A Systems Biology Starter Kit for Arenaviruses

Abstract
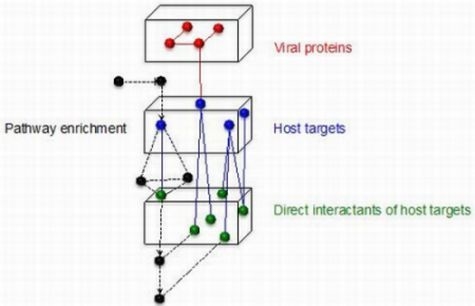
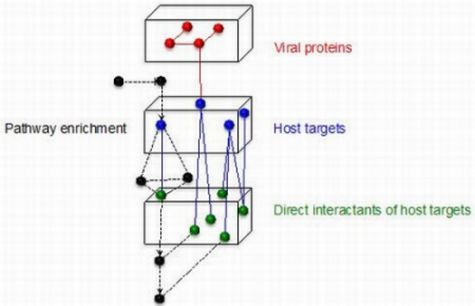
:
{kind=link}
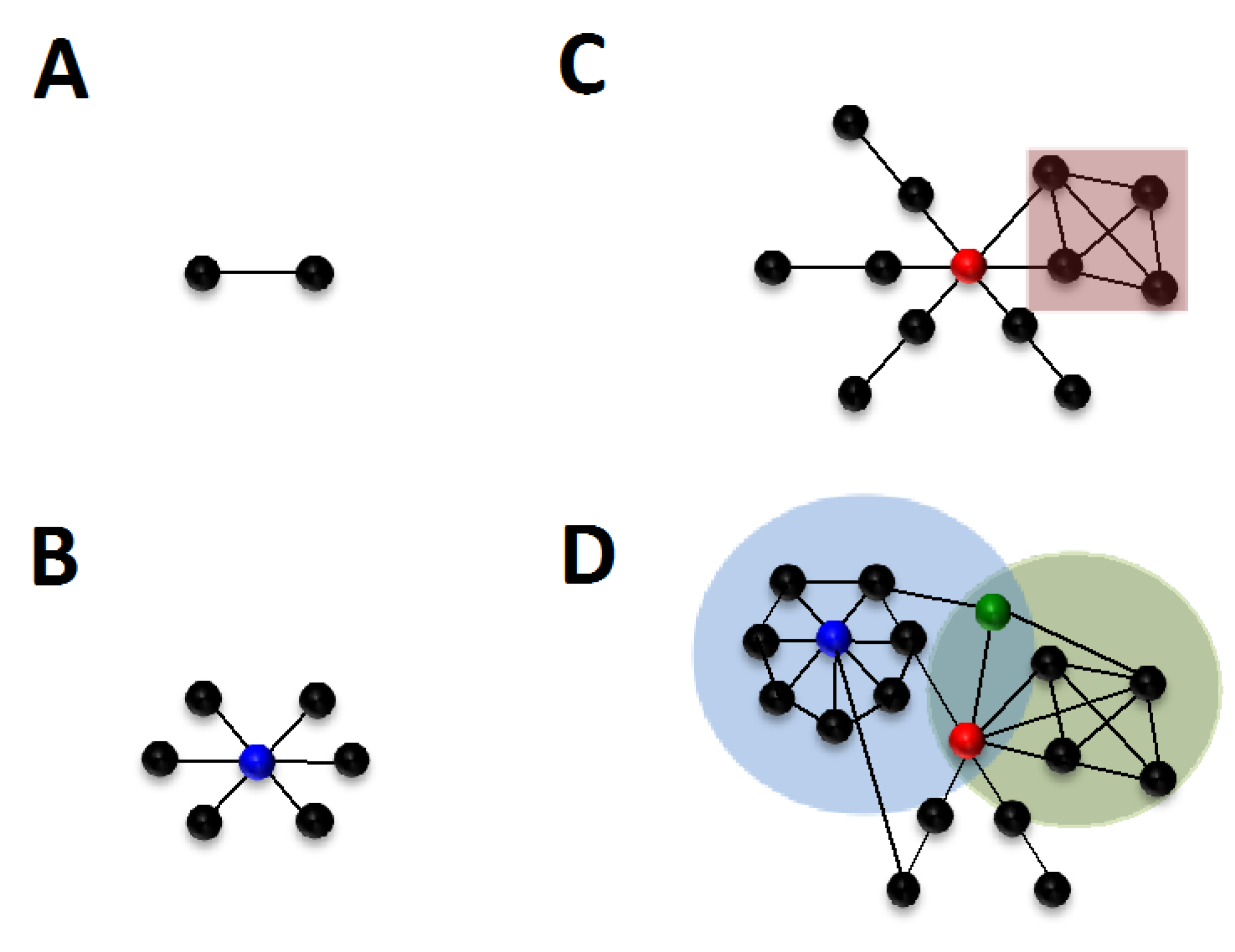{kind=link}
{kind=link}
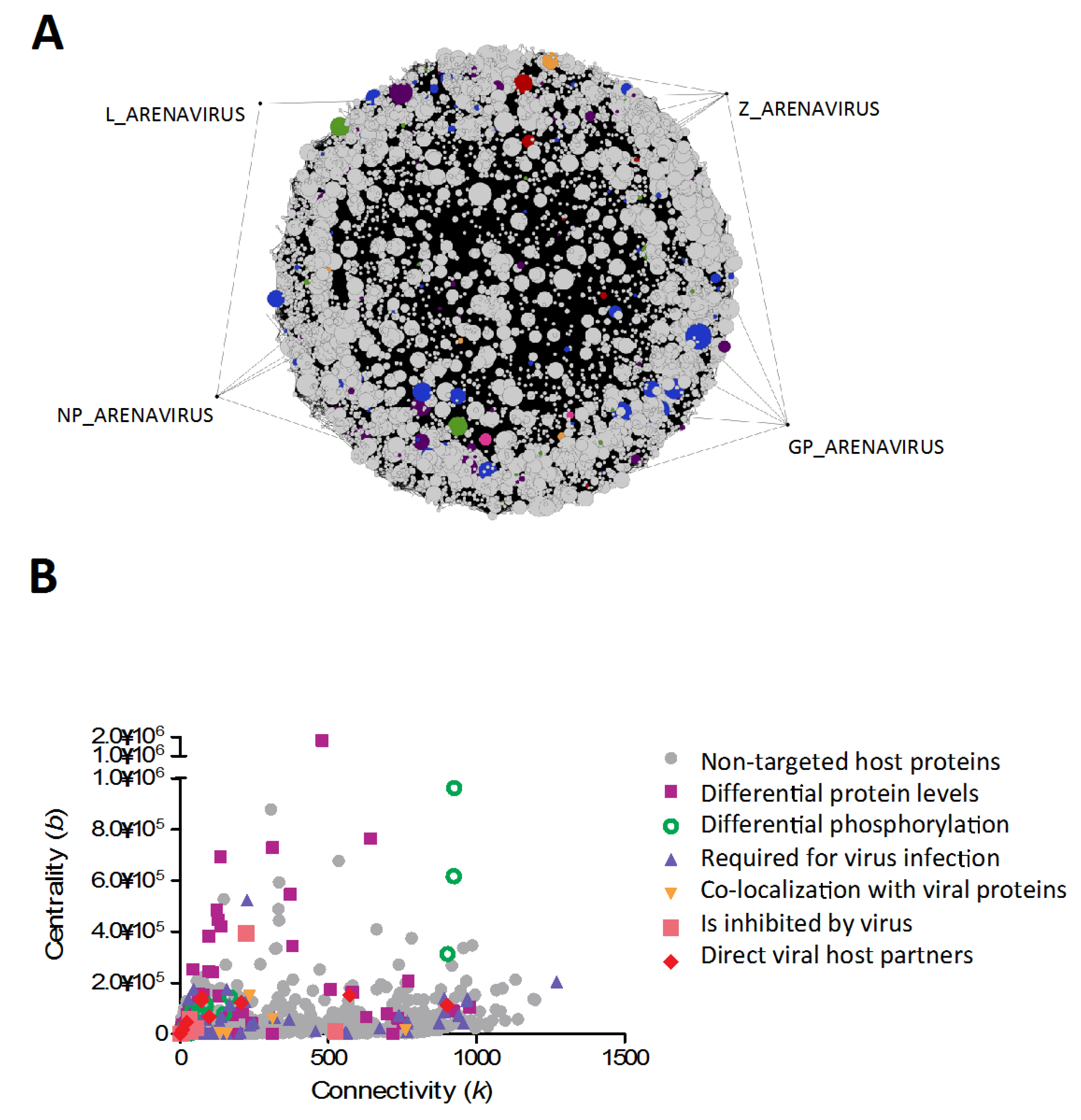{kind=link}
