakmar, T.P. Colour tuning mechanisms of visual pigments. Novartis Found. Symp. 1999, 224, 124–135, discussion 135–141, 181–190. [Google Scholar]
- Fasick, J.I.; Applebury, M.L.; Oprian, D.D. Spectral tuning in the mammalian short-wavelength sensitive cone pigments. Biochemistry 2002, 41, 6860–6865. [Google Scholar] [CrossRef]
- Kusnetzow, A.K.; Dukkipati, A.; Babu, K.R.; Ramos, L.; Knox, B.E.; Birge, R.R. Vertebrate ultraviolet visual pigments: Protonation of the retinylidene schiff base and a counterion switch during photoactivation. Proc. Natl. Acad. Sci. USA 2004, 101, 941–946. [Google Scholar]
- Yokoyama, S. Evolution of dim-light and color vision pigments. Annu. Rev. Genomics Hum. Genet. 2008, 9, 259–282. [Google Scholar] [CrossRef]
- Tsin, A.T.; Beatty, D.D.; Bridges, C.D.; Alvarez, R. Selective utilization of vitamins A1 and A2 by goldfish photoreceptors. Investig. Ophthalmol. Vis. Sci. 1983, 24, 1324–1327. [Google Scholar]
- Ma, J.X.; Kono, M.; Xu, L.; Das, J.; Ryan, J.C.; Hazard, E.S., III; Oprian, D.D.; Crouch, R.K. Salamander UV cone pigment: Sequence, expression, and spectral properties. Vis. Neurosci. 2001, 18, 393–399. [Google Scholar]
- Temple, S.E.; Plate, E.M.; Ramsden, S.; Haimberger, T.J.; Roth, W.M.; Hawryshyn, C.W. Seasonal cycle in vitamin A1/A2-based visual pigment composition during the life history of coho salmon (Oncorhynchus kisutch). J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 2006, 192, 301–313. [Google Scholar] [CrossRef]
- Ala-Laurila, P.; Donner, K.; Crouch, R.K.; Cornwall, M.C. Chromophore switch from 11-cis-dehydroretinal (A2) to 11-cis-retinal (aA1) decreases dark noise in salamander red rods. J. Physiol. 2007, 585, 57–74. [Google Scholar] [CrossRef]
- Saarinen, P.; Pahlberg, J.; Herczeg, G.; Viljanen, M.; Karjalainen, M.; Shikano, T.; Merila, J.; Donner, K. Spectral tuning by selective chromophore uptake in rods and cones of eight populations of nine-spined stickleback (Pungitius pungitius). J. Exp. Biol. 2012, 215, 2760–2773. [Google Scholar] [CrossRef]
- Provencio, I.; Loew, E.R.; Foster, R.G. Vitamin A2-based visual pigments in fully terrestrial vertebrates. Vis. Res. 1992, 32, 2201–2208. [Google Scholar] [CrossRef]
- Dowling, J.E. Chemistry of visual adaptation in the rat. Nature 1960, 188, 114–118. [Google Scholar] [CrossRef]
- Crouch, R.K.; Chader, G.J.; Wiggert, B.; Pepperberg, D.R. Retinoids and the visual process. Photochem. Photobiol. 1996, 64, 613–621. [Google Scholar] [CrossRef]
- Lamb, T.D.; Pugh, E.N., Jr. Dark adaptation and the retinoid cycle of vision. Prog. Retin. Eye Res. 2004, 23, 307–380. [Google Scholar] [CrossRef]
- Travis, G.H.; Golczak, M.; Moise, A.R.; Palczewski, K. Diseases caused by defects in the visual cycle: Retinoids as potential therapeutic agents. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 469–512. [Google Scholar] [CrossRef]
- Von Lintig, J.; Kiser, P.D.; Golczak, M.; Palczewski, K. The biochemical and structural basis for trans-to-cis isomerization of retinoids in the chemistry of vision. Trends Biochem. Sci. 2010, 35, 400–410. [Google Scholar] [CrossRef]
- Mata, N.L.; Radu, R.A.; Clemmons, R.C.; Travis, G.H. Isomerization and oxidation of vitamin A in cone-dominant retinas: A novel pathway for visual-pigment regeneration in daylight. Neuron 2002, 36, 69–80. [Google Scholar] [CrossRef]
- Fleisch, V.C.; Schonthaler, H.B.; von Lintig, J.; Neuhauss, S.C. Subfunctionalization of a retinoid-binding protein provides evidence for two parallel visual cycles in the cone-dominant zebrafish retina. J. Neurosci. 2008, 28, 8208–8216. [Google Scholar] [CrossRef]
- Wang, J.S.; Estevez, M.E.; Cornwall, M.C.; Kefalov, V.J. Intra-retinal visual cycle required for rapid and complete cone dark adaptation. Nat. Neurosci. 2009, 12, 295–302. [Google Scholar] [CrossRef]
