Evidence Showing that Tetraspanins Inhibit HIV-1-Induced Cell-Cell Fusion at a Post-Hemifusion Stage

Abstract
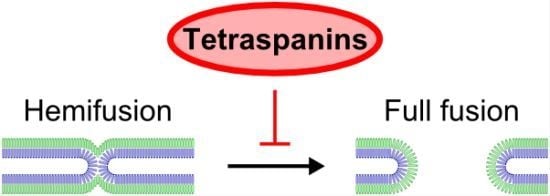
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
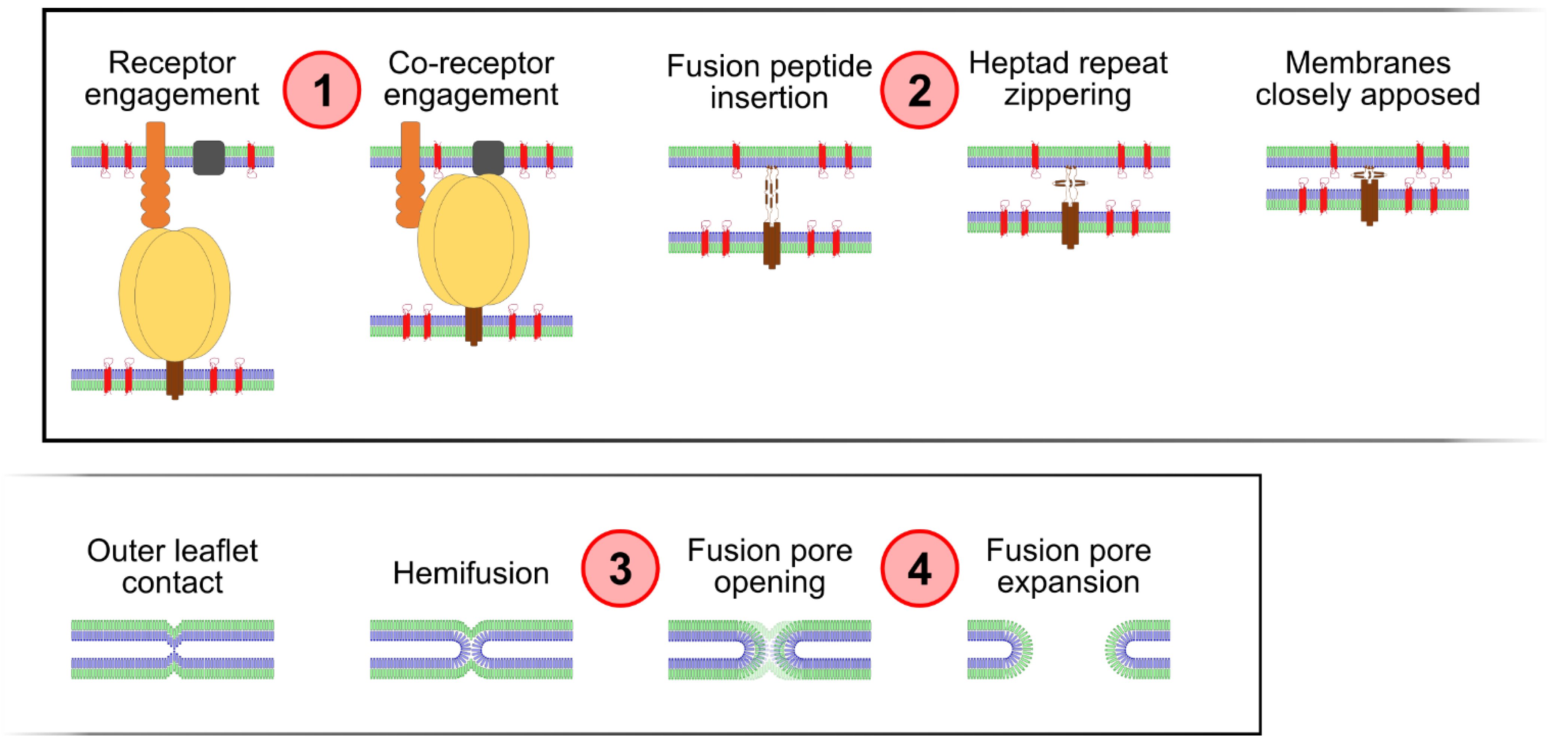
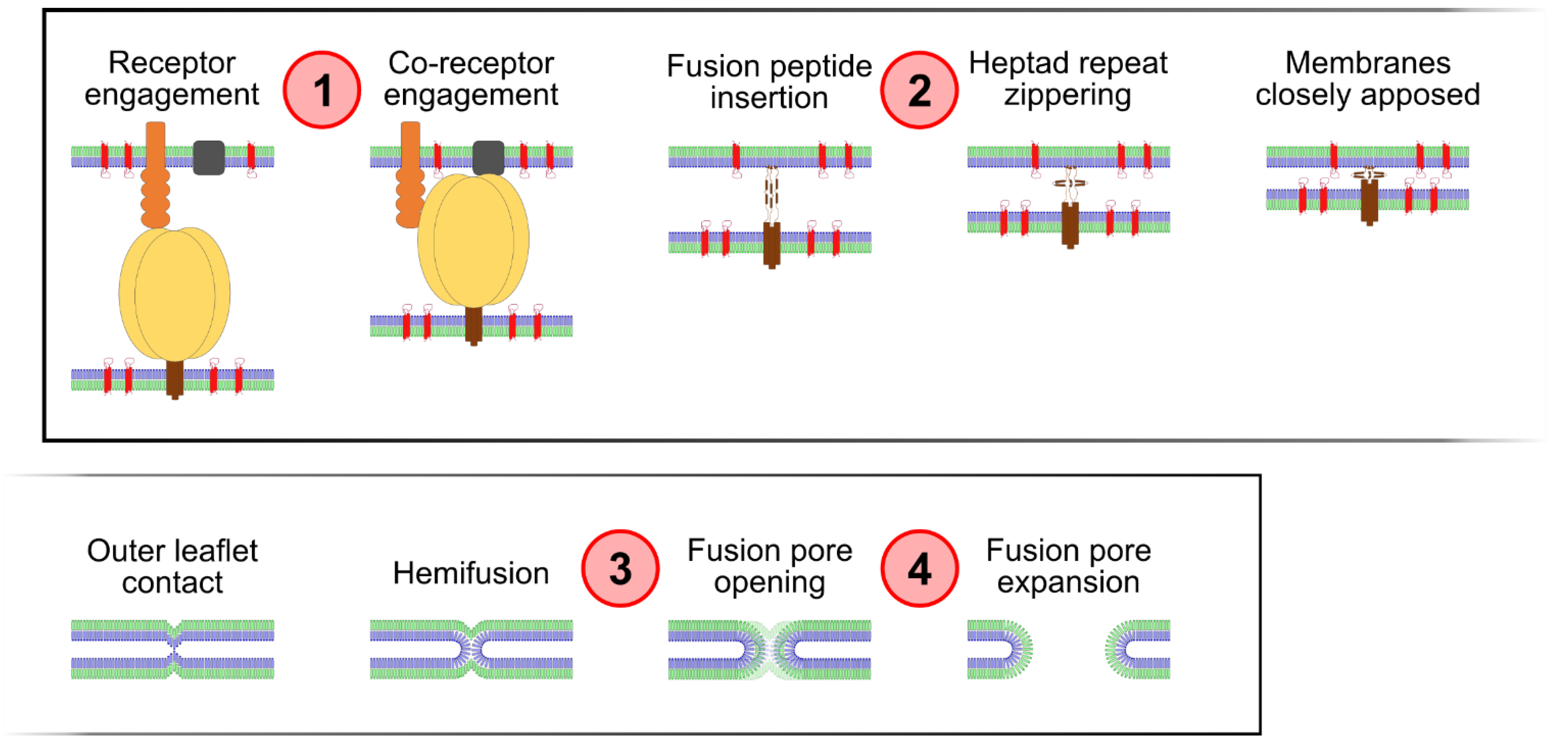
2. Results and Discussion
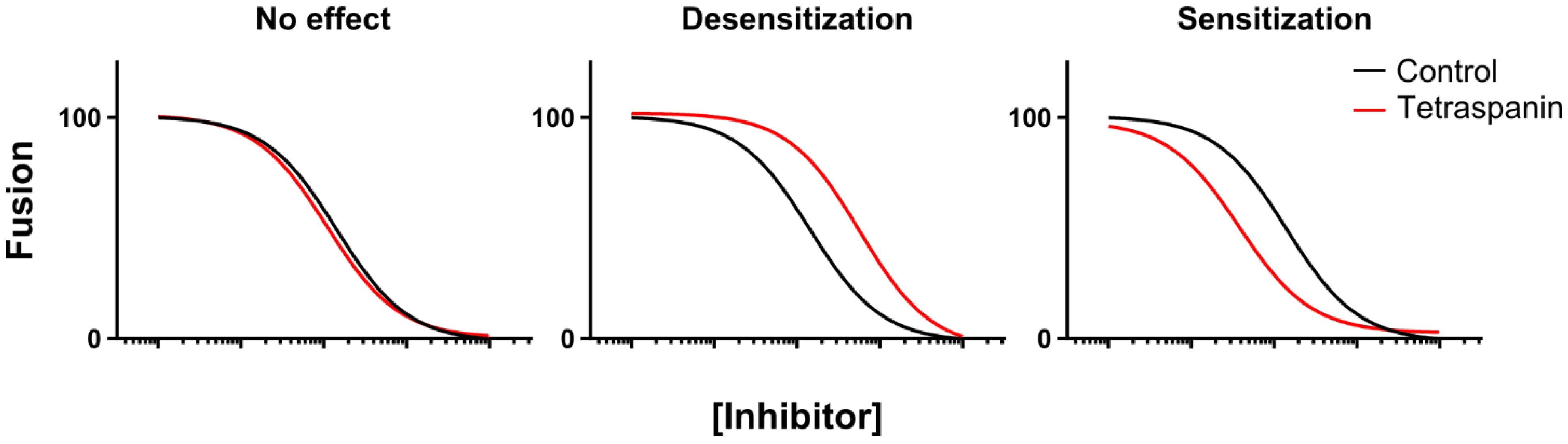
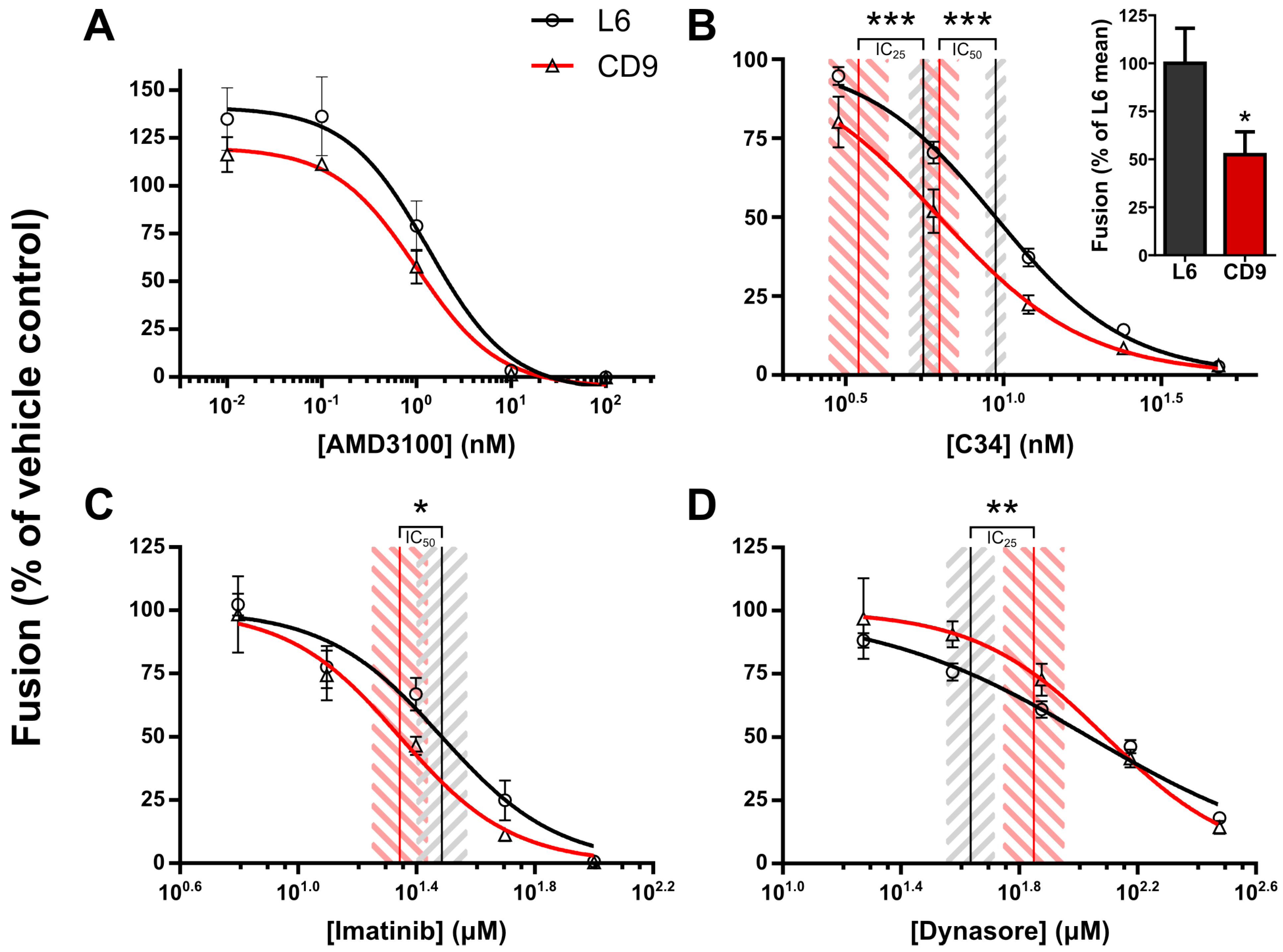
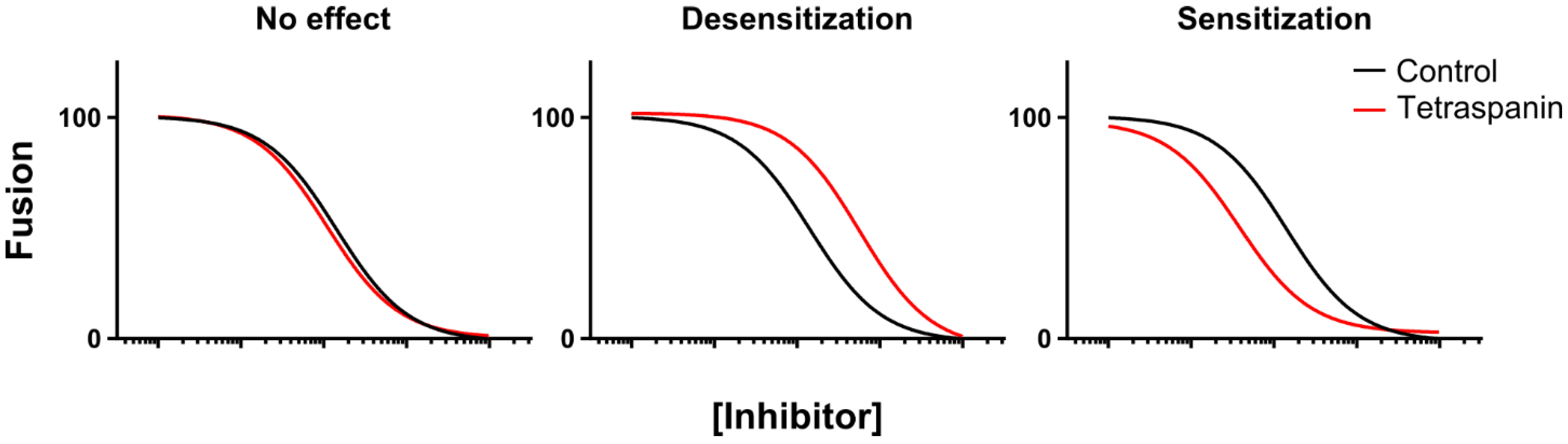
2.1. Temporal Delineation of CD9-Mediated Fusion Repression Using Chemical Epistasis

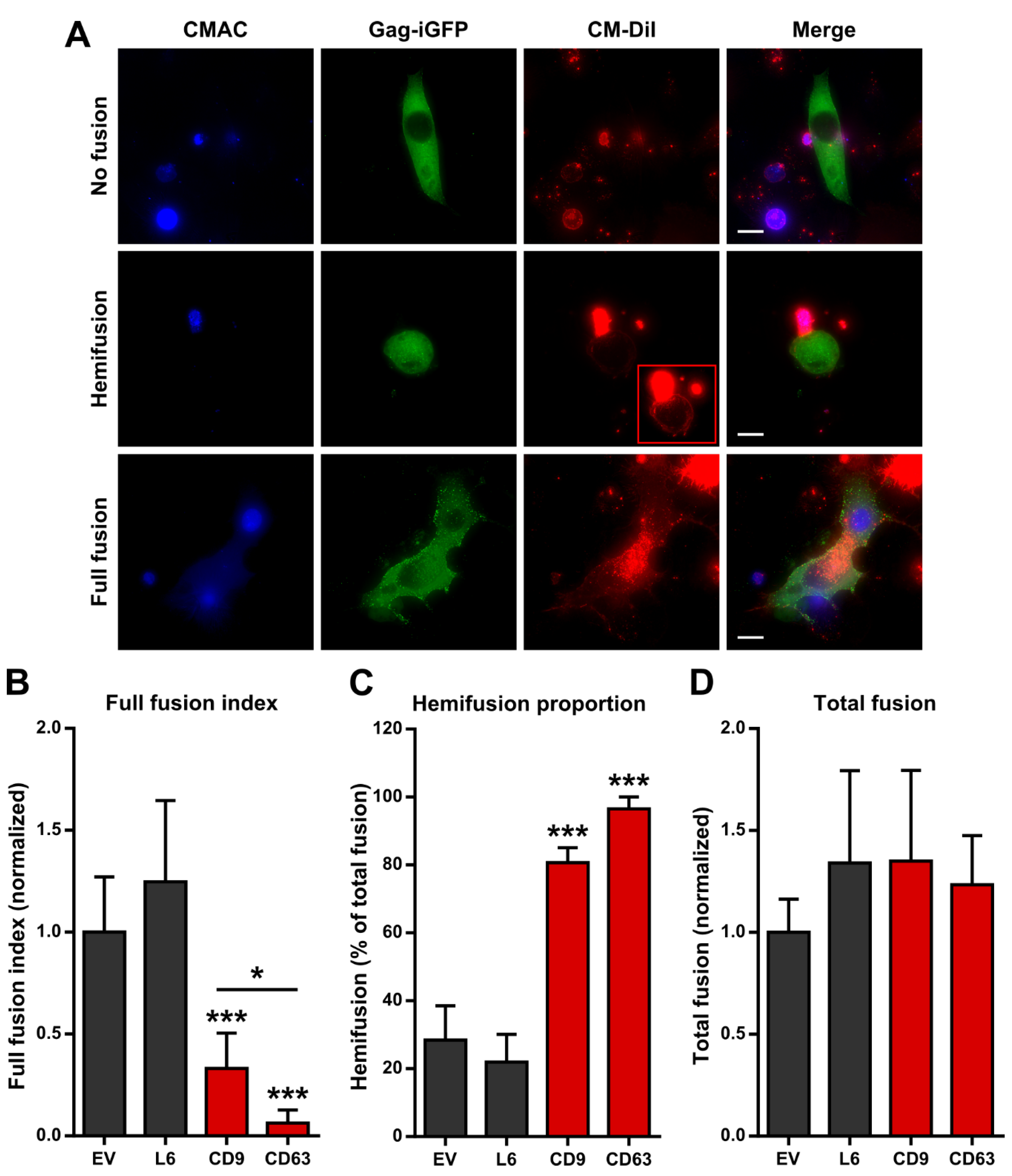
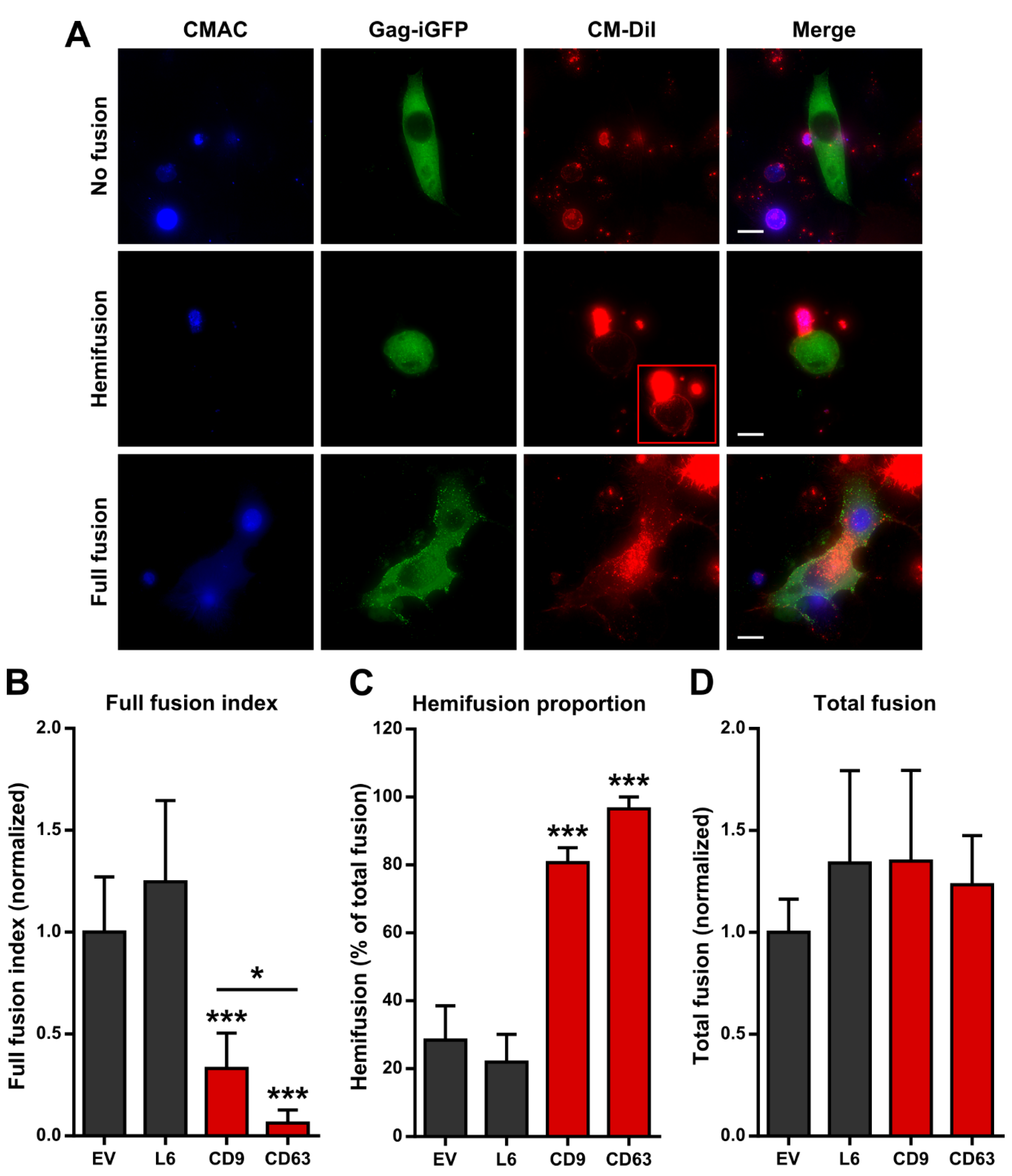
2.2. Tetraspanin Overexpression Leads to Accumulation of the Hemifusion Intermediate
3. Experimental Section
3.1. Cells, Plasmids, Antibodies, and Reagents
3.2. Dual Split Protein (DSP)-Based Cell-Cell Fusion Assay
3.3. Dye Transfer Cell-Cell Fusion Assay
3.4. Imaging
3.5. Statistical Analysis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell binding and entry. Cold Spring Harb. Perspect. Med. 2012, 2. [Google Scholar] [CrossRef]
- Koenig, S.; Gendelman, H.E.; Orenstein, J.M.; dal Canto, M.C.; Pezeshkpour, G.H.; Yungbluth, M.; Janotta, F.; Aksamit, A.; Martin, M.A.; Fauci, A.S. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science 1986, 233, 1089–1093. [Google Scholar]
- Rinfret, A.; Latendresse, H.; Lefebvre, R.; St-Louis, G.; Jolicoeur, P.; Lamarre, L. Human immunodeficiency virus-infected multinucleated histiocytes in oropharyngeal lymphoid tissues from two asymptomatic patients. Am. J. Pathol. 1991, 138, 421–426. [Google Scholar]
