2.1. Sequence Analyses and Open Reading Frames
Preliminary analysis of nucleotide sequences from a replicase 189 bp amplicon placed the two novel strains, DL52 and DL54, into a “JS-like” subgroup [
4]. Reverse-line blot hybridization failed to genotype the two strains into genogroups I or II [
4].
A total of 17 strains (
Table 1) were used to examine the relationships among nucleotides and amino acids in the
Levivirus genus. The first 9 strains in genogroup I,
Table 1,
i.e., MS2, ST4, DL1, DL2, DL13, DL16, R17, M12 and J20, were referred to as “MS2-like.”
Genogroup I MS2-like strains Open Reading Frame (ORF) start and stop codons were located at identical or very similar nucleotide positions as previously reported for strain MS2. The JS strains also had identical ORF start and stop codon positions as the MS-2 like strains (
Table 2).
Nucleotide pairwise comparisons of full-length genomes were made between all strains within the
Levivirus genome, including strains within genogroups I, JS and genogroups II. Within the nine strains of MS2-like genogroup I, full-length nucleotide sequence similarity was 91–99% [
3] whereas the two JS strains, DL52 and DL54, shared 96.73% sequence similarity to each other. In comparison, the JS nucleotide sequences were more similar to MS2-like genogroup I (80–85%) than to the genogroup I strain fr (69%) or to genogroup II strains (52–54%) (
Table 3 a).
Table 1.
Male-specific ssRNA coliphages (FRNA), family Leviviridae, genus Levivirus, strain origins and identifications.
Table 1.
Male-specific ssRNA coliphages (FRNA), family Leviviridae, genus Levivirus, strain origins and identifications.
| Strain | Genogroup | Source | Origin | Accession number |
|---|
| MS2 | I | sewage | Berkeley, CA | NC_001417 |
| M12 | I | sewage | Germany | AF195778 |
| DL1 | I | river water | Tijuana River, CA | EF107159 |
| DL2 | I | bay water | Delaware Bay, DE | N/A |
| DL13 | I | oyster | Whiskey Creek, NC | N/A |
| DL16 | I | bay water | Great Bay, NH | EF108464 |
| J20 | I | chicken litter | South Carolina | EF204939 |
| ST4 | I | unknown | unknown | EF204940 |
| R17 | I | sewage | Philadelphia, PA | EF108465 |
| fr | I | dung hill | Heidelberg, Germany | X15031 |
| DL52 | I-JS | bay water | Rachel Carson Reserve, NC | JQ966307 |
| DL54 | I-JS | bay water | Narragansett Bay, RI | JQ966308 |
| GA | II | sewage | Ookayama, Japan | NC_001426 |
| KU1 | II | sewage | Kuwait | AF227250 |
| DL10 | II | mussel | Tijuana River, CA | FJ483837 |
| DL20 | II | clam | Narragansett Bay, RI | FJ483839 |
| T72 | II | bird | Talbert Marsh sandflats, CA | FJ483838 |
Table 2.
Open Reading Frame positions and genome lengths of FRNA coliphage (family
Leviviridae, genus
Levivirus). Nucleotide positions are based on alignment. Number of amino acids for each gene is in parentheses [
3].
Table 2.
Open Reading Frame positions and genome lengths of FRNA coliphage (family Leviviridae, genus Levivirus). Nucleotide positions are based on alignment. Number of amino acids for each gene is in parentheses [3].
| Open Reading Frame Locations (amino acids) |
|---|
| Strain | Group | Full length | ORF1 | ORF2 | ORF3 | ORF4 |
|---|
| MS2 a | I | 3569 | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| M12 a,b | I | 3340b | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | ND |
| DL1 | I | 3570 | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| DL2 | I | 3491c | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| DL13 | I | 3491c | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| DL16 | I | 3569 | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| J20 | I | 3569 | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| ST4 | I | 3569 | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| R17 | I | 3569 | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398(545) |
| fr a | I | 3575 | 129-1310(393) | 1336-1728(130) | 1691-1906(71) | 1762-3399(545) |
| DL52 | JS | 3525 | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398 d (545) |
| DL54 | JS | 3398 c | 130-1311(393) | 1335-1727(130) | 1678-1905(75) | 1761-3398 d (545) |
Table 3.
(a) Pairwise nucleotide full-length genome percent similarity. (i) Levivirus JS strains DL52 and DL54 compared to genogroup I; (ii) Levivirus JS strains DL52 and DL54 compared to genogroup II. Pairwise alignments were performed in BioEdit with DAYHOFF similarity parameters.
Table 3.