- Travis, G.H.; Kaylor, J.; Yuan, Q. Analysis of the retinoid isomerase activities in the retinal pigment epithelium and retina. Methods Mol. Biol. 2010, 652, 329–339. [Google Scholar] [CrossRef]
- Wang, J.S.; Kefalov, V.J. The cone-specific visual cycle. Prog. Retin. Eye Res. 2011, 30, 115–128. [Google Scholar] [CrossRef]
- Kaylor, J.J.; Yuan, Q.; Cook, J.; Sarfare, S.; Makshanoff, J.; Miu, A.; Kim, A.; Kim, P.; Habib, S.; Roybal, C.N.; et al. Identification of DES1 as a vitamin A isomerase in muller glial cells of the retina. Nat. Chem. Biol. 2012. [Google Scholar] [CrossRef]
- Jastrzebska, B.; Palczewski, K.; Golczak, M. Role of bulk water in the hydrolysis of rhodopsin’s chromophore. J. Biol. Chem. 2011, 286, 18930–18937. [Google Scholar]
- Chen, M.H.; Kuemmel, C.; Birge, R.R.; Knox, B.E. Rapid release of retinal from a cone visual pigment following photoactivation. Biochemistry 2012, 51, 4117–4125. [Google Scholar] [CrossRef]
- Woodruff, M.L.; Wang, Z.; Chung, H.Y.; Redmond, T.M.; Fain, G.L.; Lem, J. Spontaneous activity of opsin apoprotein is a cause of Leber congenital amaurosis. Nat. Genet. 2003, 35, 158–164. [Google Scholar] [CrossRef]
- Kefalov, V.J.; Estevez, M.E.; Kono, M.; Goletz, P.W.; Crouch, R.K.; Cornwall, M.C.; Yau, K.W. Breaking the covalent bond—A pigment property that contributes to desensitization in cones. Neuron 2005, 46, 879–890. [Google Scholar] [CrossRef]
- Okawa, H.; Sampath, A.P.; Laughlin, S.B.; Fain, G.L. ATP consumption by mammalian rod photoreceptors in darkness and in light. Curr. Biol. 2008, 18, 1917–1921. [Google Scholar] [CrossRef]
- Emran, F.; Rihel, J.; Adolph, A.R.; Dowling, J.E. Zebrafish larvae lose vision at night. Proc. Natl. Acad. Sci. USA 2010, 107, 6034–6039. [Google Scholar] [CrossRef]
- Wang, X.; Wang, T.; Jiao, Y.; von Lintig, J.; Montell, C. Requirement for an enzymatic visual cycle in drosophila. Curr. Biol. 2010, 20, 93–102. [Google Scholar] [CrossRef]
- Molday, R.S.; Zhong, M.; Quazi, F. The role of the photoreceptor abc transporter ABCA4 in lipid transport and stargardt macular degeneration. Biochim. Biophys. Acta 2009, 1791, 573–583. [Google Scholar] [CrossRef]
- Tsybovsky, Y.; Molday, R.S.; Palczewski, K. The ATP-binding cassette transporter ABCA4: Structural and functional properties and role in retinal disease. Adv. Exp. Med. Biol. 2010, 703, 105–125. [Google Scholar] [CrossRef]
- Weng, J.; Mata, N.L.; Azarian, S.M.; Tzekov, R.T.; Birch, D.G.; Travis, G.H. Insights into the function of rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in ABCR knockout mice. Cell 1999, 98, 13–23. [Google Scholar] [CrossRef]
- Sun, H.; Molday, R.S.; Nathans, J. Retinal stimulates ATP hydrolysis by purified and reconstituted ABCR, the photoreceptor-specific ATP-binding cassette transporter responsible for Stargardt disease. J. Biol. Chem. 1999, 274, 8269–8281. [Google Scholar]
- Mata, N.L.; Weng, J.; Travis, G.H. Biosynthesis of a major lipofuscin fluorophore in mice and humans with ABCR-mediated retinal and macular degeneration. Proc. Natl. Acad. Sci. USA 2000, 97, 7154–7159. [Google Scholar]
- Sun, H.; Nathans, J. Mechanistic studies of ABCR, the ABC transporter in photoreceptor outer segments responsible for autosomal recessive Stargardt disease. J. Bioenergy Biomembr. 2001, 33, 523–530. [Google Scholar] [CrossRef]
- Zhong, M.; Molday, R.S. Binding of retinoids to ABCA4, the photoreceptor ABC transporter associated with Stargardt macular degeneration. Methods Mol. Biol. 2010, 652, 163–176. [Google Scholar] [CrossRef]
- Boyer, N.P.; Higbee, D.; Currin, M.B.; Blakeley, L.R.; Chen, C.; Ablonczy, Z.; Crouch, R.K.; Koutalos, Y. Lipofuscin and N-retinylidene-N-retinylethanolamine (A2E) accumulate in retinal pigment epithelium in absence of light exposure: Their origin is 11-cis-retinal. J. Biol. Chem. 2012, 287, 22276–22286. [Google Scholar]
- Quazi, F.; Lenevich, S.; Molday, R.S. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat. Commun. 2012, 3, 925. [Google Scholar] [CrossRef]