- Frankel, S.S.; Wenig, B.M.; Burke, A.P.; Mannan, P.; Thompson, L.D.; Abbondanzo, S.L.; Nelson, A.M.; Pope, M.; Steinman, R.M. Replication of HIV-1 in dendritic cell-derived syncytia at the mucosal surface of the adenoid. Science 1996, 272, 115–117. [Google Scholar]
- Murooka, T.T.; Deruaz, M.; Marangoni, F.; Vrbanac, V.D.; Seung, E.; von Andrian, U.H.; Tager, A.M.; Luster, A.D.; Mempel, T.R. HIV-infected T cells are migratory vehicles for viral dissemination. Nature 2012, 490, 283–287. [Google Scholar] [CrossRef]
- Doitsh, G.; Cavrois, M.; Lassen, K.G.; Zepeda, O.; Yang, Z.; Santiago, M.L.; Hebbeler, A.M.; Greene, W.C. Abortive HIV infection mediates CD4 T cell depletion and inflammation in human lymphoid tissue. Cell 2010, 143, 789–801. [Google Scholar] [CrossRef]
- Doitsh, G.; Galloway, N.L.; Geng, X.; Yang, Z.; Monroe, K.M.; Zepeda, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Munoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2013, 505, 509–514. [Google Scholar] [CrossRef]
- Jolly, C.; Kashefi, K.; Hollinshead, M.; Sattentau, Q.J. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 2004, 199, 283–293. [Google Scholar] [CrossRef]
- Dale, B.M.; Alvarez, R.A.; Chen, B.K. Mechanisms of enhanced HIV spread through T-cell virological synapses. Immunol. Rev. 2013, 251, 113–124. [Google Scholar] [CrossRef]