(a) Pairwise nucleotide full-length genome percent similarity. (i) Levivirus JS strains DL52 and DL54 compared to genogroup I; (ii) Levivirus JS strains DL52 and DL54 compared to genogroup II. Pairwise alignments were performed in BioEdit with DAYHOFF similarity parameters.
| (i) Genogroup I and JS strains |
|---|
| Strain | DL52 | DL54 |
|---|
| DL52 | 100 | |
| DL54 | 96.73 | 100 |
| DL1 | 81.48 | 81.87 |
| DL16 | 85.41 | 84.72 |
| ST4 | 80.30 | 80.11 |
| R17 | 80.55 | 80.53 |
| J20 | 82.00 | 82.01 |
| MS2 | 80.12 | 80.01 |
| fr | 69.18 | 69.06 |
| (ii) Genogroup II and JS strains |
| Strain | DL52 | DL54 |
| DL52 | 100 | |
| DL54 | 96.73 | 100 |
| T72 | 53.96 | 53.53 |
| DL10 | 54.07 | 53.89 |
| DL20 | 52.87 | 52.65 |
| GA | 52.44 | 52.29 |
| KU1 | 52.94 | 52.66 |
Despite their sequence similarities, genome lengths for JS strains (3525 nt) were shorter than all genogroup I strains (3569–3575 nt) (
Table 2) but longer than genogroup II (3458–3486 nt) [
3]. Numerous deletions in the 3' untranslated region and a portion of ORF4 (replicase) in JS strains accounted for the decreased genome length (data not shown) but did not alter the ORF positions when the genogroup I strains were aligned (
Table 2).
Analysis of the replicase gene revealed a 2 nt insertion at the 1374 nucleotide region when counting ORF4 start site as nucleotide 1 (
Figure 1). This insertion occurred upstream from the ORF4 stop codon. Beginning approximately 40 nt downstream from the replicase ORF4 stop codon and continuing to the 3' termini, 53 nt deletions were present in the JS strains when aligned to MS2-like genomes. Nucleotide alignment of the replicase and nontranslated regions (NTR) revealed numerous nt deletions in the JS strains when compared to genogroup I strains accounting for the change in amino acid composition. However, JS strains shared the 3' terminal “signature”, ACCACCCA, present in
Levivirus genogroups I and II [
3].
Figure 1.
Replicase recombinant region in two JS strains when compared to genogroup I strains (family Leviviridae). Alignment (BioEdit v7.0.1) of the replicase nucleotide sequences from Levivirus genogroup I strains DL1, DL2, DL13, DL16, ST4, R17, J20, MS2 with JS strains DL52 and DL54. For clarity, only a portion of the alignment is shown. Alignment of each genogroup is depicted in discontinuous blocks. The numbers along the top are the nucleotide positions within the replicase gene with the start position of ORF4 assigned as nucleotide 1. Genome sequences read 5'-3' direction. Dots indicate identity with the consensus sequence. Degenerate bases are noted in the standard IUB codes. The replicase start codon and two nucleotide insertions are highlighted in red. Dashes denote a nucleotide sequence deletion from the consensus sequence.
Figure 1.
Replicase recombinant region in two JS strains when compared to genogroup I strains (family Leviviridae). Alignment (BioEdit v7.0.1) of the replicase nucleotide sequences from Levivirus genogroup I strains DL1, DL2, DL13, DL16, ST4, R17, J20, MS2 with JS strains DL52 and DL54. For clarity, only a portion of the alignment is shown. Alignment of each genogroup is depicted in discontinuous blocks. The numbers along the top are the nucleotide positions within the replicase gene with the start position of ORF4 assigned as nucleotide 1. Genome sequences read 5'-3' direction. Dots indicate identity with the consensus sequence. Degenerate bases are noted in the standard IUB codes. The replicase start codon and two nucleotide insertions are highlighted in red. Dashes denote a nucleotide sequence deletion from the consensus sequence.
2.2. Amino Acid Analysis
Initially, nucleotide pairwise analyses of full-length genomes were made comparing all strains within the
Levivirus genome, including genogroups I, JS and II; an 80–85% nucleotide similarity between JS strains and the MS2-like strains was observed (
Table 3 a). In comparison, the amino acid sequences of the maturation, capsid and lysis proteins of the JS strains were very similar to those of the MS2-like genogroup I strains, sharing 97–100%, 98–100% and 95–100% sequence similarities, respectively (
Table 3 b). Genogroup I strain fr, when compared to MS2-like and JS genogroup I strains, only shared an amino acid similarity to the maturation, capsid and lysis proteins ranging from 75.73–91.85% (
Table 3 b). In contrast, the replicase protein sequences of the JS strains were quite dissimilar to the replicase protein sequences of the MS2-like genogroup I strains, displaying a similarity range of 79–85% (
Table 3 c). However, a similarity of 97–99% was observed among the highly conserved replicase genes for the MS2-like strains. Strain fr shared a 79% replicase similarity to JS strains and approximately 88–89% similarity to MS2-like strains. Genogroup II replicase was approximately 52–53% similar to JS strains, 50–53% to MS2-like and fr strains and 92–98% similar to other genogroup II strains (
Table 3 c).
Table 3.
(b) Percent similarity in amino acid sequences between Levivirus JS strains and genogroup I maturation, capsid and lysis proteins. Amino acid pairwise computations were performed in Bionumerics.
Table 3.
(c) Amino acid percent similarity comparisons between Levivirus JS strains, DL52 and DL54, to Levivirus genogroup I and genogroup II RNA-dependent RNA polymerase (replicase) protein. Amino acid pairwise computations were performed in Bionumerics.
Table 3.