- Allikmets, R.; Singh, N.; Sun, H.; Shroyer, N.F.; Hutchinson, A.; Chidambaram, A.; Gerrard, B.; Baird, L.; Stauffer, D.; Peiffer, A.; et al. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat. Genet. 1997, 15, 236–246. [Google Scholar] [CrossRef]
- Cremers, F.P.; van de Pol, D.J.; van Driel, M.; den Hollander, A.I.; van Haren, F.J.; Knoers, N.V.; Tijmes, N.; Bergen, A.A.; Rohrschneider, K.; Blankenagel, A.; et al. Autosomal recessive retinitis pigmentosa and cone-rod dystrophy caused by splice site mutations in the Stargardt’s disease gene ABCR. Hum. Mol. Genet. 1998, 7, 355–362. [Google Scholar] [CrossRef]
- Martinez-Mir, A.; Paloma, E.; Allikmets, R.; Ayuso, C.; del Rio, T.; Dean, M.; Vilageliu, L.; Gonzalez-Duarte, R.; Balcells, S. Retinitis pigmentosa caused by a homozygous mutation in the stargardt disease gene ABCR. Nat. Genet. 1998, 18, 11–12. [Google Scholar] [CrossRef]
- Fu, Y.; Zhong, H.; Wang, M.H.; Luo, D.G.; Liao, H.W.; Maeda, H.; Hattar, S.; Frishman, L.J.; Yau, K.W. Intrinsically photosensitive retinal ganglion cells detect light with a vitamin A-based photopigment, melanopsin. Proc. Natl. Acad. Sci. USA 2005, 102, 10339–10344. [Google Scholar]
- Panda, S.; Nayak, S.K.; Campo, B.; Walker, J.R.; Hogenesch, J.B.; Jegla, T. Illumination of the melanopsin signaling pathway. Science 2005, 307, 600–604. [Google Scholar] [CrossRef]
- Walker, M.T.; Brown, R.L.; Cronin, T.W.; Robinson, P.R. Photochemistry of retinal chromophore in mouse melanopsin. Proc. Natl. Acad. Sci. USA 2008, 105, 8861–8865. [Google Scholar] [CrossRef]
- Sexton, T.J.; Golczak, M.; Palczewski, K.; van Gelder, R.N. Melanopsin is highly resistant to light and chemical bleaching in vivo. J. Biol. Chem. 2012, 287, 20888–20897. [Google Scholar]
- Yamashita, T.; Ohuchi, H.; Tomonari, S.; Ikeda, K.; Sakai, K.; Shichida, Y. Opn5 is a UV-sensitive bistable pigment that couples with Gi subtype of G protein. Proc. Natl. Acad. Sci. USA 2010, 107, 22084–22089. [Google Scholar]
- Bi, A.; Cui, J.; Ma, Y.P.; Olshevskaya, E.; Pu, M.; Dizhoor, A.M.; Pan, Z.H. Ectopic expression of a microbial-type rhodopsin restores visual responses in mice with photoreceptor degeneration. Neuron 2006, 50, 23–33. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, L.P.; Brauner, M.; Liewald, J.F.; Kay, K.; Watzke, N.; Wood, P.G.; Bamberg, E.; Nagel, G.; Gottschalk, A.; et al. Multimodal fast optical interrogation of neural circuitry. Nature 2007, 446, 633–639. [Google Scholar] [CrossRef]
- Oberhauser, V.; Voolstra, O.; Bangert, A.; von Lintig, J.; Vogt, K. NinaB combines carotenoid oxygenase and retinoid isomerase activity in a single polypeptide. Proc. Natl. Acad. Sci. USA 2008, 105, 19000–19005. [Google Scholar] [CrossRef]
- Ross, A.C.; Gardner, E.M. The function of vitamin A in cellular growth and differentiation, and its roles during pregnancy and lactation. Adv. Exp. Med. Biol. 1994, 352, 187–200. [Google Scholar]
- Napoli, J.L. Biochemical pathways of retinoid transport, metabolism, and signal transduction. Clin. Immunol. Immunopathol. 1996, 80, S52–S62. [Google Scholar] [CrossRef]
- Stephensen, C.B. Vitamin A, infection, and immune function. Annu. Rev. Nutr. 2001, 21, 167–192. [Google Scholar]
- Drager, U.C. Retinoic acid signaling in the functioning brain. Sci. STKE 2006, 2006, pe10. [Google Scholar] [CrossRef]
- Maden, M. Retinoic acid in the development, regeneration and maintenance of the nervous system. Nat. Rev. Neurosci. 2007, 8, 755–765. [Google Scholar] [CrossRef]
- Duester, G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef]
- Niederreither, K.; Dolle, P. Retinoic acid in development: Towards an integrated view. Nat. Rev. Genet. 2008, 9, 541–553. [Google Scholar] [CrossRef]
- Takahashi, J.; Palmer, T.D.; Gage, F.H. Retinoic acid and neurotrophins collaborate to regulate neurogenesis in adult-derived neural stem cell cultures. J. Neurobiol. 1999, 38, 65–81. [Google Scholar] [CrossRef]