- Sylwester, A.; Murphy, S.; Shutt, D.; Soll, D.R. HIV-induced T cell syncytia are self-perpetuating and the primary cause of T cell death in culture. J. Immunol. 1997, 158, 3996–4007. [Google Scholar]
- Perfettini, J.L.; Castedo, M.; Roumier, T.; Andreau, K.; Nardacci, R.; Piacentini, M.; Kroemer, G. Mechanisms of apoptosis induction by the HIV-1 envelope. Cell Death Differ. 2005, 12, 916–923. [Google Scholar] [CrossRef]
- Lemaire, C.; Perfettini, J.L.; Kroemer, G.; Brenner, C. Molecular mechanisms of HIV-1 syncytial apoptosis. In Cell Death during HIV Infection; Badley, A.D., Ed.; CRC Press: Boca Raton, FL, USA, 2007; pp. 271–278. [Google Scholar]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1. [Google Scholar] [CrossRef]
- Byland, R.; Marsh, M. Trafficking of viral membrane proteins. Curr. Top. Microbiol. Immunol. 2005, 285, 219–254. [Google Scholar]
- Murakami, T.; Ablan, S.; Freed, E.O.; Tanaka, Y. Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J. Virol. 2004, 78, 1026–1031. [Google Scholar] [CrossRef]
- Wyma, D.J.; Jiang, J.; Shi, J.; Zhou, J.; Lineberger, J.E.; Miller, M.D.; Aiken, C. Coupling of human immunodeficiency virus type 1 fusion to virion maturation: A novel role of the gp41 cytoplasmic tail. J. Virol. 2004, 78, 3429–3435. [Google Scholar] [CrossRef]
- Jiang, J.; Aiken, C. Maturation-dependent human immunodeficiency virus type 1 particle fusion requires a carboxyl-terminal region of the gp41 cytoplasmic tail. J. Virol. 2007, 81, 9999–10008. [Google Scholar] [CrossRef]