(c) Amino acid percent similarity comparisons between Levivirus JS strains, DL52 and DL54, to Levivirus genogroup I and genogroup II RNA-dependent RNA polymerase (replicase) protein. Amino acid pairwise computations were performed in Bionumerics.
| Replicase Protein |
|---|
![Viruses 04 01548 i004]() |
All genogroup I strains, including fr, and the two JS strains had a replicase protein length of 545 amino acids (
Table 2) [
3]. However, JS replicase differed from genogroup I replicase as it had one amino acid insertion at replicase position 467 and one amino acid deletion at the 3' termini of the stop codon, but maintained a total of 545 amino acids (data not shown). Identical to genogroup I strains, the replicase catalytic domain in the JS strains occurred between amino acid positions 243–373, thereby adding confidence to placing the grouping of JS into genogroup I [
3]. Beginning at amino acid number 455 within the replicase gene, JS strains were unique in amino acid composition and diverged from the MS2-like strains.
2.4. Phylogenetic and Recombination Analyses
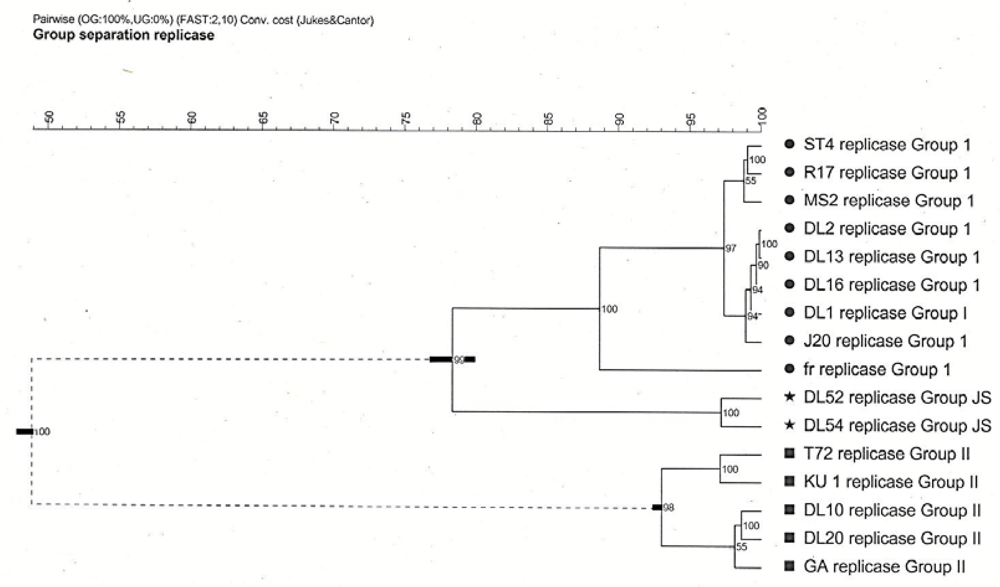
Cophenetic correlations showed the genogroup I strains, the JS subgroup strains, and the genogroup II strains all formed faithful clusters with correlations of 100, 90 and 98, respectively. The cluster cutoff method, however, showed only two relevant clusters being the genogroup I strains, which included fr and JS, and genogroup II strains (
Figure 2).
Figure 2.
Cophenetic cluster analysis of Levivirus (family Leviviridae) genogroups I and II strains generated from pairwise similarities of the replicase amino acid sequences. Horizontal bars at three of the branches show the standard deviations of the average similarities of the clusters. Numbers at each branch are the cophenetic correlations which represent the faithfulness of the clusters. Two relevant clusters, as determined by the cluster Cutoff method, are grouped as dictated by the dashed lines. Analysis performed in Bionumerics.
Figure 2.
Cophenetic cluster analysis of Levivirus (family Leviviridae) genogroups I and II strains generated from pairwise similarities of the replicase amino acid sequences. Horizontal bars at three of the branches show the standard deviations of the average similarities of the clusters. Numbers at each branch are the cophenetic correlations which represent the faithfulness of the clusters. Two relevant clusters, as determined by the cluster Cutoff method, are grouped as dictated by the dashed lines. Analysis performed in Bionumerics.
When referring to nucleotide or amino acid positions within the replicase gene, the numbering is in reference to the start codon as being position 1. In all analysis programs, the nucleotide or amino acid sequences were aligned to other strains and were therefore approximate positions on the replicase gene.
All recombination programs used, SimPlot, RAT, RDP3 and Recco, statistically predicted recombination in both JS strains, DL52 and DL54, when compared to genogroup I MS2-like strains. No recombination, however, was detected when DL52 and DL54 were compared to genogroup I strain fr and all genogroup II strains.