- Evans, R.M. The molecular basis of signaling by vitamin A and its metabolites. Harvey Lect. 1994, 90, 105–117. [Google Scholar]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoid nuclear receptors: Lessons from genetic and pharmacological dissections of the retinoic acid signaling pathway during mouse embryogenesis. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 451–480. [Google Scholar] [CrossRef]
- Wolbach, S.R.; Howe, P.R. Tissue change following deprivation of fat-soluble A vitamin. J. Exp. Med. 1925, 42, 753–777. [Google Scholar] [CrossRef]
- West, K.P., Jr. Vitamin A deficiency: Its epidemiology and relation to child mortality and morbidity. In Vitamin A in Health and Disease; Blomhoff, R., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1994. [Google Scholar]
- Dowling, J.E. Night blindness. Sci. Am. 1966, 215, 78–84. [Google Scholar] [CrossRef]
- Sommer, A. Vitamin A: Its effect on childhood sight and life. Nutr. Rev. 1994, 52, S60–S66. [Google Scholar] [CrossRef]
- Misner, D.L.; Jacobs, S.; Shimizu, Y.; de Urquiza, A.M.; Solomin, L.; Perlmann, T.; de Luca, L.M.; Stevens, C.F.; Evans, R.M. Vitamin A deprivation results in reversible loss of hippocampal long-term synaptic plasticity. Proc. Natl. Acad. Sci. USA 2001, 98, 11714–11719. [Google Scholar]
- Cocco, S.; Diaz, G.; Stancampiano, R.; Diana, A.; Carta, M.; Curreli, R.; Sarais, L.; Fadda, F. Vitamin A deficiency produces spatial learning and memory impairment in rats. Neuroscience 2002, 115, 475–482. [Google Scholar] [CrossRef]
- Biesalski, H.K. The significance of vitamin A for the development and function of the lung. Forum Nutr. 2003, 56, 37–40. [Google Scholar]
- Ross, A.C. On the sources of retinoic acid in the lung: Understanding the local conversion of retinol to retinoic acid. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 286, L247–L248. [Google Scholar] [CrossRef]
- Vahlquist, A. Role of retinoids in normal and diseased skin. In Vitamin A in Health and Disease; Blomhoff, R., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1994; pp. 365–424. [Google Scholar]
- Morley, J.E.; Damassa, D.A.; Gordon, J.; Pekary, A.E.; Hershman, J.M. Thyroid function and vitamin A deficiency. Life Sci. 1978, 22, 1901–1905. [Google Scholar] [CrossRef]
- Livera, G.; Rouiller-Fabre, V.; Pairault, C.; Levacher, C.; Habert, R. Regulation and perturbation of testicular functions by vitamin A. Reproduction 2002, 124, 173–180. [Google Scholar] [CrossRef]
- Chen, L.; Khillan, J.S. Promotion of feeder-independent self-renewal of embryonic stem cells by retinol (vitamin A). Stem Cells 2008, 26, 1858–1864. [Google Scholar] [CrossRef]
- Ziouzenkova, O.; Orasanu, G.; Sharlach, M.; Akiyama, T.E.; Berger, J.P.; Viereck, J.; Hamilton, J.A.; Tang, G.; Dolnikowski, G.G.; Vogel, S.; et al. Retinaldehyde represses adipogenesis and diet-induced obesity. Nat. Med. 2007, 13, 695–702. [Google Scholar]
- Chen, N.; Onisko, B.; Napoli, J.L. The nuclear transcription factor RARα associates with neuronal RNA granules and suppresses translation. J. Biol. Chem. 2008, 283, 20841–20847. [Google Scholar] [CrossRef]
- Aoto, J.; Nam, C.I.; Poon, M.M.; Ting, P.; Chen, L. Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 2008, 60, 308–320. [Google Scholar] [CrossRef]
- Chytil, F.; Ong, D.E. Mediation of retinoic acid-induced growth and anti-tumour activity. Nature 1976, 260, 49–51. [Google Scholar] [CrossRef]
- Love, J.M.; Gudas, L.J. Vitamin A, differentiation and cancer. Curr. Opin. Cell Biol. 1994, 6, 825–831. [Google Scholar] [CrossRef]
- Yang, Q.; Graham, T.E.; Mody, N.; Preitner, F.; Peroni, O.D.; Zabolotny, J.M.; Kotani, K.; Quadro, L.; Kahn, B.B. Serum retinol binding protein 4 contributes to insulin resistance in obesity and type 2 diabetes. Nature 2005, 436, 356–362. [Google Scholar] [CrossRef]