- Chojnacki, J.; Staudt, T.; Glass, B.; Bingen, P.; Engelhardt, J.; Anders, M.; Schneider, J.; Muller, B.; Hell, S.W.; Krausslich, H.G. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science 2012, 338, 524–528. [Google Scholar] [CrossRef]
- Roy, N.H.; Chan, J.; Lambelé, M.; Thali, M. Clustering and mobility of HIV-1 Env at viral assembly sites predict its propensity to induce cell-cell fusion. J. Virol. 2013, 87, 7516–7525. [Google Scholar] [CrossRef]
- Hogue, I.B.; Grover, J.R.; Soheilian, F.; Nagashima, K.; Ono, A. Gag induces the coalescence of clustered lipid rafts and tetraspanin-enriched microdomains at HIV-1 assembly sites on the plasma membrane. J. Virol. 2011, 85, 9749–9766. [Google Scholar] [CrossRef]
- Krementsov, D.N.; Rassam, P.; Margeat, E.; Roy, N.H.; Schneider-Schaulies, J.; Milhiet, P.E.; Thali, M. HIV-1 assembly differentially alters dynamics and partitioning of tetraspanins and raft components. Traffic 2010, 11, 1401–1414. [Google Scholar] [CrossRef]
- Weng, J.; Krementsov, D.N.; Khurana, S.; Roy, N.H.; Thali, M. Formation of syncytia is repressed by tetraspanins in human immunodeficiency virus type 1-producing cells. J. Virol. 2009, 83, 7467–7474. [Google Scholar] [CrossRef]
- Sato, K.; Aoki, J.; Misawa, N.; Daikoku, E.; Sano, K.; Tanaka, Y.; Koyanagi, Y. Modulation of human immunodeficiency virus type 1 infectivity through incorporation of tetraspanin proteins. J. Virol. 2008, 82, 1021–1033. [Google Scholar] [CrossRef]
