Detection of Coronaviruses in Bats of Various Species in Italy



Abstract
:1. Introduction
2. Results and Discussion

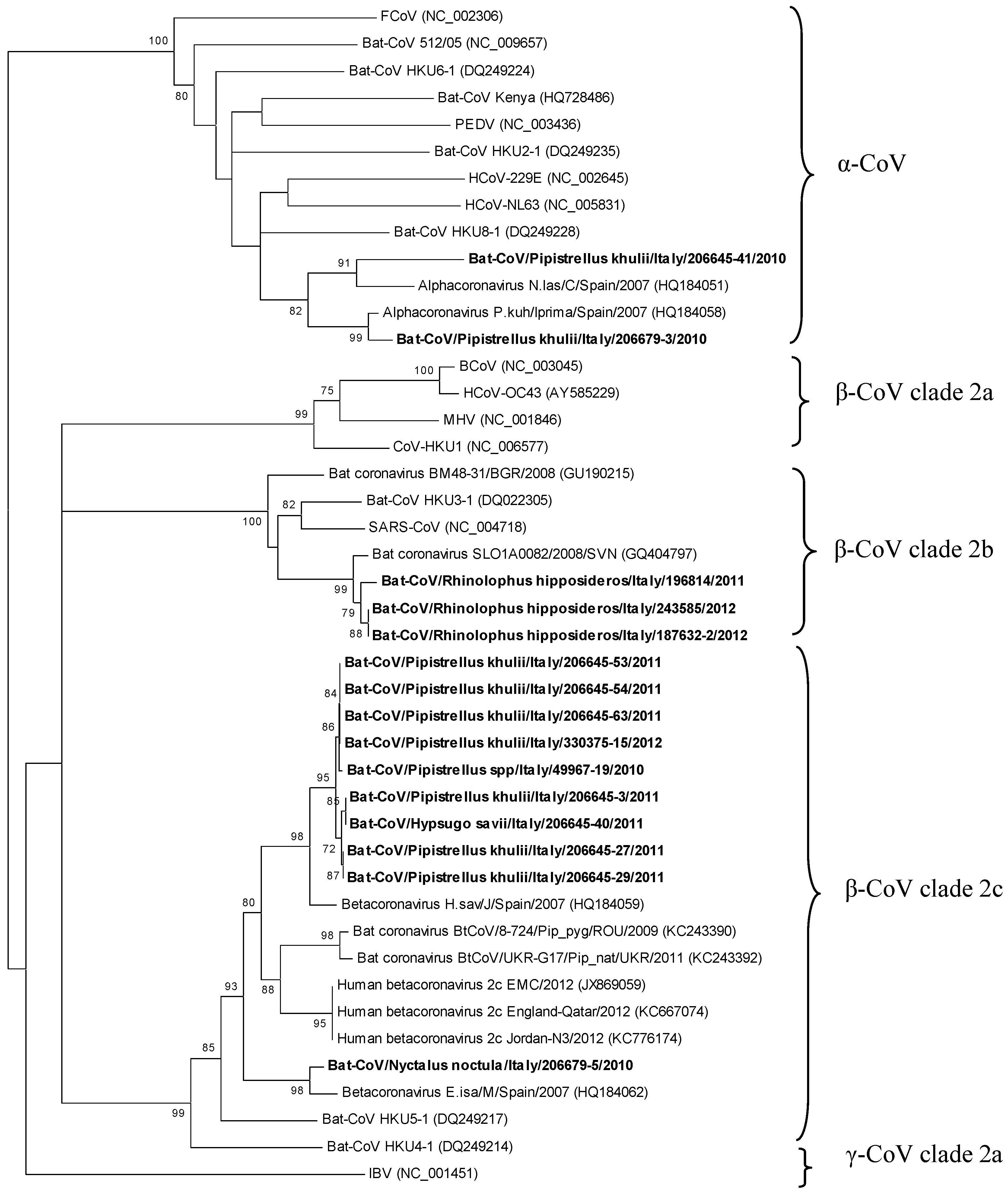{kind=link}
| Host | Carcasses | Faeces | ||
|---|---|---|---|---|
| N | % | N | % | |
| Kuhl’s pipistrelle (Pipistrellus kuhlii) | 68 | 54 | 28 | 40.7 |
| Pipistrelle bat (Pipistrellus spp.) | 48 | 38 | 30 | 43.5 |
| European free-tailed bat (Tadarida teniotis) | 2 | 1.6 | 1 | 1.4 |
| Common noctule (Nyctalus noctula) | 2 | 1.6 | 1 | 1.4 |
| Savi’s pipistrelle (Hypsugo savii) | 1 | 0.8 | 2 | 2.9 |
| Common long-eared bat (Plecotus auritus) | 3 | 2.4 | 0 | - |
| Parti-colored bat (Vespertilio murinus) | 2 | 1.6 | 1 | 1.4 |
| Lesser horseshoe bat (Rhinolophus hipposideros) | 0 | - | 6 | 8.7 |
| Total samples | 126 | 100 | 69 | 100 |
| N ° ID | Host species | Sample type | Sex | Age | CoV strain | Group | GenBank accession number | Nucleotide similarity (%) |
|---|---|---|---|---|---|---|---|---|
| 206679-3/2010 | Kuhl’s pipistrelle | Faeces | F | Adult | Bat-CoV/Pipistrellus khulii/Italy/206679-3/2010 | α | KF500949 | 97% Alphacoronavirus P.kuh/Iprima/Spain/2007 |
| 206679-5/2010 | Common noctule | Faeces | F | Adult | Bat-CoV/Nyctalus noctula/Italy/206679-5/2010 | β 2c | KF500941 | 96% Betacoronavirus E.isa/M/Spain/2007 |
| 206645-3/2011 | Kuhl’s pipistrelle | Carcass * | M | Juvenile | Bat-CoV/Pipistrellus khulii/Italy/206645-3/2011 | β 2c | KF500942 | 94% Betacoronavirus H.sav/J/Spain/2007 |
| 206645-27/2011 | Kuhl’s pipistrelle | Carcass * | F | Adult | Bat-CoV/Pipistrellus khulii/Italy/206645-27/2011 | β 2c | KF500943 | 94% Betacoronavirus H.sav/J/Spain/2007 |
| 206645-29/2011 | Kuhl’s pipistrelle | Carcass * | F | Adult | Bat-CoV/Pipistrellus khulii/Italy/206645-29/2011 | β 2c | KF500944 | 94% Betacoronavirus H.sav/J/Spain/2007 |
| 206645-40/2011 | Hypsugo savii | Carcass * | F | Adult | Bat-CoV/Hypsugo savii/206645-40/2011 | β 2c | KF500940 | 94% Betacoronavirus H.sav/J/Spain/2007 |
| 206645-41/2010 | Kuhl’s pipistrelle | Carcass * | M | Juvenile | Bat-CoV/Pipistrellus khulii/Italy/206645-41/2010 | α | KF500945 | 86% Alphacoronavirus N.las/C/Spain/2007 |
| 206645-53/2011 | Kuhl’s pipistrelle | Carcass * | nd | nd | Bat-CoV/Pipistrellus khulii/Italy/206645-53/2011 | β 2c | KF500946 | 94% Betacoronavirus H.sav/J/Spain/2007 |
| 206645-54/2011 | Kuhl’s pipistrelle | Carcass ^ | nd | nd | Bat-CoV/Pipistrellus khulii/Italy/206645-54/2011 | β 2c | KF500947 | 94% Betacoronavirus H.sav/J/Spain/2007 |
| 206645-63/2011 | Kuhl’s pipistrelle | Carcass * | nd | nd | Bat-CoV/Pipistrellus khulii/Italy/206645-63/2011 | β 2c | KF500948 | 94% Betacoronavirus H.sav/J/Spain/2007 |
| 143488/2011 | Pipistrelle bat | Faeces | nd | nd | Bat-CoV/Rhinolophus hipposideros/Italy/143488/2011 | β 2b ˜ | NA | |