- Basu, T.K.; Basualdo, C. Vitamin A homeostasis and diabetes mellitus. Nutrition 1997, 13, 804–806. [Google Scholar] [CrossRef]
- Nau, H.; Chahoud, I.; Dencker, L.; Lammer, E.J.; Scott, W.J. Teratogenicity of vitamin A and retinoids. In Vitamin A in Health and Disease; Blomhoff, R., Ed.; Marcel Dekker, Inc.: New York, NY, USA, 1994; pp. 615–664. [Google Scholar]
- Orfanos, C.E.; Zouboulis, C.C.; Almond-Roesler, B.; Geilen, C.C. Current use and future potential role of retinoids in dermatology. Drugs 1997, 53, 358–388. [Google Scholar] [CrossRef]
- Smith, F.R.; Goodman, D.S. Vitamin A transport in human vitamin A toxicity. N. Engl. J. Med. 1976, 294, 805–808. [Google Scholar] [CrossRef]
- Collins, M.D.; Mao, G.E. Teratology of retinoids. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 399–430. [Google Scholar] [CrossRef]
- Penniston, K.L.; Tanumihardjo, S.A. The acute and chronic toxic effects of vitamin A. Am. J. Clin. Nutr. 2006, 83, 191–201. [Google Scholar]
- Myhre, A.M.; Carlsen, M.H.; Bohn, S.K.; Wold, H.L.; Laake, P.; Blomhoff, R. Water-miscible, emulsified, and solid forms of retinol supplements are more toxic than oil-based preparations. Am. J. Clin. Nutr. 2003, 78, 1152–1159. [Google Scholar]
- Adams, J. Structure-activity and dose-response relationships in the neural and behavioral teratogenesis of retinoids. Neurotoxicol. Teratol. 1993, 15, 193–202. [Google Scholar] [CrossRef]
- Nau, H. Teratogenicity of isotretinoin revisited: Species variation and the role of all-trans-retinoic acid. J. Am. Acad. Dermatol. 2001, 45, S183–S187. [Google Scholar] [CrossRef]
- Crandall, J.; Sakai, Y.; Zhang, J.; Koul, O.; Mineur, Y.; Crusio, W.E.; McCaffery, P. 13-cis-retinoic acid suppresses hippocampal cell division and hippocampal-dependent learning in mice. Proc. Natl. Acad. Sci. USA 2004, 101, 5111–5116. [Google Scholar]
- Sieving, P.A.; Chaudhry, P.; Kondo, M.; Provenzano, M.; Wu, D.; Carlson, T.J.; Bush, R.A.; Thompson, D.A. Inhibition of the visual cycle in vivo by 13-cis retinoic acid protects from light damage and provides a mechanism for night blindness in isotretinoin therapy. Proc. Natl. Acad. Sci. USA 2001, 98, 1835–1840. [Google Scholar]
- Voolstra, O.; Oberhauser, V.; Sumser, E.; Meyer, N.E.; Maguire, M.E.; Huber, A.; von Lintig, J. NinaB is essential for drosophila vision but induces retinal degeneration in opsin-deficient photoreceptors. J. Biol. Chem. 2010, 285, 2130–2139. [Google Scholar]
- Sun, H.; Nathans, J. ABCR, the ATP-binding cassette transporter responsible for Stargardt macular dystrophy, is an efficient target of all-trans-retinal-mediated photooxidative damage in vitro. Implications for retinal disease. J. Biol. Chem. 2001, 276, 11766–11774. [Google Scholar] [CrossRef]
- Kanan, Y.; Moiseyev, G.; Agarwal, N.; Ma, J.X.; Al-Ubaidi, M.R. Light induces programmed cell death by activating multiple independent proteases in a cone photoreceptor cell line. Investig. Ophthalmol. Vis. Sci. 2007, 48, 40–51. [Google Scholar] [CrossRef]
- Masutomi, K.; Chen, C.; Nakatani, K.; Koutalos, Y. All-trans retinal mediates light-induced oxidation in single living rod photoreceptors (dagger). Photochem. Photobiol. 2012, 88, 1356–1361. [Google Scholar] [CrossRef]
- Maeda, A.; Maeda, T.; Golczak, M.; Palczewski, K. Retinopathy in mice induced by disrupted all-trans-retinal clearance. J. Biol. Chem. 2008, 283, 26684–26693. [Google Scholar]
- Sparrow, J.R.; Cai, B.; Jang, Y.P.; Zhou, J.; Nakanishi, K. A2E, a fluorophore of RPE lipofuscin, can destabilize membrane. Adv. Exp. Med. Biol. 2006, 572, 63–68. [Google Scholar]
- Sparrow, J.R.; Boulton, M. RPE lipofuscin and its role in retinal pathobiology. Exp. Eye Res. 2005, 80, 595–606. [Google Scholar]
- De, S.; Sakmar, T.P. Interaction of A2E with model membranes. Implications to the pathogenesis of age-related macular degeneration. J. Gen. Physiol. 2002, 120, 147–157. [Google Scholar]
