NF-κB and IRF7 Pathway Activation by Epstein-Barr Virus Latent Membrane Protein 1

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. LMP1 Structure
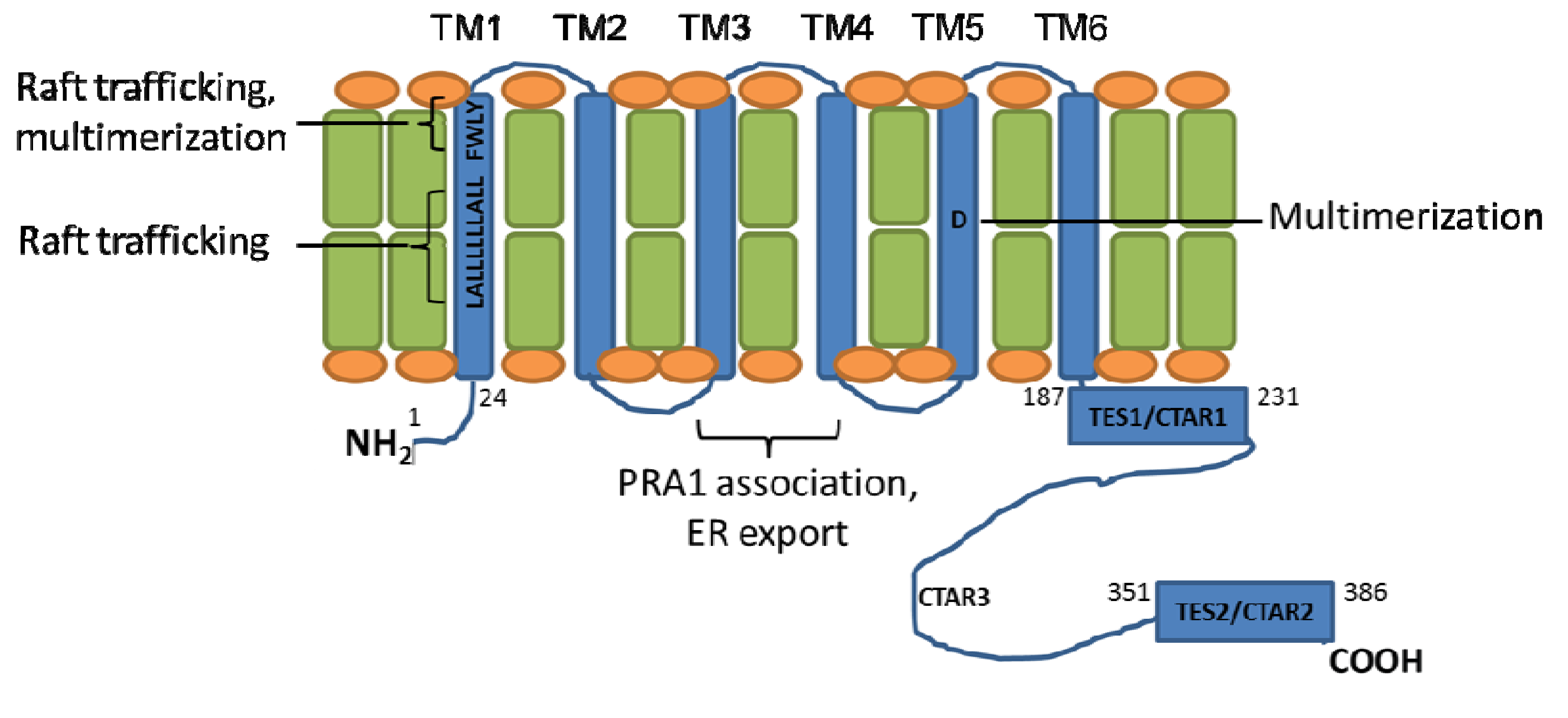
3. LMP1 Transmembrane Domains Enable Constitutive LMP1 C-terminal Tail Signaling
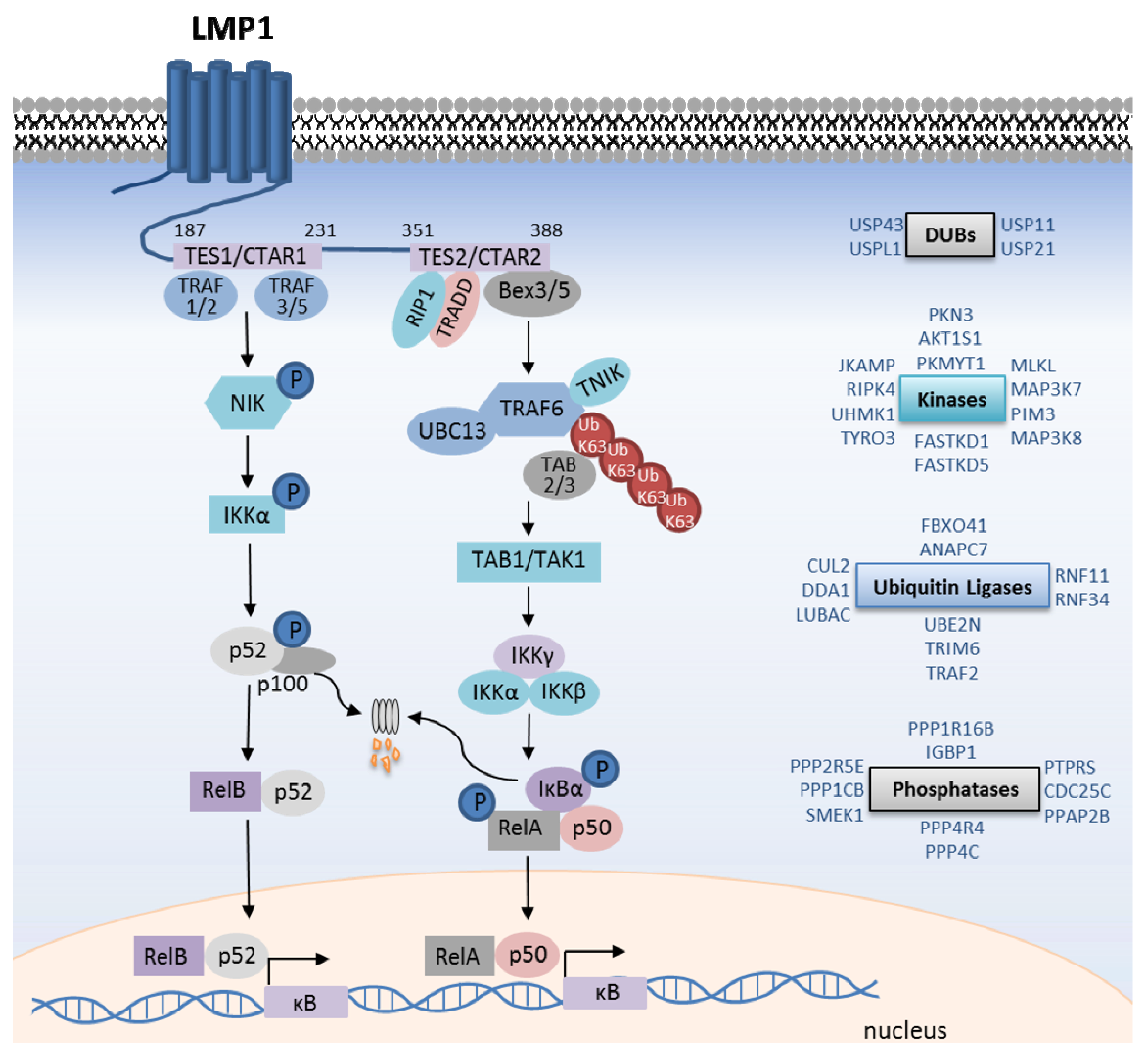
4. LMP1-Mediated Canonical NF-κB Activation
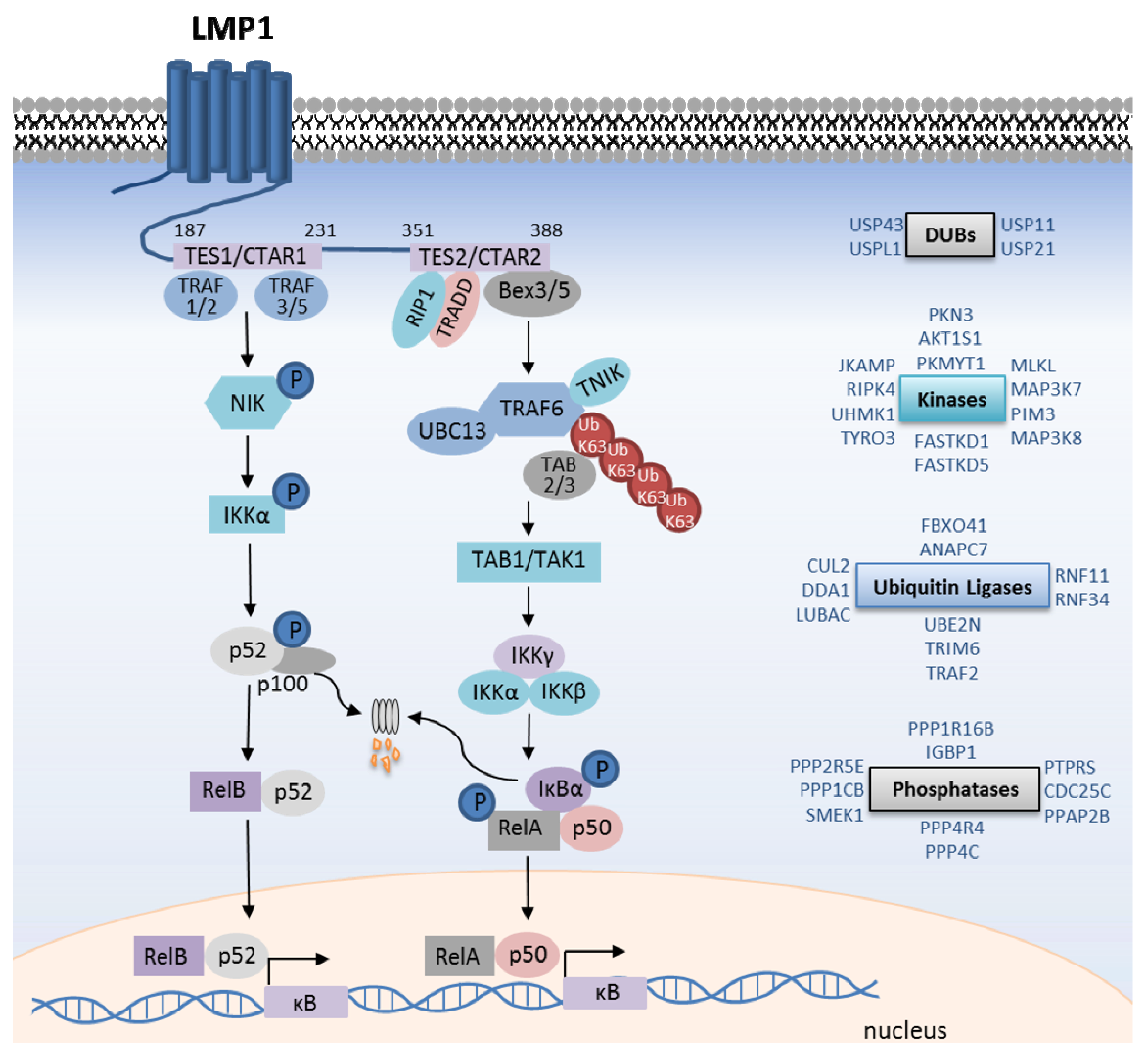
5. LMP1 Canonical NF-κB Gene Targets
6. LMP1-Mediated Non-Canonical NF-κB Activation
7. LMP1/Atypical NF-κB Pathway Activation
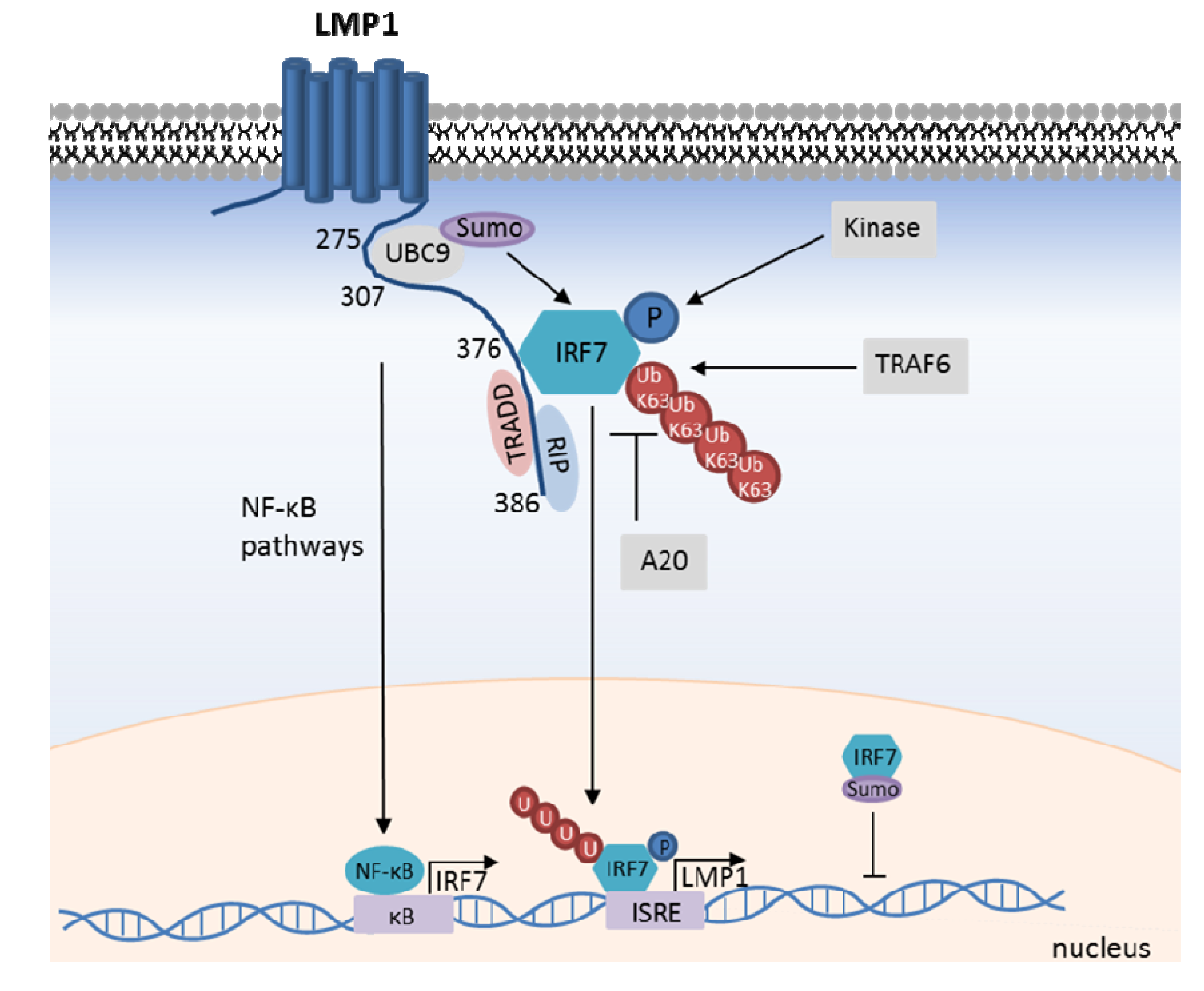
8. The LMP1/IRF7 Pathway
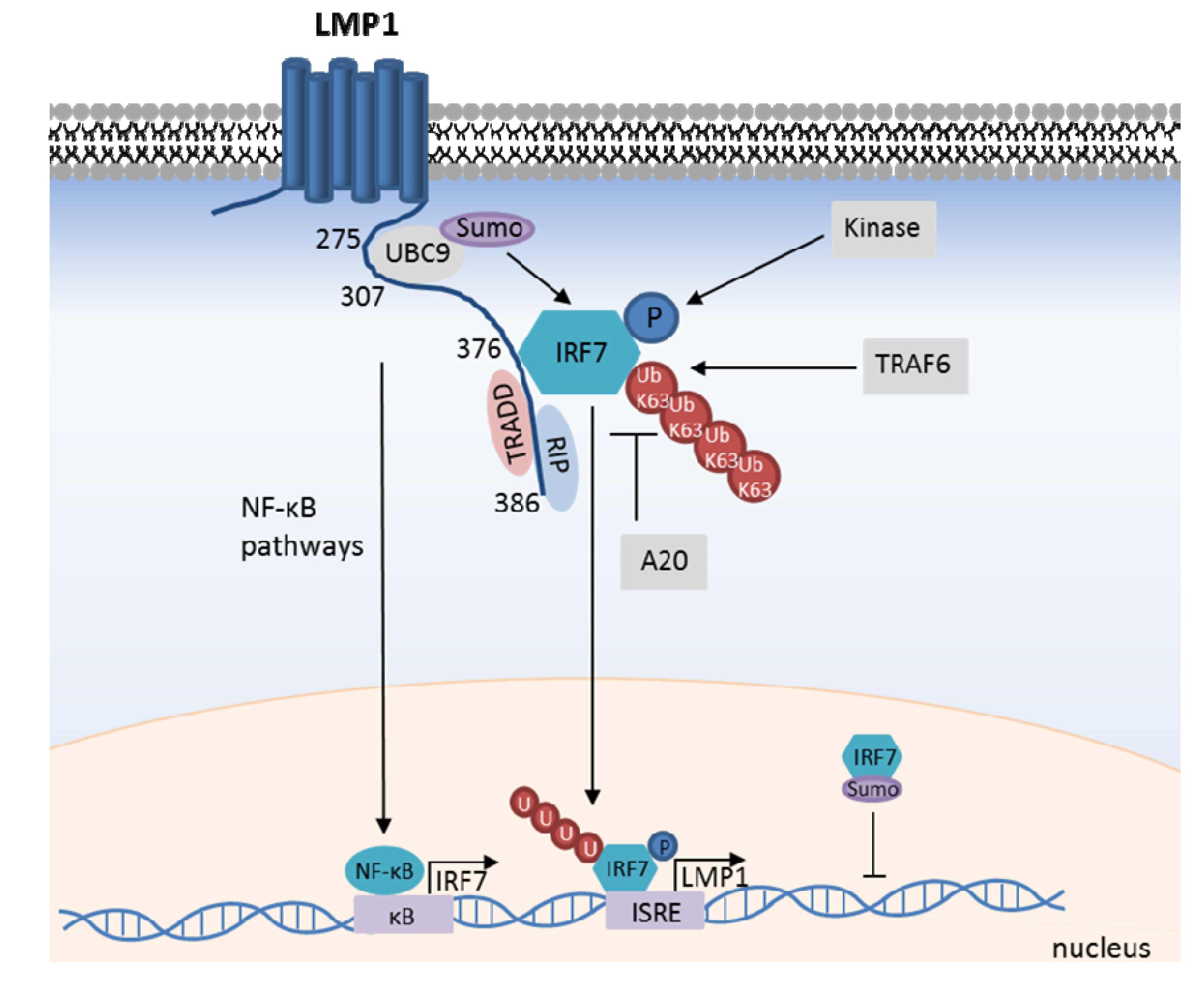
9. Concluding Remarks
Acknowledgements
Conflict of Interest
References and Notes
- Sixbey, J.W.; Nedrud, J.G.; Raab-Traub, N.; Hanes, R.A.; Pagano, J.S. Epstein-Barr virus replication in oropharyngeal epithelial cells. N. Engl. J. Med. 1984, 310, 1225–1230. [Google Scholar] [CrossRef]
- Sixbey, J.W.; Vesterinen, E.H.; Nedrud, J.G.; Raab-Traub, N.; Walton, L.A.; Pagano, J.S. Replication of Epstein-Barr virus in human epithelial cells infected in vitro. Nature 1983, 306, 480–483. [Google Scholar] [CrossRef]
- Forte, E.; Luftig, M.A. The role of micrornas in Epstein-Barr virus latency and lytic reactivation. Microbes Infect. 2011, 13, 1156–1167. [Google Scholar] [CrossRef]
- Rickinson, A.B. Epstein-Barr virus. In Fields Virology, 5th; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2007; Volume 2, pp. 2655–2700. [Google Scholar]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef]
- Swaminathan, S.; Kenney, S. The Epstein-Barr virus lytic life cycle. In DNA Tumor Viruses; Damania, B., Pipas, J.M., Eds.; Springer: New York, NY, USA, 2009; pp. 285–315. [Google Scholar]
- Wang, D.; Liebowitz, D.; Kieff, E. An ebv membrane protein expressed in immortalized lymphocytes transforms established rodent cells. Cell 1985, 43, 831–840. [Google Scholar] [CrossRef]
- Zhang, B.; Kracker, S.; Yasuda, T.; Casola, S.; Vanneman, M.; Homig-Holzel, C.; Wang, Z.; Derudder, E.; Li, S.; Chakraborty, T.; et al. Immune surveillance and therapy of lymphomas driven by Epstein-Barr virus protein LMP1 in a mouse model. Cell 2012, 148, 739–751. [Google Scholar] [CrossRef]
- Thornburg, N.J.; Kulwichit, W.; Edwards, R.H.; Shair, K.H.; Bendt, K.M.; Raab-Traub, N. LMPLMP1 signaling and activation of NF-kappaB in LMP1 transgenic mice. Oncogene 2006, 25, 288–297. [Google Scholar]
- Uchida, J.; Yasui, T.; Takaoka-Shichijo, Y.; Muraoka, M.; Kulwichit, W.; Raab-Traub, N.; Kikutani, H. Mimicry of CD40 signals by Epstein-Barr virus LMP1 in b lymphocyte responses. Science 1999, 286, 300–303. [Google Scholar] [CrossRef]