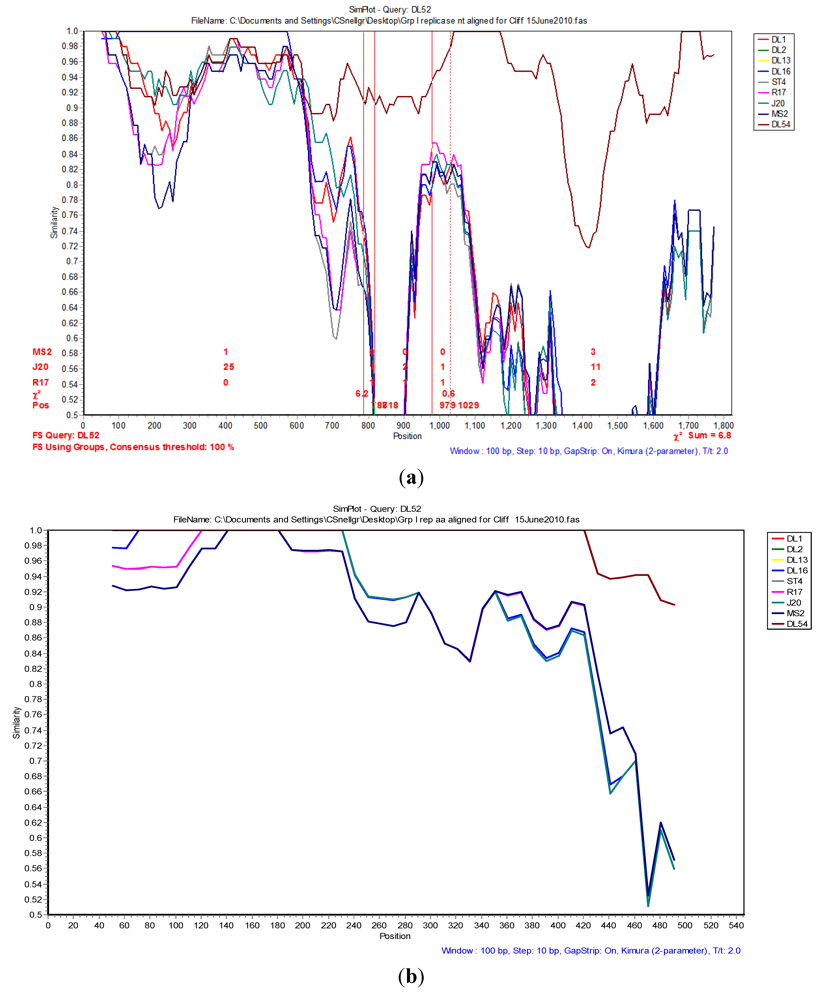
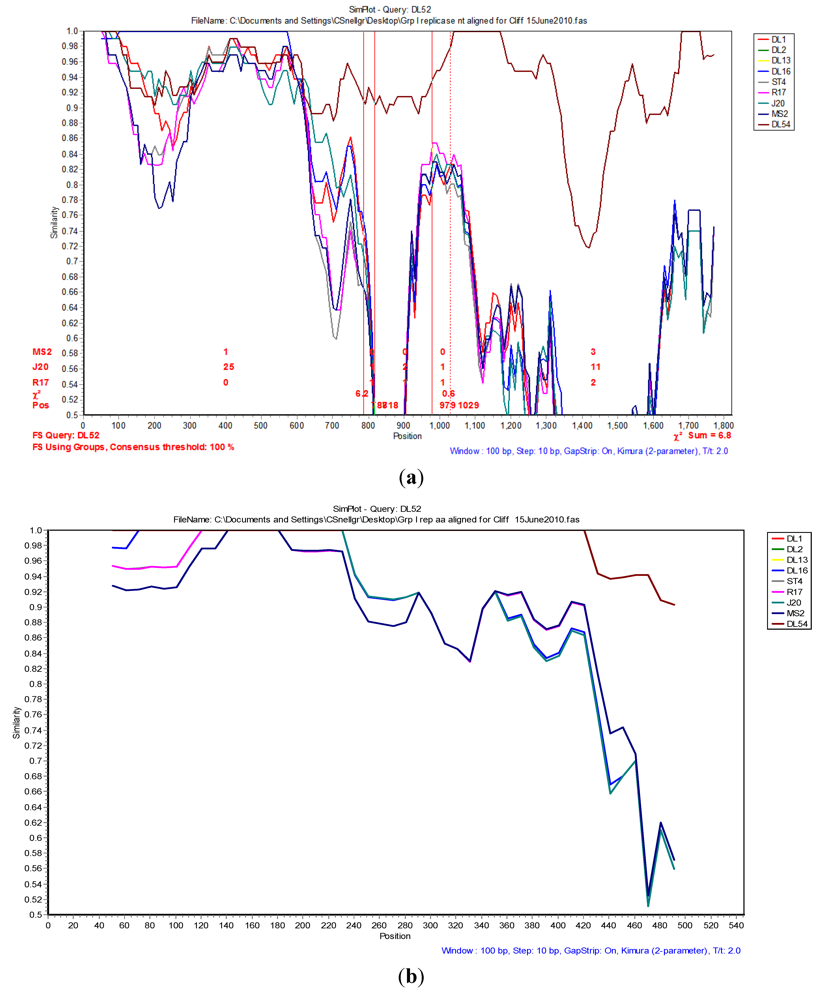
The Simplot and bootscan analyses of the replicase nucleotides from JS strains DL52 compared to
Levivirus genogroup I strains DL54, DL1, DL3, DL13, DL16, ST4, R17, J20 and MS2 is shown in
Figure 3A. Since the replicase nucleotide sequences in strain DL54 were 97% similar to strain DL52, DL52 was chosen as the query. The SimPlot analysis revealed the first recombination breakpoint occurred in the replicase from strain DL52 at nt positions 787–818 (approximate amino acid 262–273) where the χ
2 changes from 0.8 to 6.3 (sum χ
2 of 7.1). The second breakpoint occurred at nt positions 979–1029 (approximate amino acid 326–343) where the χ
2 changes from 0.6 to 7.0 (sum χ
2 of 7.6). However, Simplot amino acid analysis (
Figure 3 b) with strain DL52 showed a divergence at approximate amino acid position 460 region which is in agreement with the manual alignment (
Figure 1).