- Vives-Bauza, C.; Anand, M.; Shirazi, A.K.; Magrane, J.; Gao, J.; Vollmer-Snarr, H.R.; Manfredi, G.; Finnemann, S.C. The age lipid A2E and mitochondrial dysfunction synergistically impair phagocytosis by retinal pigment epithelial cells. J. Biol. Chem. 2008, 283, 24770–24780. [Google Scholar]
- Zhou, J.; Kim, S.R.; Westlund, B.S.; Sparrow, J.R. Complement activation by bisretinoid constituents of RPE lipofuscin. Investig. Ophthalmol. Vis. Sci. 2009, 50, 1392–1399. [Google Scholar]
- Moiseyev, G.; Nikolaeva, O.; Chen, Y.; Farjo, K.; Takahashi, Y.; Ma, J.X. Inhibition of the visual cycle by A2E through direct interaction with RPE65 and implications in stargardt disease. Proc. Natl. Acad. Sci. USA 2010, 107, 17551–17556. [Google Scholar]
- Radu, R.A.; Hu, J.; Yuan, Q.; Welch, D.L.; Makshanoff, J.; Lloyd, M.; McMullen, S.; Travis, G.H.; Bok, D. Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J. Biol. Chem. 2011, 286, 18593–18601. [Google Scholar]
- Goodman, D.S. Plasma retinol-binding protein. In The Retinoids; Sporn, M.B., Boberts, A.B., Goodman, D.S., Eds.; Academic Press, Inc.: Orlando, FL, USA, 1984; Volume 2, pp. 41–88. [Google Scholar]
- Rask, L.; Anundi, H.; Bohme, J.; Eriksson, U.; Fredriksson, A.; Nilsson, S.F.; Ronne, H.; Vahlquist, A.; Peterson, P.A. The retinol-binding protein. Scand. J. Clin. Lab. Investig. Suppl. 1980, 154, 45–61. [Google Scholar]
- Blomhoff, R.; Green, M.H.; Berg, T.; Norum, K.R. Transport and storage of vitamin A. Science 1990, 250, 399–404. [Google Scholar]
- Quadro, L.; Hamberger, L.; Colantuoni, V.; Gottesman, M.E.; Blaner, W.S. Understanding the physiological role of retinol-binding protein in vitamin A metabolism using transgenic and knockout mouse models. Mol. Aspects Med. 2003, 24, 421–430. [Google Scholar]
- Zanotti, G.; Berni, R. Plasma retinol-binding protein: Structure and interactions with retinol, retinoids, and transthyretin. Vitam. Horm. 2004, 69, 271–295. [Google Scholar]
- Newcomer, M.E.; Ong, D.E. Plasma retinol binding protein: Structure and function of the prototypic lipocalin. Biochim. Biophys. Acta 2000, 1482, 57–64. [Google Scholar]
- Kawaguchi, R.; Yu, J.; Honda, J.; Hu, J.; Whitelegge, J.; Ping, P.; Wiita, P.; Bok, D.; Sun, H. A membrane receptor for retinol binding protein mediates cellular uptake of vitamin A. Science 2007, 315, 820–825. [Google Scholar]
- Sun, H.; Kawaguchi, R. The membrane receptor for plasma retinol-binding protein, a new type of cell-surface receptor. Int. Rev. Cell Mol. Biol. 2011, 288, 1–41. [Google Scholar]
- Kawaguchi, R.; Yu, J.; Ter-Stepanian, M.; Zhong, M.; Cheng, G.; Yuan, Q.; Jin, M.; Travis, G.H.; Ong, D.; Sun, H. Receptor-mediated cellular uptake mechanism that couples to intracellular storage. ACS Chem. Biol. 2011, 6, 1041–1051. [Google Scholar]
- Kawaguchi, R.; Zhong, M.; Kassai, M.; Ter-Stepanian, M.; Sun, H. STRA6-catalyzed vitamin A influx, efflux and exchange. J. Membr. Biol. 2012, 245, 731–745. [Google Scholar]
- Golczak, M.; Maeda, A.; Bereta, G.; Maeda, T.; Kiser, P.D.; Hunzelmann, S.; von Lintig, J.; Blaner, W.S.; Palczewski, K. Metabolic basis of visual cycle inhibition by retinoid and nonretinoid compounds in the vertebrate retina. J. Biol. Chem. 2008, 283, 9543–9554. [Google Scholar]
- Isken, A.; Golczak, M.; Oberhauser, V.; Hunzelmann, S.; Driever, W.; Imanishi, Y.; Palczewski, K.; von Lintig, J. RBP4 disrupts vitamin A uptake homeostasis in a STRA6-deficient animal model for Matthew-Wood syndrome. Cell Metab. 2008, 7, 258–268. [Google Scholar]
- Kawaguchi, R.; Yu, J.; Wiita, P.; Ter-Stepanian, M.; Sun, H. Mapping the membrane topology and extracellular ligand binding domains of the retinol binding protein receptor. Biochemistry 2008, 47, 5387–5395. [Google Scholar]
- Kawaguchi, R.; Yu, J.; Wiita, P.; Honda, J.; Sun, H. An essential ligand-binding domain in the membrane receptor for retinol-binding protein revealed by large-scale mutagenesis and a human polymorphism. J. Biol. Chem. 2008, 283, 15160–15168. [Google Scholar]