- Shair, K.H.; Bendt, K.M.; Edwards, R.H.; Nielsen, J.N.; Moore, D.T.; Raab-Traub, N. Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) and LMP2a function cooperatively to promote carcinoma development in a mouse carcinogenesis model. J. Virol. 2012, 86, 5352–5365. [Google Scholar] [CrossRef]
- Cesarman, E. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett. 2011, 305, 163–174. [Google Scholar] [CrossRef]
- Castillo, J.J.; Beltran, B.E.; Miranda, R.N.; Paydas, S.; Winer, E.S.; Butera, J.N. Epstein-Barr virus-positive diffuse large b-cell lymphoma of the elderly: What we know so far. Oncologist 2011, 16, 87–96. [Google Scholar] [CrossRef]
- Jarrett, R.F. Viruses and hodgkin's lymphoma. Ann. Oncol. 2002, 13, 23–29. [Google Scholar] [CrossRef]
- Pagano, J.S. EBV diseases. In DNA Tumor Viruses; Damania, B., Pipas, J.M., Eds.; Springer: New York, NY, USA, 2009; Volume 1, pp. 217–240. [Google Scholar]
- Vockerodt, M.; Morgan, S.L.; Kuo, M.; Wei, W.; Chukwuma, M.B.; Arrand, J.R.; Kube, D.; Gordon, J.; Young, L.S.; Woodman, C.B.; et al. The Epstein-Barr virus oncoprotein, latent membrane protein-1, reprograms germinal centre b cells towards a Hodgkin's Reed-sternberg-like phenotype. J. Pathol. 2008, 216, 83–92. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Abramson, J.S. Hiv-associated hodgkin's lymphoma: Prognosis and therapy in the era of cart. Adv. Hematol. 2012, 2012, 507257. [Google Scholar]
- Raab-Traub, N. Epstein-Barr virus in the pathogenesis of npc. Semin. Cancer Biol. 2002, 12, 431–441. [Google Scholar] [CrossRef]
- Dawson, C.W.; Port, R.J.; Young, L.S. The role of the EBV-encoded latent membrane proteins LMP1 and LMP2 in the pathogenesis of nasopharyngeal carcinoma (npc). Semin. Cancer Biol. 2012, 22, 144–153. [Google Scholar] [CrossRef]
- Kutok, J.L.; Wang, F. Spectrum of Epstein-Barr virus-associated diseases. Annu. Rev. Pathol. 2006, 1, 375–404. [Google Scholar] [CrossRef]
- Bei, J.X.; Jia, W.H.; Zeng, Y.X. Familial and large-scale case-control studies identify genes associated with nasopharyngeal carcinoma. Semin. Cancer Biol. 2012, 22, 96–106. [Google Scholar] [CrossRef]
- Strong, M.J.; Xu, G.; Coco, J.; Baribault, C.; Vinay, D.S.; Lacey, M.R.; Strong, A.L.; Lehman, T.A.; Seddon, M.B.; Lin, Z.; et al. Differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: Implications for possible immune adjuvant therapy. PLoS Pathog. 2013, 9, e1003341. [Google Scholar] [CrossRef]
- Huen, D.S.; Henderson, S.A.; Croom-Carter, D.; Rowe, M. The Epstein-Barr virus latent membrane protein-1 (LMP1) mediates activation of nf-kappa b and cell surface phenotype via two effector regions in its carboxy-terminal cytoplasmic domain. Oncogene 1995, 10, 549–560. [Google Scholar]
- Mitchell, T.; Sugden, B. Stimulation of nf-kappa b-mediated transcription by mutant derivatives of the latent membrane protein of Epstein-Barr virus. J. Virol. 1995, 69, 2968–2976. [Google Scholar]