Figure 3.
(a) The Simplot and bootscan analyses of the replicase nucleotides from JS strain DL52 queried to DL54, DL1, DL3, DL13, DL16, ST4, R17, J20 and MS2. The breakpoints are shown by the vertical red lines. The first recombination breakpoint occurred in the replicase gene in strain DL52 at nucleotide positions 787–818 where the χ2 changed from 0.8 to 6.3 (sum χ2 of 7.1). The second breakpoint occurred at nucleotide positions 979–1029 where the χ2 changed from 0.6 to 7.0 (sum χ2 of 7.6); (b) The Simplot and bootscan analyses of the replicase amino acids from JS strain DL52 queried to DL54, DL1, DL3, DL13, DL16, ST4, R17, J20 and MS2.
Figure 3.
(a) The Simplot and bootscan analyses of the replicase nucleotides from JS strain DL52 queried to DL54, DL1, DL3, DL13, DL16, ST4, R17, J20 and MS2. The breakpoints are shown by the vertical red lines. The first recombination breakpoint occurred in the replicase gene in strain DL52 at nucleotide positions 787–818 where the χ2 changed from 0.8 to 6.3 (sum χ2 of 7.1). The second breakpoint occurred at nucleotide positions 979–1029 where the χ2 changed from 0.6 to 7.0 (sum χ2 of 7.6); (b) The Simplot and bootscan analyses of the replicase amino acids from JS strain DL52 queried to DL54, DL1, DL3, DL13, DL16, ST4, R17, J20 and MS2.
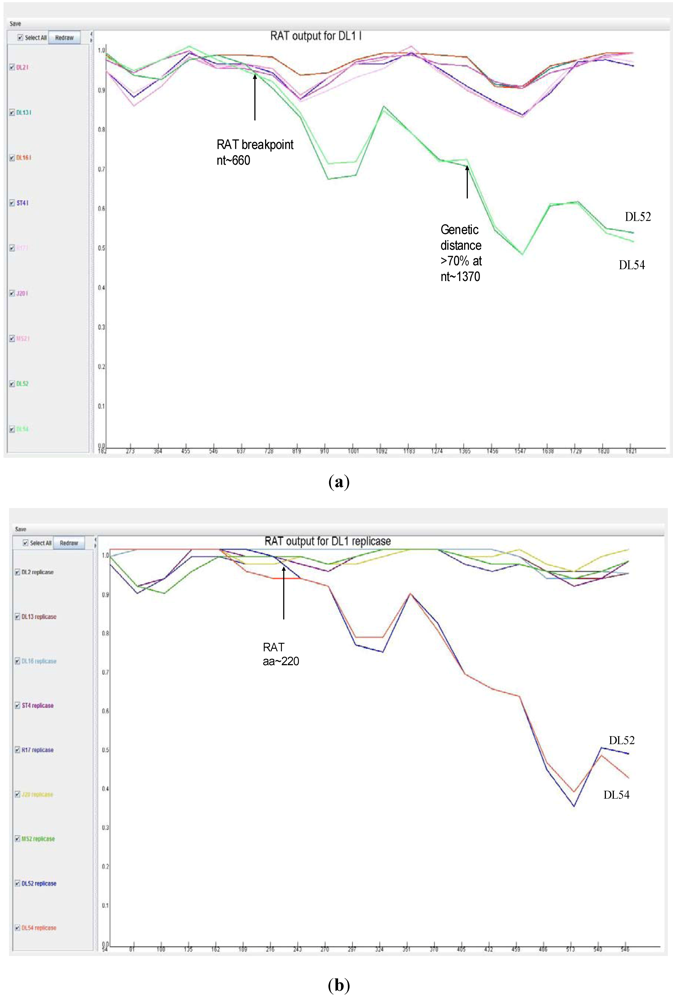
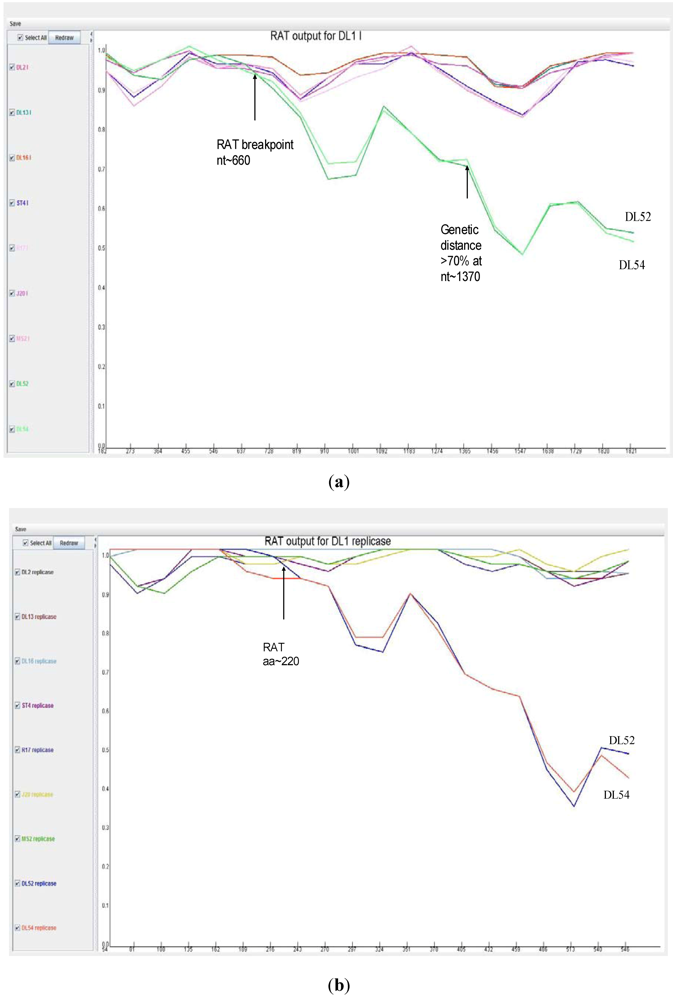
When analyzed with RAT, the nucleotide breakpoint (crossover) positions occurred at approximately nt 660 (
Figure 4 a) or amino acid 220 (
Figure 4 b) within the replicase gene. This crossover occurred when the two recombinant strains, DL52 and DL54, crossed the lines of the other MS-2 like strains and diverged by increasing genetic distances.