- Seeliger, M.W.; Biesalski, H.K.; Wissinger, B.; Gollnick, H.; Gielen, S.; Frank, J.; Beck, S.; Zrenner, E. Phenotype in retinol deficiency due to a hereditary defect in retinol binding protein synthesis. Investig. Ophthalmol. Vis. Sci. 1999, 40, 3–11. [Google Scholar]
- Folli, C.; Viglione, S.; Busconi, M.; Berni, R. Biochemical basis for retinol deficiency induced by the I41N and G75D mutations in human plasma retinol-binding protein. Biochem. Biophys. Res. Commun. 2005, 336, 1017–1022. [Google Scholar]
- Quadro, L.; Hamberger, L.; Gottesman, M.E.; Wang, F.; Colantuoni, V.; Blaner, W.S.; Mendelsohn, C.L. Pathways of vitamin A delivery to the embryo: Insights from a new tunable model of embryonic vitamin A deficiency. Endocrinology 2005, 146, 4479–4490. [Google Scholar]
- Quadro, L.; Blaner, W.S.; Salchow, D.J.; Vogel, S.; Piantedosi, R.; Gouras, P.; Freeman, S.; Cosma, M.P.; Colantuoni, V.; Gottesman, M.E. Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J. 1999, 18, 4633–4644. [Google Scholar]
- Pasutto, F.; Sticht, H.; Hammersen, G.; Gillessen-Kaesbach, G.; Fitzpatrick, D.R.; Nurnberg, G.; Brasch, F.; Schirmer-Zimmermann, H.; Tolmie, J.L.; Chitayat, D.; et al. Mutations in STRA6 cause a broad spectrum of malformations including anophthalmia, congenital heart defects, diaphragmatic hernia, alveolar capillary dysplasia, lung hypoplasia, and mental retardation. Am. J. Hum. Genet. 2007, 80, 550–560. [Google Scholar]
- Golzio, C.; Martinovic-Bouriel, J.; Thomas, S.; Mougou-Zrelli, S.; Grattagliano-Bessieres, B.; Bonniere, M.; Delahaye, S.; Munnich, A.; Encha-Razavi, F.; Lyonnet, S.; et al. Matthew-Wood syndrome is caused by truncating mutations in the retinol-binding protein receptor gene STRA6. Am. J. Hum. Genet. 2007, 80, 1179–1187. [Google Scholar]
- Ruiz, A.; Mark, M.; Jacobs, H.; Klopfenstein, M.; Hu, J.; Lloyd, M.; Habib, S.; Tosha, C.; Radu, R.A.; Ghyselinck, N.B.; et al. Retinoid content, visual responses and ocular morphology are compromised in the retinas of mice lacking the retinol-binding protein receptor, STRA6. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3027–3039. [Google Scholar]
- Znoiko, S.L.; Rohrer, B.; Lu, K.; Lohr, H.R.; Crouch, R.K.; Ma, J.X. Downregulation of cone-specific gene expression and degeneration of cone photoreceptors in the Rpe65−/− mouse at early ages. Investig. Ophthalmol. Vis. Sci. 2005, 46, 1473–1479. [Google Scholar]
- Rohrer, B.; Lohr, H.R.; Humphries, P.; Redmond, T.M.; Seeliger, M.W.; Crouch, R.K. Cone opsin mislocalization in Rpe65−/− mice: A defect that can be corrected by 11-cis retinal. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3876–3882. [Google Scholar]
- Rohrer, B.; Crouch, R. Rod and cone pigment regeneration in Rpe65−/− mice. Adv. Exp. Med. Biol. 2006, 572, 101–107. [Google Scholar]
- Zhang, H.; Fan, J.; Li, S.; Karan, S.; Rohrer, B.; Palczewski, K.; Frederick, J.M.; Crouch, R.K.; Baehr, W. Trafficking of membrane-associated proteins to cone photoreceptor outer segments requires the chromophore 11-cis-retinal. J. Neurosci. 2008, 28, 4008–4014. [Google Scholar]
- Quadro, L.; Hamberger, L.; Gottesman, M.E.; Colantuoni, V.; Ramakrishnan, R.; Blaner, W.S. Transplacental delivery of retinoid: The role of retinol-binding protein and lipoprotein retinyl ester. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E844–E851. [Google Scholar]
- Ruiz, A.; Ghyselinck, N.B.; Mata, N.; Nusinowitz, S.; Lloyd, M.; Dennefeld, C.; Chambon, P.; Bok, D. Somatic ablation of the Lrat gene in the mouse retinal pigment epithelium drastically reduces its retinoid storage. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5377–5387. [Google Scholar]
- Harrison, E.H. Mechanisms of digestion and absorption of dietary vitamin A. Annu. Rev. Nutr. 2005, 25, 87–103. [Google Scholar]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A metabolism: An update. Nutrients 2011, 3, 63–103. [Google Scholar]