- Kaye, K.M.; Izumi, K.M.; Li, H.; Johannsen, E.; Davidson, D.; Longnecker, R.; Kieff, E. An Epstein-Barr virus that expresses only the first 231 LMP1 amino acids efficiently initiates primary b-lymphocyte growth transformation. J. Virol. 1999, 73, 10525–10530. [Google Scholar]
- Kaye, K.M.; Izumi, K.M.; Mosialos, G.; Kieff, E. The Epstein-Barr virus LMP1 cytoplasmic carboxy terminus is essential for b-lymphocyte transformation; Fibroblast cocultivation complements a critical function within the terminal 155 residues. J. Virol. 1995, 69, 675–683. [Google Scholar]
- Raab-Traub, N. Novel mechanisms of EBV-induced oncogenesis. Curr. Opin. Virol. 2012, 2, 453–458. [Google Scholar] [CrossRef]
- Soni, V.; Cahir-McFarland, E.; Kieff, E. LMP1 trafficking activates growth and survival pathways. Adv. Exp. Med. Biol. 2007, 597, 173–187. [Google Scholar] [CrossRef]
- Raab-Traub, N. Epstein-Barr virus transfrming proteins: Biologic properties and contribution to oncogenesis. In DNA Tumor Viruses; Damania, B., Pipas, J.M., Eds.; Springer: New York, NY, USA, 2009; Volume 1, pp. 259–284. [Google Scholar]
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef]
- Ghosh, S.; Hayden, M.S. New regulators of NF-kappaB in inflammation. Nat. Rev. Immunol. 2008, 8, 837–848. [Google Scholar] [CrossRef]
- Honda, K.; Ohba, Y.; Yanai, H.; Negishi, H.; Mizutani, T.; Takaoka, A.; Taya, C.; Taniguchi, T. Spatiotemporal regulation of myd88-irf-7 signalling for robust type-i interferon induction. Nature 2005, 434, 1035–1040. [Google Scholar] [CrossRef]
- Nguyen, H.; Hiscott, J.; Pitha, P.M. The growing family of interferon regulatory factors. Cytokine Growth Factor Rev. 1997, 8, 293–312. [Google Scholar] [CrossRef]
- Nehyba, J.; Hrdlickova, R.; Bose, H.R. Dynamic evolution of immune system regulators: The history of the interferon regulatory factor family. Mol. Biol. Evol. 2009, 26, 2539–2550. [Google Scholar] [CrossRef]
- Ning, S.; Pagano, J.S.; Barber, G.N. Irf7: Activation, regulation, modification and function. Genes Immun. 2011, 12, 399–414. [Google Scholar] [CrossRef]
- Sabbah, A.; Chang, T.H.; Harnack, R.; Frohlich, V.; Tominaga, K.; Dube, P.H.; Xiang, Y.; Bose, S. Activation of innate immune antiviral responses by Nod2. Nat. Immunol. 2009, 10, 1073–1080. [Google Scholar] [CrossRef]
- Farlik, M.; Rapp, B.; Marie, I.; Levy, D.E.; Jamieson, A.M.; Decker, T. Contribution of a TANK-binding kinase 1-interferon (IFN) regulatory factor 7 pathway to IFN-γ-induced gene expression. Mol. Cell. Biol. 2012, 32, 1032–1043. [Google Scholar] [CrossRef]
- Gires, O.; Kohlhuber, F.; Kilger, E.; Baumann, M.; Kieser, A.; Kaiser, C.; Zeidler, R.; Scheffer, B.; Ueffing, M.; Hammerschmidt, W. Latent membrane protein 1 of Epstein-Barr virus interacts with JAK3 and activates STAT proteins. EMBO J. 1999, 18, 3064–3073. [Google Scholar] [CrossRef]