- Krementsov, D.N.; Weng, J.; Lambelé, M.; Roy, N.H.; Thali, M. Tetraspanins regulate cell-to-cell transmission of HIV-1. Retrovirology 2009, 6. [Google Scholar] [CrossRef]
- Barraud-Lange, V.; Boucheix, C. The role of tetraspanin complexes in egg-sperm fusion. In Tetraspanins; Berditchevski, F., Rubinstein, E., Eds.; Springer: Dordrecht, Netherlands, 2013; pp. 203–231. [Google Scholar]
- Takeda, Y.; Tachibana, I.; Miyado, K.; Kobayashi, M.; Miyazaki, T.; Funakoshi, T.; Kimura, H.; Yamane, H.; Saito, Y.; Goto, H.; et al. Tetraspanins CD9 and CD81 function to prevent the fusion of mononuclear phagocytes. J. Cell Biol. 2003, 161, 945–956. [Google Scholar] [CrossRef]
- Parthasarathy, V.; Martin, F.; Higginbottom, A.; Murray, H.; Moseley, G.W.; Read, R.C.; Mal, G.; Hulme, R.; Monk, P.N.; Partridge, L.J. Distinct roles for tetraspanins CD9, CD63 and CD81 in the formation of multinucleated giant cells. Immunology 2009, 127, 237–248. [Google Scholar] [CrossRef]
- Tachibana, I.; Hemler, M.E. Role of transmembrane 4 superfamily (TM4SF) proteins CD9 and CD81 in muscle cell fusion and myotube maintenance. J. Cell Biol. 1999, 146, 893–904. [Google Scholar] [CrossRef]