Figure 4.
Recombination analysis of the replicase nucleotide sequences from Leviviridae genogroup I strains DL13, DL16, ST4, R17, J20, MS2 and JS strains DL54, DL52 queried to DL1. Recombination Analysis Tool (RAT) was used to generate graphics with a window of 182 nt and step increments of 92 nt. The Y-axis represents the genetic distance and the X-axis is the sequence location along the genome. (a) The JS strains, depicted in green, diverged from the other genogroup I strains at approximate nucleotide (nt) position 660; (b) Recombination analysis of the replicase amino acid sequences from Leviviridae genogroup I strains DL13, DL16, ST4, R17, J20, MS2 and JS strains DL54, DL52 queried to DL1. Recombination Analysis Tool (RAT) was used to generate graphics with a window of 54 aa and step increments of 27 aa. The JS strains, DL52 and DL54, diverged from the other genogroup I strains at approximate amino acid 220 within the replicase gene.
Figure 4.
Recombination analysis of the replicase nucleotide sequences from Leviviridae genogroup I strains DL13, DL16, ST4, R17, J20, MS2 and JS strains DL54, DL52 queried to DL1. Recombination Analysis Tool (RAT) was used to generate graphics with a window of 182 nt and step increments of 92 nt. The Y-axis represents the genetic distance and the X-axis is the sequence location along the genome. (a) The JS strains, depicted in green, diverged from the other genogroup I strains at approximate nucleotide (nt) position 660; (b) Recombination analysis of the replicase amino acid sequences from Leviviridae genogroup I strains DL13, DL16, ST4, R17, J20, MS2 and JS strains DL54, DL52 queried to DL1. Recombination Analysis Tool (RAT) was used to generate graphics with a window of 54 aa and step increments of 27 aa. The JS strains, DL52 and DL54, diverged from the other genogroup I strains at approximate amino acid 220 within the replicase gene.
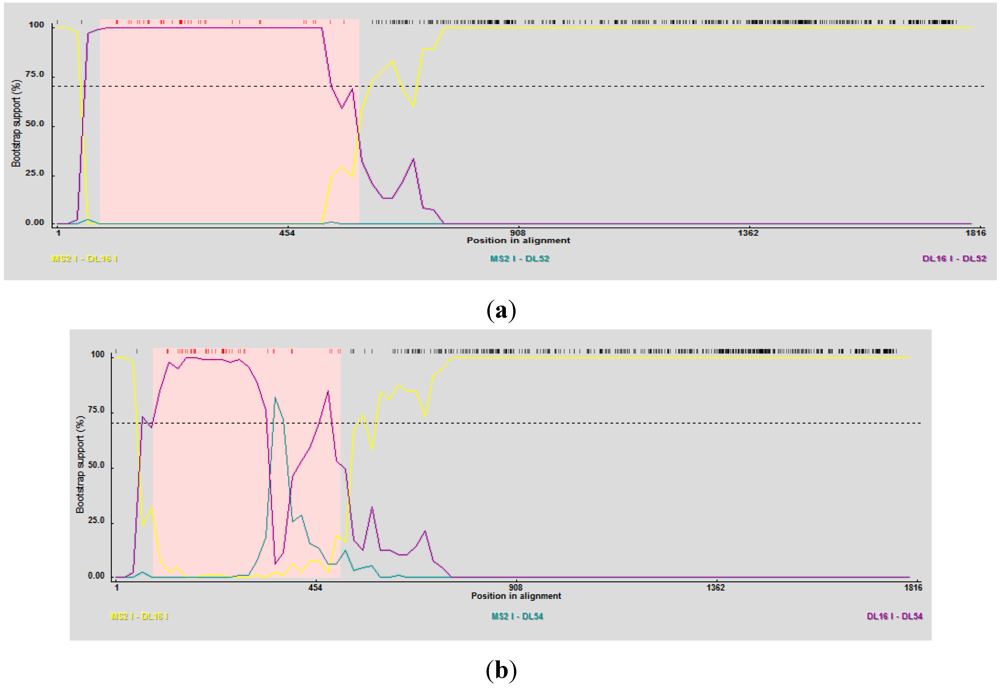
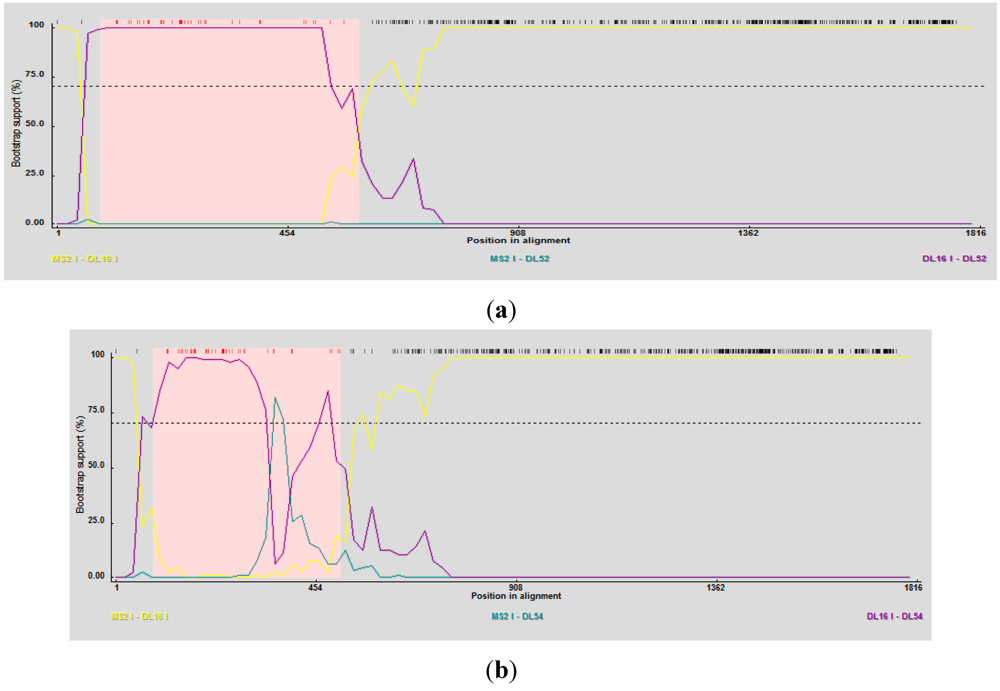
RDP3 predicted DL52 and DL54 as the recombinant strains using several detection methods and analysis algorithms (
Table 4) and suggested DL16 as a minor parent strain. Breakpoint nucleotides for strains DL52 and DL54 (when aligned to genogroup I strains) occurred between nt 84–592 and 84–401, respectively (
Figure 5 a, b), corresponding to the approximate amino acid breakpoint positions of 133–197 within the replicase gene.