- Higuchi, M.; Kieff, E.; Izumi, K.M. The Epstein-Barr virus latent membrane protein 1 putative janus kinase 3 (JAK3) binding domain does not mediate JAK3 association or activation in B-lymphoma or lymphoblastoid cell lines. J. Virol. 2002, 76, 455–459. [Google Scholar] [CrossRef]
- Izumi, K.M.; Cahir McFarland, E.D.; Riley, E.A.; Rizzo, D.; Chen, Y.; Kieff, E. The residues between the two transformation effector sites of Epstein-Barr virus latent membrane protein 1 are not critical for B-lymphocyte growth transformation. J. Virol. 1999, 73, 9908–9916. [Google Scholar]
- Bentz, G.L.; Shackelford, J.; Pagano, J.S. Epstein-Barr virus latent membrane protein 1 regulates the function of interferon regulatory factor 7 by inducing its sumoylation. J. Virol. 2012, 86, 12251–12261. [Google Scholar] [CrossRef]
- Bentz, G.L.; Whitehurst, C.B.; Pagano, J.S. Epstein-Barr virus latent membrane protein 1 (LMP1) c-terminal-activating region 3 contributes to LMP1-mediated cellular migration via its interaction with Ubc9. J. Virol. 2011, 85, 10144–10153. [Google Scholar] [CrossRef]
- Hatzivassiliou, E.; Miller, W.E.; Raab-Traub, N.; Kieff, E.; Mosialos, G. A fusion of the EBV latent membrane protein-1 (LMP1) transmembrane domains to the CD40 cytoplasmic domain is similar to LMP1 in constitutive activation of epidermal growth factor receptor expression, nuclear factor-kappa b, and stress-activated protein kinas. J. Immunol. 1998, 160, 1116–1121. [Google Scholar]
- Lam, N.; Sugden, B. LMP1, a viral relative of the TNF receptor family, signals principally from intracellular compartments. EMBO J. 2003, 22, 3027–3038. [Google Scholar] [CrossRef]
- Gires, O.; Ueffing, M.; Hammerschmidt, W. Chimeric and mutated variants of LMP1. A helpful tool to analyze the structure-function relationship of a pseudoreceptor. Methods Mol. Biol 2001, 174, 313–323. [Google Scholar]
- Floettmann, J.E.; Rowe, M. Epstein-Barr virus latent membrane protein-1 (LMP1) c-terminus activation region 2 (CTAR2) maps to the far c-terminus and requires oligomerisation for NF-kappaB activation. Oncogene 1997, 15, 1851–1858. [Google Scholar]
- Yasui, T.; Luftig, M.; Soni, V.; Kieff, E. Latent infection membrane protein transmembrane FWLY is critical for intermolecular interaction, raft localization, and signaling. Proc. Natl. Acad. Sci. USA 2004, 101, 278–283. [Google Scholar] [CrossRef]
- Gurezka, R.; Laage, R.; Brosig, B.; Langosch, D. A heptad motif of leucine residues found in membrane proteins can drive self-assembly of artificial transmembrane segments. J. Biol. Chem. 1999, 274, 9265–9270. [Google Scholar] [CrossRef]