- Charrin, S.; Latil, M.; Soave, S.; Polesskaya, A.; Chretien, F.; Boucheix, C.; Rubinstein, E. Normal muscle regeneration requires tight control of muscle cell fusion by tetraspanins CD9 and CD81. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Ooi, Y.S.; Stiles, K.M.; Liu, C.Y.; Taylor, G.M.; Kielian, M. Genome-wide RNAi screen identifies novel host proteins required for alphavirus entry. PLoS Pathog. 2013, 9, e1003835. [Google Scholar] [CrossRef]
- Blumenthal, R.; Durell, S.; Viard, M. HIV entry and envelope glycoprotein-mediated fusion. J. Biol. Chem. 2012, 287, 40841–40849. [Google Scholar] [CrossRef]
- Melikyan, G.B. Common principles and intermediates of viral protein-mediated fusion: The HIV-1 paradigm. Retrovirology 2008, 5. [Google Scholar] [CrossRef]
- Lekishvili, T.; Fromm, E.; Mujoomdar, M.; Berditchevski, F. The tumour-associated antigen L6 (L6-Ag) is recruited to the tetraspanin-enriched microdomains: Implication for tumour cell motility. J. Cell Sci. 2008, 121, 685–694. [Google Scholar] [CrossRef]
- Harmon, B.; Campbell, N.; Ratner, L. Role of Abl kinase and the Wave2 signaling complex in HIV-1 entry at a post-hemifusion step. PLoS Pathog. 2010, 6, e1000956. [Google Scholar] [CrossRef]
- Lai, W.; Huang, L.; Ho, P.; Montefiori, D.; Chen, C.H. The role of dynamin in HIV type 1 Env-mediated cell-cell fusion. AIDS Res. Hum. Retroviruses 2011, 27, 1013–1017. [Google Scholar] [CrossRef]
- Richard, J.P.; Leikina, E.; Langen, R.; Henne, W.M.; Popova, M.; Balla, T.; McMahon, H.T.; Kozlov, M.M.; Chernomordik, L.V. Intracellular curvature-generating proteins in cell-to-cell fusion. Biochem. J. 2011, 440, 185–193. [Google Scholar] [CrossRef]