| 49967-19/2010 | Pipistrelle bat | Carcass ° | nd | Juvenile | Bat-CoV/Pipistrellus spp/Italy/49967-19/2010 | β 2c | KF500951 | 94% Betacoronavirus H.sav/J/Spain/2007 |
| 187632-2/2012 | Lesser horseshoe bat | Faeces | F | nd | Bat-CoV/Rhinolophus hipposideros/Italy/187632-2/2012 | β 2b | KF500952 | 97% Bat coronavirus SLO1A0082/2008/SVN |
| 243585/2012 | Lesser horseshoe bat | Faeces | F | nd | Bat-CoV/Rhinolophus hipposideros/Italy/243585/2012 | β 2b | KF500953 | 97% Bat coronavirus SLO1A0082/2008/SVN |
| 196814/2011 | Lesser horseshoe bat | Faeces | nd | nd | Bat-CoV/Rhinolophus hipposideros/Italy/196814/2011 | β 2b | KF500954 | 97% Bat coronavirus SLO1A0082/2008/SVN |
| 330375-15/2012 | Kuhl’s pipistrelle | Faeces | M | Adult | Bat-CoV/Pipistrellus khulii/Italy/330375-15/2012 | β 2c | KF500950 | 94% Betacoronavirus H.sav/J/Spain/2007 |

3. Experimental
3.1. Sampling
3.2. Genome Detection and Sequencing
3.3. Virus Isolation
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Woo, P.C.; Lau, S.K.; Huang, Y.; Yuen, K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef]
- De Groot, R.J.; Baker, S.C.; Baric, R.; Enjuanes, L.; Gorbalenya, A.; Holmes, K.V.; Perlman, S.; Poon, L.; Rottier, P.J.; Talbot, P.J.; et al. Coronaviridae. In Virus Taxonomy, Classification and Nomenclature of Viruses; Elsevier Academic Press: Philadelphia, PA, USA, 2011; pp. 806–828. [Google Scholar]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wnag, M.; et al. Discovery of seven novel mammalian and avian coronaviruses in the genus Deltacoronavirus supports bat coronaviruses as the gene source of Alphacoronavirus and Betacoronavirus and avian coronaviruses as the gene source of Gammacoronavirus and Deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef]
- Decaro, N.; Buonavoglia, C. An update on canine coronaviruses: Viral evolution and pathobiology. Vet. Microbiol. 2008, 132, 221–234. [Google Scholar] [CrossRef]
- Decaro, N.; Buonavoglia, C. Canine coronavirus: Not only an enteric pathogen. Vet. Clin. Small Anim. 2008, 41, 1121–1132. [Google Scholar] [CrossRef]
- Cheng, V.C.; Lau, S.K.; Woo, P.C.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus as an agent of emerging and reemerging infection. Clin. Microbiol. Rev. 2007, 20, 660–694. [Google Scholar] [CrossRef]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Tsoi, H.W.; Wong, B.H.; Wong, S.S.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Poon, L.L.; Chu, D.K.; Chan, K.H.; Wong, O.K.; Ellis, T.N.; Leung, Y.H.; Lau, S.K.; Woo, P.C.; Suen, K.Y.; Yuen, K.Y.; et al. Identification of a novel coronavirus in bats. J. Virol. 2005, 79, 2001–2009. [Google Scholar] [CrossRef]
- Tang, X.C.; Zhang, J.X.; Zhang, S.Y.; Wang, P.; Fan, X.H.; Li, L.F.; Li, G.; Dong, B.Q.; Liu, W.; Cheung, C.L.; et al. Prevalence and genetic diversity of coronaviruses in bats from China. J. Virol. 2006, 80, 7481–7490. [Google Scholar] [CrossRef]