- Kaykas, A.; Worringer, K.; Sugden, B. LMP-1's transmembrane domains encode multiple functions required for LMP-1's efficient signaling. J. Virol. 2002, 76, 11551–11560. [Google Scholar] [CrossRef]
- Lee, J.; Sugden, B. A membrane leucine heptad contributes to trafficking, signaling, and transformation by latent membrane protein 1. J. Virol. 2007, 81, 9121–9130. [Google Scholar] [CrossRef]
- Liu, H.P.; Wu, C.C.; Chang, Y.S. Pra1 promotes the intracellular trafficking and NF-kappaB signaling of EBV latent membrane protein 1. EMBO J. 2006, 25, 4120–4130. [Google Scholar] [CrossRef]
- Sammond, D.W.; Joce, C.; Takeshita, R.; McQuate, S.E.; Ghosh, N.; Martin, J.M.; Yin, H. Transmembrane peptides used to investigate the homo-oligomeric interface and binding hotspot of latent membrane protein 1. Biopolymers 2011, 95, 772–784. [Google Scholar]
- Zuercher, E.; Butticaz, C.; Wyniger, J.; Martinez, R.; Battegay, M.; Boffi El Amari, E.; Dang, T.; Egger, J.F.; Fehr, J.; Mueller-Garamvogyi, E.; et al. Genetic diversity of EBV-encoded LMP1 in the swiss HIV cohort study and implication for NF-kappaB activation. PLoS One 2012, 7, e32168. [Google Scholar] [CrossRef]
- Schultheiss, U.; Puschner, S.; Kremmer, E.; Mak, T.W.; Engelmann, H.; Hammerschmidt, W.; Kieser, A. TRAF6 is a critical mediator of signal transduction by the viral oncogene latent membrane protein 1. EMBO J. 2001, 20, 5678–5691. [Google Scholar] [CrossRef]
- Wu, L.; Nakano, H.; Wu, Z. The c-terminal activating region 2 of the Epstein-Barr virus-encoded latent membrane protein 1 activates NF-kappaB through TRAF6 and TAK1. J. Biol. Chem. 2006, 281, 2162–2169. [Google Scholar]
- Luftig, M.; Prinarakis, E.; Yasui, T.; Tsichritzis, T.; Cahir-McFarland, E.; Inoue, J.; Nakano, H.; Mak, T.W.; Yeh, W.C.; Li, X.; et al. Epstein-Barr virus latent membrane protein 1 activation of NF-kappaB through IRAK1 and TRAF6. Proc. Natl. Acad. Sci. USA 2003, 100, 15595–15600. [Google Scholar] [CrossRef]
- Boehm, D.; Gewurz, B.E.; Kieff, E.; Cahir-McFarland, E. Epstein-Barr latent membrane protein 1 transformation site 2 activates NF-kappaB in the absence of NF-kappaB essential modifier residues 133–224 or 373–419. Proc. Natl. Acad. Sci. USA 2010, 107, 18103–18108. [Google Scholar] [CrossRef]
- Arcipowski, K.M.; Bishop, G.A. TRAF binding is required for a distinct subset of in vivo B cell functions of the oncoprotein LMP1. J. Immunol. 2012, 189, 5165–5170. [Google Scholar] [CrossRef]
- Izumi, K.M.; Kieff, E.D. The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc. Natl. Acad. Sci. USA 1997, 94, 12592–12597. [Google Scholar] [CrossRef]
- Schneider, F.; Neugebauer, J.; Griese, J.; Liefold, N.; Kutz, H.; Briseno, C.; Kieser, A. The viral oncoprotein LMP1 exploits tradd for signaling by masking its apoptotic activity. PLoS Biol. 2008, 6, e8. [Google Scholar] [CrossRef]