- Ishikawa, H.; Meng, F.; Kondo, N.; Iwamoto, A.; Matsuda, Z. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng. Des. Sel. 2012, 25, 813–820. [Google Scholar] [CrossRef]
- Kondo, N.; Miyauchi, K.; Meng, F.; Iwamoto, A.; Matsuda, Z. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J. Biol. Chem. 2010, 285, 14681–14688. [Google Scholar] [CrossRef]
- Weiss, C.D.; Barnett, S.W.; Cacalano, N.; Killeen, N.; Littman, D.R.; White, J.M. Studies of HIV-1 envelope glycoprotein-mediated fusion using a simple fluorescence assay. AIDS 1996, 10, 241–246. [Google Scholar] [CrossRef]
- Muñoz-Barroso, I.; Salzwedel, K.; Hunter, E.; Blumenthal, R. Role of the membrane-proximal domain in the initial stages of human immunodeficiency virus type 1 envelope glycoprotein-mediated membrane fusion. J. Virol. 1999, 73, 6089–6092. [Google Scholar]
- Roy, N.H.; Lambelé, M.; Chan, J.; Symeonides, M.; Thali, M. Ezrin is a component of the HIV-1 virological presynapse and contributes to the inhibition of cell-cell fusion. The University of Vermont: Burlington, VT, USA, Unpublished data. 2014. [Google Scholar]
- Kol, N.; Shi, Y.; Tsvitov, M.; Barlam, D.; Shneck, R.Z.; Kay, M.S.; Rousso, I. A stiffness switch in human immunodeficiency virus. Biophys. J. 2007, 92, 1777–1783. [Google Scholar] [CrossRef]
- Pang, H.B.; Hevroni, L.; Kol, N.; Eckert, D.M.; Tsvitov, M.; Kay, M.S.; Rousso, I. Virion stiffness regulates immature HIV-1 entry. Retrovirology 2013, 10. [Google Scholar] [CrossRef]