- Carrington, C.V.; Foster, J.E.; Zhu, H.C.; Zhang, J.X.; Smith, G.J.; Thompson, N.; Auguste, A.J.; Ramkissoon, V.; Adesiyun, A.A.; Guan, Y. Detection and phylogenetic analysisof group 1 coronaviruses in South American bats. Emerg. Infect. Dis. 2008, 14, 1890–1893. [Google Scholar] [CrossRef]
- Chu, D.K.; Peiris, J.S.; Chen, H.; Guan, Y.; Poon, L.L. Genomic characterizations of batcoronaviruses (1A, 1B and HKU8) and evidence for co-infections in Miniopterus bats. J. Gen. Virol. 2008, 89, 1282–1287. [Google Scholar] [CrossRef]
- Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Gottsche, M.; Panning, M.; Drexler, F.J.; Petersen, N.; Annan, A.; Grywna, K.; Muller, M.; et al. Detection and prevalencepatterns of group I coronaviruses in bats, Northern Germany. Emerg. Infect. Dis. 2008, 14, 626–631. [Google Scholar] [CrossRef]
- Reusken, C.B.; Lina, P.H.; Pielaat, A.; de Vries, A.; Dam-Deisz, C.; Adema, J.; Drexler, J.F.; Drosten, C.; Kooi, E.A. Circulation of group 2 coronaviruses in a bat speciescommon to urban areas in Western Europe. Vector Borne Zoonotic Dis. 2010, 10, 785–791. [Google Scholar]
- Chan, J.F.; To, K.K.; Tse, H.; Jin, D.Y.; Yuen, K.Y. Interspecies transmission and emergence of novel viruses: Lessons from bats and birds. Trends Microbiol. 2013, 21, 544–555. [Google Scholar] [CrossRef]
- Falcón, A.; Vázquez-Morón, S.; Casas, I.; Aznar, C.; Ruiz, G.; Pozo, F.; Perez-Breña, P.; Juste, J.; Ibáñez, C.; Garin, I.; et al. Detection of alpha and betacoronaviruses in multiple Iberian bat species. Arch. Virol. 2011, 156, 1883–1890. [Google Scholar] [CrossRef]
- Annan, A.; Baldwin, H.J.; Corman, V.M.; Klose, S.M.; Owusu, M.; Nkrumah, E.E.; Badu, E.K.; Anti, P.; Agbenyega, O.; Meyer, B.; et al. Human betacoronavirus 2c EMC/2012-related viruses in bats, Ghana and Europe. Emerg. Infect. Dis. 2013, 19, 456–459. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar]
- Chan, J.F.; Li, K.S.; To, K.K.; Cheng, V.C.; Chen, H.; Yuen, K.Y. Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoV-EMC) the beginning of another SARS-like pandemic? J. Infect. 2012, 65, 477–489. [Google Scholar] [CrossRef]
- Woo, P.C.; Lau, S.K.; Li, K.S.; Tsang, A.K.; Yuen, K.Y. Genetic relatedness of the novel human group C betacoronavirus to Tylonycteris bat coronavirus HKU4 and Pipistrellus bat coronavirus HKU5. Emerg. Microbes Infect. 2012, 1, e35. [Google Scholar] [CrossRef]
- Reusken, C.B.; Bart, L.; Haagmans, B.L.; Müller, M.A.; Gutierrez, C.; Godeke, G.; Meyer, B.; Muth, D.; Raj, V.S.; Smits-De Vries, L.; et al. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: A comparative serological study. Lancet Infect. Dis. 2013, 13, 859–866. [Google Scholar] [CrossRef]
- Drexler, J.F.; Gloza-Rausch, F.; Glende, J.; Corman, V.M.; Muth, D.; Goettsche, M. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol. 2010, 84, 11336–11349. [Google Scholar] [CrossRef]