- Gewurz, B.E.; Towfic, F.; Mar, J.C.; Shinners, N.P.; Takasaki, K.; Zhao, B.; Cahir-McFarland, E.D.; Quackenbush, J.; Xavier, R.J.; Kieff, E. Genome-wide sirna screen for mediators of NF-kappaB activation. Proc. Natl. Acad. Sci. USA 2012, 109, 2467–2472. [Google Scholar] [CrossRef]
- Izumi, K.M.; Cahir McFarland, E.D.; Ting, A.T.; Riley, E.A.; Seed, B.; Kieff, E.D. The Epstein-Barr virus oncoprotein latent membrane protein 1 engages the tumor necrosis factor receptor-associated proteins TRADD and receptor-interacting protein (RIP) but does not induce apoptosis or require rip for NF-kappaB activation. Mol. Cell. Biol. 1999, 19, 5759–5767. [Google Scholar]
- Wan, J.; Zhang, W.; Wu, L.; Bai, T.; Zhang, M.; Lo, K.W.; Chui, Y.L.; Cui, Y.; Tao, Q.; Yamamoto, M.; et al. BS69, a specific adaptor in the latent membrane protein 1-mediated c-Jun n-terminal kinase pathway. Mol. Cell. Biol. 2006, 26, 448–456. [Google Scholar] [CrossRef]
- Mukai, J.; Hachiya, T.; Shoji-Hoshino, S.; Kimura, M.T.; Nadano, D.; Suvanto, P.; Hanaoka, T.; Li, Y.; Irie, S.; Greene, L.A.; et al. NADE, a p75NTR-associated cell death executor, is involved in signal transduction mediated by the common neurotrophin receptor p75NTR. J. Biol. Chem. 2000, 275, 17566–17570. [Google Scholar] [CrossRef]
- Naderi, A.; Teschendorff, A.E.; Beigel, J.; Cariati, M.; Ellis, I.O.; Brenton, J.D.; Caldas, C. BEX2 is overexpressed in a subset of primary breast cancers and mediates nerve growth factor/nuclear factor-kappaB inhibition of apoptosis in breast cancer cell lines. Cancer Res. 2007, 67, 6725–6736. [Google Scholar] [CrossRef]
- Shkoda, A.; Town, J.A.; Griese, J.; Romio, M.; Sarioglu, H.; Knofel, T.; Giehler, F.; Kieser, A. The germinal center kinase TNIK is required for canonical NF-kappaB and jnk signaling in B-cells by the EBV oncoprotein LMP1 and the 4CD0 receptor. PLoS Biol. 2012, 10, e1001376. [Google Scholar] [CrossRef]
- Song, Y.J.; Jen, K.Y.; Soni, V.; Kieff, E.; Cahir-McFarland, E. Il-1 receptor-associated kinase 1 is critical for latent membrane protein 1-induced p65/rela serine 536 phosphorylation and NF-kappaB activation. Proc. Natl. Acad. Sci. USA 2006, 103, 2689–2694. [Google Scholar]
- Soni, V.; Kieff, E. Brigham and Women’s Hospital: Boston, MA, USA, 2007; Unpublished work.
- Rieser, E.; Cordier, S.M.; Walczak, H. Linear ubiquitination: A newly discovered regulator of cell signalling. Trends BioChem. Sci. 2013, 38, 94–102. [Google Scholar] [CrossRef]
- Tokunaga, F.; Iwai, K. Linear ubiquitination: A novel NF-kappaB regulatory mechanism for inflammatory and immune responses by the lubac ubiquitin ligase complex. Endocr. J. 2012, 59, 641–652. [Google Scholar] [CrossRef]
- Gewurz, B.; Kieff, E. Brigham and Women’s Hospital: Boston, MA, USA, 2012; Unpublished work.