Table 4.
Prediction of DL52 and DL54 as recombinant strains by analysis of Levivirus (family Leviviridae) genogroup I using Recombination Detection Program (RDP3).
Table 4.
Prediction of DL52 and DL54 as recombinant strains by analysis of Levivirus (family Leviviridae) genogroup I using Recombination Detection Program (RDP3).
| Confirmation Table of Recombination Events |
|---|
| Methods | Events | Average p-value |
|---|
| RDP | 2 | 2.199 × 10−15 |
| GENECONV | 1 | 3.031 × 10−27 |
| Bootscan | 2 | 7.867 × 10−19 |
| MaxChi | 2 | 1.445 × 10−10 |
| Chimaera | 2 | 3.536 × 10−11 |
| SiScan | 1 | 1.168 × 10−13 |
| 3Seq | 1 | 4.486 × 10−8 |
Figure 5.
(a) RDP3 analyses prediction of DL52 as a recombinant strain. Recombination area within the replicase gene is shown in pink beginning at nucleotide 84 and crossing over at 592, upstream from the catalytic domain. DL52 was queried to all Levivirus (family Leviviridae) genogroup I FRNA Levivirus strains. RDP3 suggested DL16 as the minor parental strain; (b) RDP3 analyses predicted DL54 as a recombinant strain. Recombination area within the replicase gene is shown in pink beginning at nucleotide 84 and crossing over at 401, upstream from the catalytic domain. RDP3 suggested DL16 as the minor parental strain.
Figure 5.
(a) RDP3 analyses prediction of DL52 as a recombinant strain. Recombination area within the replicase gene is shown in pink beginning at nucleotide 84 and crossing over at 592, upstream from the catalytic domain. DL52 was queried to all Levivirus (family Leviviridae) genogroup I FRNA Levivirus strains. RDP3 suggested DL16 as the minor parental strain; (b) RDP3 analyses predicted DL54 as a recombinant strain. Recombination area within the replicase gene is shown in pink beginning at nucleotide 84 and crossing over at 401, upstream from the catalytic domain. RDP3 suggested DL16 as the minor parental strain.
Manual alignment in BioEdit of the replicase nucleotides, counting the ATG start codon of the replicase gene as nt 1, showed an insertion of the nucleotides YA beginning at position 1374 (
Figure 1) whereas the amino acid composition of the JS strains diverged from the other genogroup I strains slightly upstream from this insertion at amino acid position 455 (nucleotide 1366). Alignment also revealed numerous nt deletions as discussed in the “Sequence analyses and ORF” section.