- Rihtaric, D.; Hostnik, P.; Steyer, A.; Grom, J.; Toplak, I. Identification of SARS-like coronaviruses in horseshoe bats (Rhinolophus hipposideros) in Slovenia. Arch. Virol. 2010, 155, 507–514. [Google Scholar] [CrossRef]
- Balboni, A.; Gallina, L.; Palladini, A.; Prosperi, S.; Battilani, M. A real-time PCR assay for bat SARS-like coronavirus detection and its application to Italian greater horseshoe bat faecal sample surveys. Scientific World J. 2012, e989514. [Google Scholar]
- Dietz, C.; von Helversen, O. Illustrated Identification Key to the Bats of Europe. Available online: http://biocenosi.dipbsf.uninsubria.it/didattica/bat_key1.pdf (accessed on 30 October 2013).
- Dominguez, S.R.; O’Shea, T.J.; Oko, L.M.; Holmes, K.V. Detection of group 1 coronavirus in bats in north America. Emerg. Infect. Dis. 2007, 13, e9. [Google Scholar]
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important reservoir hosts of emerging virus. Clin. Microbiol. 2006, 19, 531–545. [Google Scholar] [CrossRef]
- Pfefferle, S.; Oppong, S.; Drexler, J.F.; Gloza-Rausch, F.; Ipsen, A.; Seebens, A. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg. Infect. Dis. 2009, 15, 1377–1384. [Google Scholar] [CrossRef]
- Lau, S.K.; Li, K.S.; Tsang, A.K.; Lam, C.S.; Ahmed, S.; Chen, H.; Chan, K.H.; Woo, P.C.; Yuen, K.Y. Genetic characterization of betacoronavirus lineage C viruses in bats reveals marked sequence divergence in the spike protein of pipistrellus bat coronavirus HKU5 in Japanese pipistrelle: Implications for the origin of the novel Middle East respiratory syndrome coronavirus. J. Virol. 2013, 87, 8638–8650. [Google Scholar]
- Medellín, R.A. Sustaining transboundary ecosystem services provided by bats. In Conservation of Shared Environments: Learning from the United States and Mexico; López-Hoffman, L., McGovern, E., Varady, R., Flessa, K., Eds.; University of Arizona Press: Tucson, AZ, USA, 2009; pp. 170–187. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lelli, D.; Papetti, A.; Sabelli, C.; Rosti, E.; Moreno, A.; Boniotti, M.B. Detection of Coronaviruses in Bats of Various Species in Italy. Viruses 2013, 5, 2679-2689. https://doi.org/10.3390/v5112679
Lelli D, Papetti A, Sabelli C, Rosti E, Moreno A, Boniotti MB. Detection of Coronaviruses in Bats of Various Species in Italy. Viruses. 2013; 5(11):2679-2689. https://doi.org/10.3390/v5112679
Chicago/Turabian StyleLelli, Davide, Alice Papetti, Cristiano Sabelli, Enrica Rosti, Ana Moreno, and Maria B. Boniotti. 2013. "Detection of Coronaviruses in Bats of Various Species in Italy" Viruses 5, no. 11: 2679-2689. https://doi.org/10.3390/v5112679
APA StyleLelli, D., Papetti, A., Sabelli, C., Rosti, E., Moreno, A., & Boniotti, M. B. (2013). Detection of Coronaviruses in Bats of Various Species in Italy. Viruses, 5(11), 2679-2689. https://doi.org/10.3390/v5112679