- Hostager, B.S.; Kashiwada, M.; Colgan, J.D.; Rothman, P.B. Hoil-1l interacting protein (HOIP) is essential for CD40 signaling. PLoS One 2011, 6, e23061. [Google Scholar]
- Iwai, K. Diverse ubiquitin signaling in NF-kappaB activation. Trends Cell Biol. 2012, 22, 355–364. [Google Scholar] [CrossRef]
- Walczak, H.; Iwai, K.; Dikic, I. Generation and physiological roles of linear ubiquitin chains. BMC Biol. 2012, 10, 23. [Google Scholar] [CrossRef]
- Boisson, B.; Laplantine, E.; Prando, C.; Giliani, S.; Israelsson, E.; Xu, Z.; Abhyankar, A.; Israel, L.; Trevejo-Nunez, G.; Bogunovic, D.; et al. Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL-1 and LUBAC deficiency. Nat. Immunol. 2012, 13, 1178–1186. [Google Scholar] [CrossRef]
- Matsushita, K.; Takeuchi, O.; Standley, D.M.; Kumagai, Y.; Kawagoe, T.; Miyake, T.; Satoh, T.; Kato, H.; Tsujimura, T.; Nakamura, H.; et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature 2009, 458, 1185–1190. [Google Scholar] [CrossRef]
- Gewurz, B.E.; Mar, J.C.; Padi, M.; Zhao, B.; Shinners, N.P.; Takasaki, K.; Bedoya, E.; Zou, J.Y.; Cahir-McFarland, E.; Quackenbush, J.; et al. Canonical NF-kappaB activation is essential for Epstein-Barr virus latent membrane protein 1 TES2/CTAR2 gene regulation. J. Virol. 2011, 85, 6764–6773. [Google Scholar] [CrossRef]
- Pegtel, D.M.; van de Garde, M.D.; Middeldorp, J.M. Viral mirnas exploiting the endosomal-exosomal pathway for intercellular cross-talk and immune evasion. Biochim. Biophys. Acta 2011, 1809, 715–721. [Google Scholar]
- Meckes, D.G., Jr.; Shair, K.H.; Marquitz, A.R.; Kung, C.P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar]
- Verweij, F.J.; van Eijndhoven, M.A.; Hopmans, E.S.; Vendrig, T.; Wurdinger, T.; Cahir-McFarland, E.; Kieff, E.; Geerts, D.; van der Kant, R.; Neefjes, J.; et al. LMP1 association with CD63 in endosomes and secretion via exosomes limits constitutive NF-kappaB activation. EMBO J. 2011, 30, 2115–2129. [Google Scholar] [CrossRef]
- Meckes, D.G., Jr.; Raab-Traub, N. Microvesicles and viral infection. J. Virol. 2011, 85, 12844–12854. [Google Scholar] [CrossRef]
- Shair, K.H.; Raab-Traub, N. Transcriptome changes induced by Epstein-Barr virus LMP1 and LMP2a in transgenic lymphocytes and lymphoma. MBio 2012, 3, e00288–12. [Google Scholar]
- Morris, M.A.; Dawson, C.W.; Wei, W.; O'Neil, J.D.; Stewart, S.E.; Jia, J.; Bell, A.I.; Young, L.S.; Arrand, J.R. Epstein-Barr virus-encoded LMP1 induces a hyperproliferative and inflammatory gene expression programme in cultured keratinocytes. J. Gen. Virol. 2008, 89, 2806–2820. [Google Scholar] [CrossRef]
- Cahir-McFarland, E.D.; Carter, K.; Rosenwald, A.; Giltnane, J.M.; Henrickson, S.E.; Staudt, L.M.; Kieff, E. Role of NF-kappa B in cell survival and transcription of latent membrane protein 1-expressing or Epstein-Barr virus latency III-infected cells. J. Virol. 2004, 78, 4108–4119. [Google Scholar] [CrossRef]
- Natoli, G. Maintaining cell identity through global control of genomic organization. Immunity 2010, 33, 12–24. [Google Scholar] [CrossRef]
- Sommermann, T.G.; O'Neill, K.; Plas, D.R.; Cahir-McFarland, E. IKKβ and NF-kappaB transcription govern lymphoma cell survival through akt-induced plasma membrane trafficking of GLUT1. Cancer Res. 2011, 71, 7291–7300. [Google Scholar] [CrossRef]
- Forte, E.; Salinas, R.E.; Chang, C.; Zhou, T.; Linnstaedt, S.D.; Gottwein, E.; Jacobs, C.; Jima, D.; Li, Q.J.; Dave, S.S.; et al. The Epstein-Barr virus (EBV)-induced tumor suppressor microRNA miR-34a is growth promoting in EBV-infected B cells. J. Virol. 2012, 86, 6889–6898. [Google Scholar] [CrossRef]
- Sun, S.C. Non-canonical NF-kappaB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Matsuzawa, A.; Zhang, W.; Tseng, P.H.; Keats, J.J.; Wang, H.; Vignali, D.A.; Bergsagel, P.L.; Karin, M. Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat. Immunol. 2008, 9, 1364–1370. [Google Scholar] [CrossRef]
- Devergne, O.; Hatzivassiliou, E.; Izumi, K.M.; Kaye, K.M.; Kleijnen, M.F.; Kieff, E.; Mosialos, G. Association of TRAF1, TRAF2, and TRAF3 with an Epstein-Barr virus LMP1 domain important for b-lymphocyte transformation: Role in NF-kappaB activation. Mol. Cell. Biol. 1996, 16, 7098–7108. [Google Scholar]
- Ye, H.; Park, Y.C.; Kreishman, M.; Kieff, E.; Wu, H. The structural basis for the recognition of diverse receptor sequences by TRAF2. Mol. Cell 1999, 4, 321–330. [Google Scholar] [CrossRef]
- Brown, K.D.; Hostager, B.S.; Bishop, G.A. Differential signaling and tumor necrosis factor receptor-associated factor (TRAF) degradation mediated by CD40 and the Epstein-Barr virus oncoprotein latent membrane protein 1 (LMP1). J. Exp. Med. 2001, 193, 943–954. [Google Scholar] [CrossRef]
- de Jong, S.J.; Albrecht, J.C.; Giehler, F.; Kieser, A.; Sticht, H.; Biesinger, B. Noncanonical NF-kappaB activation by the oncoprotein tio occurs through a nonconserved TRAF3-binding motif. Sci. Signal. 2013, 6, ra27. [Google Scholar] [CrossRef]
- Xie, P.; Hostager, B.S.; Bishop, G.A. Requirement for TRAF3 in signaling by LMP1 but not CD40 in b lymphocytes. J. Exp. Med. 2004, 199, 661–671. [Google Scholar] [CrossRef]
- Luftig, M.A.; Cahir-McFarland, E.; Mosialos, G.; Kieff, E. Effects of the nik aly mutation on NF-kappaB activation by the Epstein-Barr virus latent infection membrane protein, lymphotoxin beta receptor, and CD40. J. Biol. Chem. 2001, 276, 14602–14606. [Google Scholar]