- Blanco, J.; Barretina, J.; Ferri, K.F.; Jacotot, E.; Gutierrez, A.; Armand-Ugon, M.; Cabrera, C.; Kroemer, G.; Clotet, B.; Este, J.A. Cell-surface-expressed HIV-1 envelope induces the death of CD4 T cells during GP41-mediated hemifusion-like events. Virology 2003, 305, 318–329. [Google Scholar] [CrossRef]
- Garg, H.; Blumenthal, R. Role of HIV Gp41 mediated fusion/hemifusion in bystander apoptosis. Cell. Mol. Life Sci. 2008, 65, 3134–3144. [Google Scholar] [CrossRef]
- Garg, H.; Mohl, J.; Joshi, A. HIV-1 induced bystander apoptosis. Viruses 2012, 4, 3020–3043. [Google Scholar] [CrossRef]
- FlowJo, v.X.0.6, TreeStar Software, Inc.: Ashland, OR, USA, 2014.
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Symeonides, M.; Lambelé, M.; Roy, N.H.; Thali, M. Evidence Showing that Tetraspanins Inhibit HIV-1-Induced Cell-Cell Fusion at a Post-Hemifusion Stage. Viruses 2014, 6, 1078-1090. https://doi.org/10.3390/v6031078
Symeonides M, Lambelé M, Roy NH, Thali M. Evidence Showing that Tetraspanins Inhibit HIV-1-Induced Cell-Cell Fusion at a Post-Hemifusion Stage. Viruses. 2014; 6(3):1078-1090. https://doi.org/10.3390/v6031078
Chicago/Turabian StyleSymeonides, Menelaos, Marie Lambelé, Nathan H. Roy, and Markus Thali. 2014. "Evidence Showing that Tetraspanins Inhibit HIV-1-Induced Cell-Cell Fusion at a Post-Hemifusion Stage" Viruses 6, no. 3: 1078-1090. https://doi.org/10.3390/v6031078