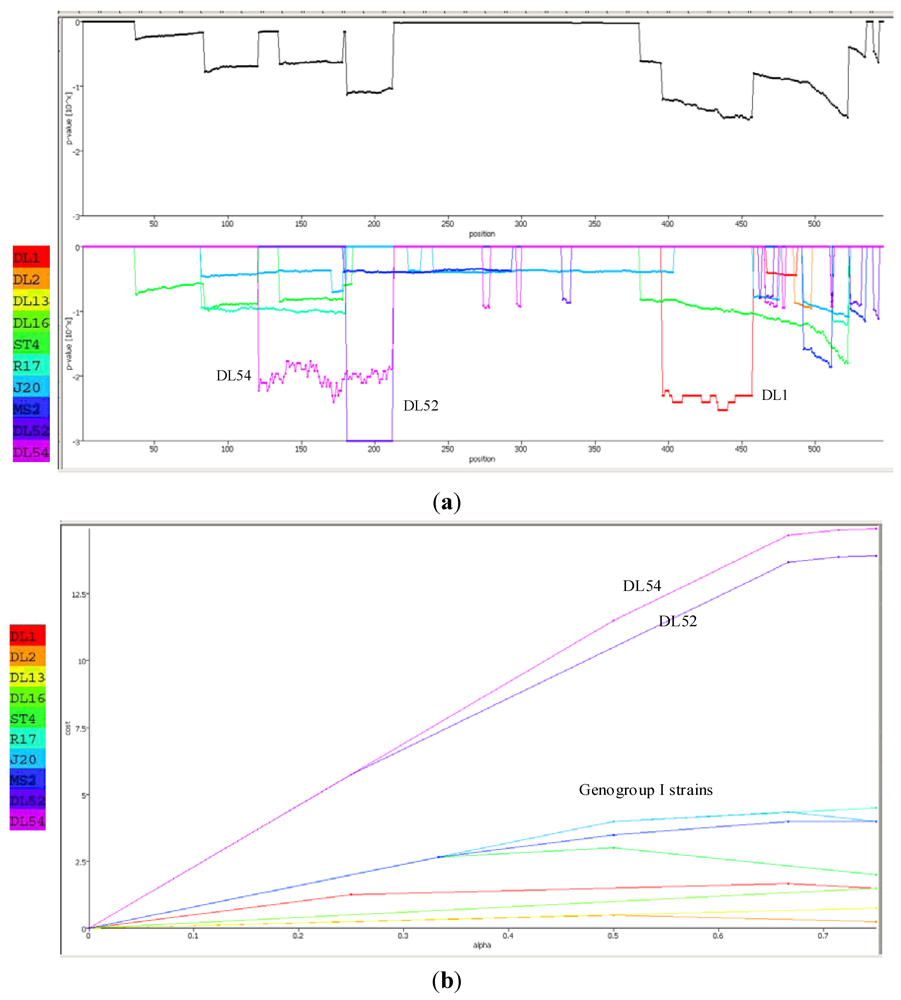
The Recco p-value inspector predicted strain DL52 had recombined with strain DL1 (
Figure 6a). In DL52, the recombinant region spanned from amino acids 181–212 whereas the DL1 region spanned from 396–457 with resulting sequence p-values of 0.000999 and 0.004995, respectively. Recco parametric cost curves predicted the highest preference for recombination in strains DL52 and DL54 (cost of 12.5–13) whereas the remaining genogroup I strains did not show a preference for recombination (cost of 0–3) (
Figure 6 b).
RAT, RDP3 and Recco all predicted recombination breakpoints ranging from amino acid positions 181–252 whereas Simplot agreed most closely with the manual alignment of 460 and 455, respectively. Also in agreement with the manual alignment was the crossover region between DL52 and DL1 occurring in the approximate amino acid region of 396–457 (
Figure 6 a). The predicted breakpoint regions occurred either upstream or downstream from the highly conserved catalytic domain amino acid positions 243–373 in
Levivirus genogroup I [
3].
Figure 6.
(a) Recco analysis of the RNA-dependent RNA polymerase (replicase) amino acid sequences in Levivirus genogroup I male-specific coliphages (FRNA). Recombination events are displayed by downward peaks in the graphics dataset. The upper graph represents the p-value for recombination at each position along the replicase gene. The lower graph is the breakpoint p-values for the entire set of Levivirus genogroup I and JS strains DL52 and DL54; (b) Recco parametric cost curve analysis of the RNA-dependent RNA polymerase (replicase) amino acid sequences for each FRNA strain in Levivirus genogroup I and JS strains DL52 and DL54. The y-axis corresponds to the cost curve and the x-axis represents α (0–1).
Figure 6.
(a) Recco analysis of the RNA-dependent RNA polymerase (replicase) amino acid sequences in Levivirus genogroup I male-specific coliphages (FRNA). Recombination events are displayed by downward peaks in the graphics dataset. The upper graph represents the p-value for recombination at each position along the replicase gene. The lower graph is the breakpoint p-values for the entire set of Levivirus genogroup I and JS strains DL52 and DL54; (b) Recco parametric cost curve analysis of the RNA-dependent RNA polymerase (replicase) amino acid sequences for each FRNA strain in Levivirus genogroup I and JS strains DL52 and DL54. The y-axis corresponds to the cost curve and the x-axis represents α (0–1).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}