- Song, Y.J.; Kang, M.S. Roles of TRAF2 and TRAF3 in Epstein-Barr virus latent membrane protein 1-induced alternative NF-kappaB activation. Virus Genes 2010, 41, 174–180. [Google Scholar] [CrossRef]
- Saito, N.; Courtois, G.; Chiba, A.; Yamamoto, N.; Nitta, T.; Hironaka, N.; Rowe, M.; Yamaoka, S. Two carboxyl-terminal activation regions of Epstein-Barr virus latent membrane protein 1 activate NF-kappaB through distinct signaling pathways in fibroblast cell lines. J. Biol. Chem. 2003, 278, 46565–46575. [Google Scholar]
- Jin, X.; Jin, H.R.; Jung, H.S.; Lee, S.J.; Lee, J.H.; Lee, J.J. An atypical E3 ligase zinc finger protein 91 stabilizes and activates NF-kappaB-inducing kinase via Lys63-linked ubiquitination. J. Biol. Chem. 2010, 285, 30539–30547. [Google Scholar]
- Kung, C.P.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 1 modulates distinctive nf- kappab pathways through c-terminus-activating region 1 to regulate epidermal growth factor receptor expression. J. Virol. 2010, 84, 6605–6614. [Google Scholar] [CrossRef]
- Zhang, L.; Pagano, J.S. Irf-7, a new interferon regulatory factor associated with Epstein-Barr virus latency. Mol. Cell. Biol. 1997, 17, 5748–5757. [Google Scholar]
- Huye, L.E.; Ning, S.; Kelliher, M.; Pagano, J.S. Interferon regulatory factor 7 is activated by a viral oncoprotein through RIP-dependent ubiquitination. Mol. Cell. Biol. 2007, 27, 2910–2918. [Google Scholar] [CrossRef]
- Kim, I.W.; Park, H.S. Colocalization of interferon regulatory factor 7 (IRF7) with latent membrane protein 1 (LMP1) of Epstein-Barr virus. J. Kor. Med. Sci. 2006, 21, 379–384. [Google Scholar] [CrossRef]
- Song, Y.J.; Izumi, K.M.; Shinners, N.P.; Gewurz, B.E.; Kieff, E. Irf7 activation by Epstein-Barr virus latent membrane protein 1 requires localization at activation sites and TRAF6, but not TRAF2 or TRAF3. Proc. Natl. Acad. Sci. USA 2008, 105, 18448–18453. [Google Scholar]
- Ning, S.; Campos, A.D.; Darnay, B.G.; Bentz, G.L.; Pagano, J.S. TRAF6 and the three c-terminal lysine sites on IRF7 are required for its ubiquitination-mediated activation by the tumor necrosis factor receptor family member latent membrane protein 1. Mol. Cell. Biol. 2008, 28, 6536–6546. [Google Scholar]
- Lin, R.; Mamane, Y.; Hiscott, J. Multiple regulatory domains control Irf-7 activity in response to virus infection. J. Biol. Chem. 2000, 275, 34320–34327. [Google Scholar]
- Wathelet, M.G.; Lin, C.H.; Parekh, B.S.; Ronco, L.V.; Howley, P.M.; Maniatis, T. Virus infection induces the assembly of coordinately activated transcription factors on the Ifn-β enhancer in vivo. Mol. Cell. 1998, 1, 507–518. [Google Scholar] [CrossRef]
- Yang, H.; Lin, C.H.; Ma, G.; Baffi, M.O.; Wathelet, M.G. Interferon regulatory factor-7 synergizes with other transcription factors through multiple interactions with p300/CBP coactivators. J. Biol. Chem. 2003, 278, 15495–15504. [Google Scholar]
- Au, W.C.; Moore, P.A.; LaFleur, D.W.; Tombal, B.; Pitha, P.M. Characterization of the interferon regulatory factor-7 and its potential role in the transcription activation of interferon A genes. J. Biol. Chem. 1998, 273, 29210–29217. [Google Scholar]
- Zhang, L.; Wu, L.; Hong, K.; Pagano, J.S. Intracellular signaling molecules activated by Epstein-Barr virus for induction of interferon regulatory factor 7. J. Virol. 2001, 75, 12393–12401. [Google Scholar] [CrossRef]
- Ning, S.; Hahn, A.M.; Huye, L.E.; Pagano, J.S. Interferon regulatory factor 7 regulates expression of Epstein-Barr virus latent membrane protein 1: A regulatory circuit. J. Virol. 2003, 77, 9359–9368. [Google Scholar] [CrossRef]
- Lu, R.; Au, W.C.; Yeow, W.S.; Hageman, N.; Pitha, P.M. Regulation of the promoter activity of interferon regulatory factor-7 gene. Activation by interferon snd silencing by hyperm ethylation. J. Biol. Chem. 2000, 275, 31805–31812. [Google Scholar]
- Zhang, L.; Zhang, J.; Lambert, Q.; Der, C.J.; Del Valle, L.; Miklossy, J.; Khalili, K.; Zhou, Y.; Pagano, J.S. Interferon regulatory factor 7 is associated with Epstein-Barr virus-transformed central nervous system lymphoma and has oncogenic properties. J. Virol. 2004, 78, 12987–12995. [Google Scholar] [CrossRef]
- Ning, S.; Pagano, J.S. The a20 deubiquitinase activity negatively regulates LMP1 activation of IRF7. J. Virol. 2010, 84, 6130–6138. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ersing, I.; Bernhardt, K.; Gewurz, B.E. NF-κB and IRF7 Pathway Activation by Epstein-Barr Virus Latent Membrane Protein 1. Viruses 2013, 5, 1587-1606. https://doi.org/10.3390/v5061587
Ersing I, Bernhardt K, Gewurz BE. NF-κB and IRF7 Pathway Activation by Epstein-Barr Virus Latent Membrane Protein 1. Viruses. 2013; 5(6):1587-1606. https://doi.org/10.3390/v5061587
Chicago/Turabian StyleErsing, Ina, Katharina Bernhardt, and Benjamin E. Gewurz. 2013. "NF-κB and IRF7 Pathway Activation by Epstein-Barr Virus Latent Membrane Protein 1" Viruses 5, no. 6: 1587-1606. https://doi.org/10.3390/v5061587
APA StyleErsing, I., Bernhardt, K., & Gewurz, B. E. (2013). NF-κB and IRF7 Pathway Activation by Epstein-Barr Virus Latent Membrane Protein 1. Viruses, 5(6), 1587-1606. https://doi.org/10.3390/v5061587