- Mallia, A.K.; Smith, J.E.; Goodman, D.W. Metabolism of retinol-binding protein and vitamin A during hypervitaminosis A in the rat. J. Lipid Res. 1975, 16, 180–188. [Google Scholar]
- Wyatt, N.; Ponting, C.; Dorin, J.; Fitzpatrick, D.; Hill, R. STRA6.2: A novel member of the STRA6 gene family. Mech. Dev. 2009, 126, S259. [Google Scholar]
- Sun, H. Membrane receptors and transporters involved in the function and transport of vitamin A and its derivatives. Biochim. Biophys. Acta 2012, 1821, 99–112. [Google Scholar]
- Alapatt, P.; Guo, F.; Komanetsky, S.M.; Wang, S.; Cai, J.; Sargsyan, A.; Díaz, E.R.; Bacon, B.T.; Aryal, P.; Graham, T.E. Liver retinol transporter and receptor for serum retinol binding protein (RBP4). J. Biol. Chem. 2012. [Google Scholar] [CrossRef]
- Dowling, J.E.; Wald, G. Vitamin A deficiency and night blindness. Proc. Natl. Acad. Sci. USA 1958, 44, 648–661. [Google Scholar]
- Hu, Y.; Chen, Y.; Moiseyev, G.; Takahashi, Y.; Mott, R.; Ma, J.X. Comparison of ocular pathologies in vitamin A-deficient mice and RPE65 gene knockout mice. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5507–5514. [Google Scholar]
- Takahashi, J.S.; DeCoursey, P.J.; Bauman, L.; Menaker, M. Spectral sensitivity of a novel photoreceptive system mediating entrainment of mammalian circadian rhythms. Nature 1984, 308, 186–188. [Google Scholar]
- Dowling, J.E.; Wald, G. The biological function of vitamin A acid. Proc. Natl. Acad. Sci. USA 1960, 46, 587–608. [Google Scholar]
- Marsh-Armstrong, N.; McCaffery, P.; Gilbert, W.; Dowling, J.E.; Drager, U.C. Retinoic acid is necessary for development of the ventral retina in zebrafish. Proc. Natl. Acad. Sci. USA 1994, 91, 7286–7290. [Google Scholar]
- Hyatt, G.A.; Schmitt, E.A.; Fadool, J.M.; Dowling, J.E. Retinoic acid alters photoreceptor development in vivo. Proc. Natl. Acad. Sci. USA 1996, 93, 13298–13303. [Google Scholar]
- Kelley, M.W.; Turner, J.K.; Reh, T.A. Retinoic acid promotes differentiation of photoreceptors in vitro. Development 1994, 120, 2091–2102. [Google Scholar]
- Hyatt, G.A.; Dowling, J.E. Retinoic acid. A key molecule for eye and photoreceptor development. Investig. Ophthalmol. Vis. Sci. 1997, 38, 1471–1475. [Google Scholar]
- Duester, G. Keeping an eye on retinoic acid signaling during eye development. Chem. Biol. Interact. 2009, 178, 178–181. [Google Scholar]
- Casey, J.; Kawaguchi, R.; McGettigan, P.; Sun, H.; Morrissey, M.; Nielsen, J.; Conroy, J.; Regan, R.; Tormey, P.; Ni Chroinin, M.; et al. First implication of STRA6 mutations in isolated anophthalmia, microphthalmia and coloboma: Adding a new dimension to the STRA6 phenotype. Hum. Mutat. 2011, 32, 1417–1426. [Google Scholar]
- Rask, L.; Geijer, C.; Bill, A.; Peterson, P.A. Vitamin A supply of the cornea. Exp. Eye Res. 1980, 31, 201–211. [Google Scholar]
- Tielsch, J.M.; Sommer, A. The epidemiology of vitamin A deficiency and xerophthalmia. Annu. Rev. Nutr. 1984, 4, 183–205. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhong, M.; Kawaguchi, R.; Kassai, M.; Sun, H. Retina, Retinol, Retinal and the Natural History of Vitamin A as a Light Sensor. Nutrients 2012, 4, 2069-2096. https://doi.org/10.3390/nu4122069
Zhong M, Kawaguchi R, Kassai M, Sun H. Retina, Retinol, Retinal and the Natural History of Vitamin A as a Light Sensor. Nutrients. 2012; 4(12):2069-2096. https://doi.org/10.3390/nu4122069
Chicago/Turabian StyleZhong, Ming, Riki Kawaguchi, Miki Kassai, and Hui Sun. 2012. "Retina, Retinol, Retinal and the Natural History of Vitamin A as a Light Sensor" Nutrients 4, no. 12: 2069-2096. https://doi.org/10.3390/nu4122069
APA StyleZhong, M., Kawaguchi, R., Kassai, M., & Sun, H. (2012). Retina, Retinol, Retinal and the Natural History of Vitamin A as a Light Sensor. Nutrients, 4(12), 2069-2096. https://doi.org/10.3390/nu4122069