1. Introduction
1.1. Systems Biology as a Tool for Virus Research
2. General Concepts in Systems Biology
2.1. Mathematical Concepts to Describe Biological Systems
2.2. Basic Principles in Biological Networks
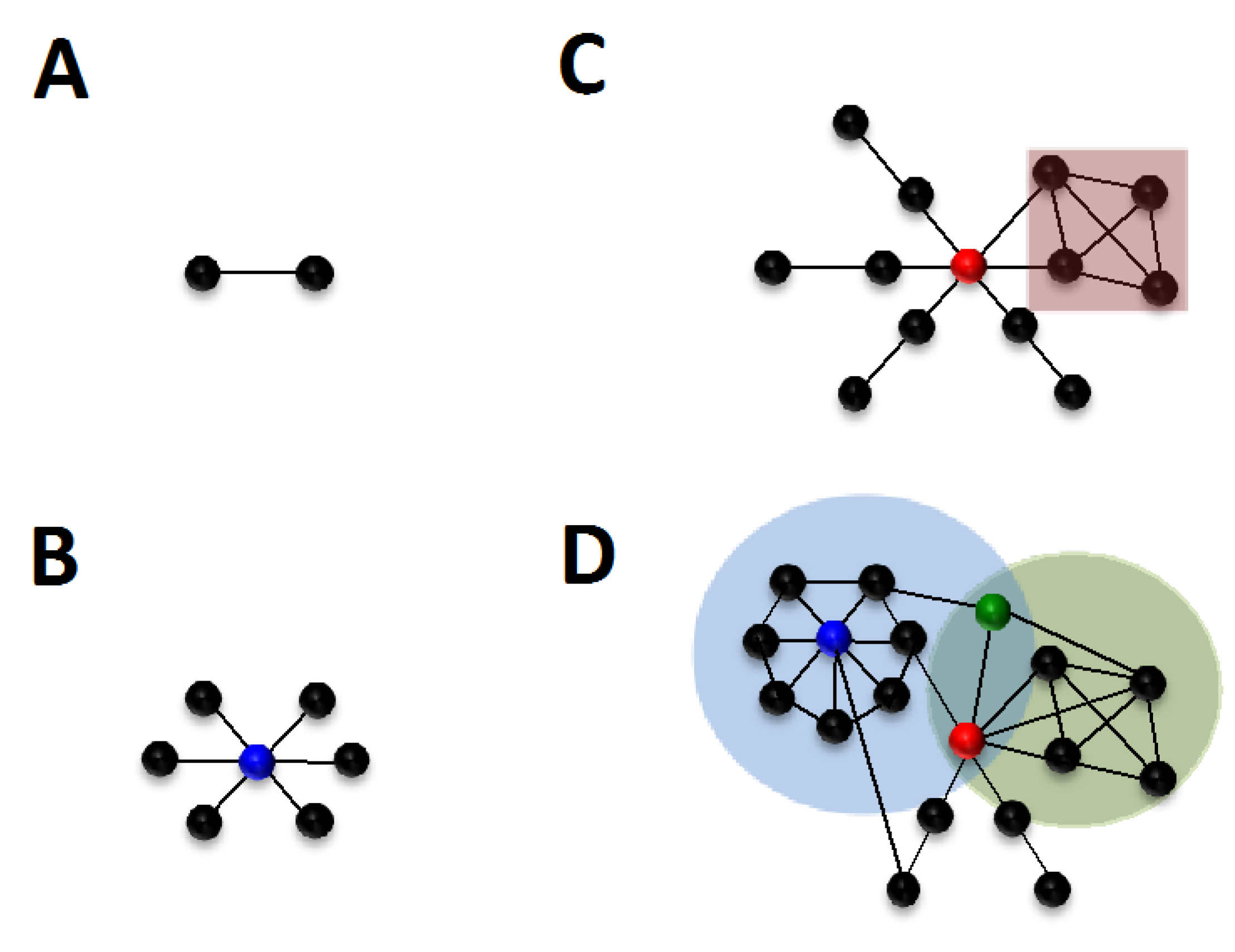
2.3. Local Properties in Biological Networks
2.4. Global Properties of Nodes and Edges
2.5. Understanding Network Structure: Defining Communities
3. Building and Analyzing an Arenavirus-Host Network
3.1. Datasets for the Construction of the Arenavirus-Host Network
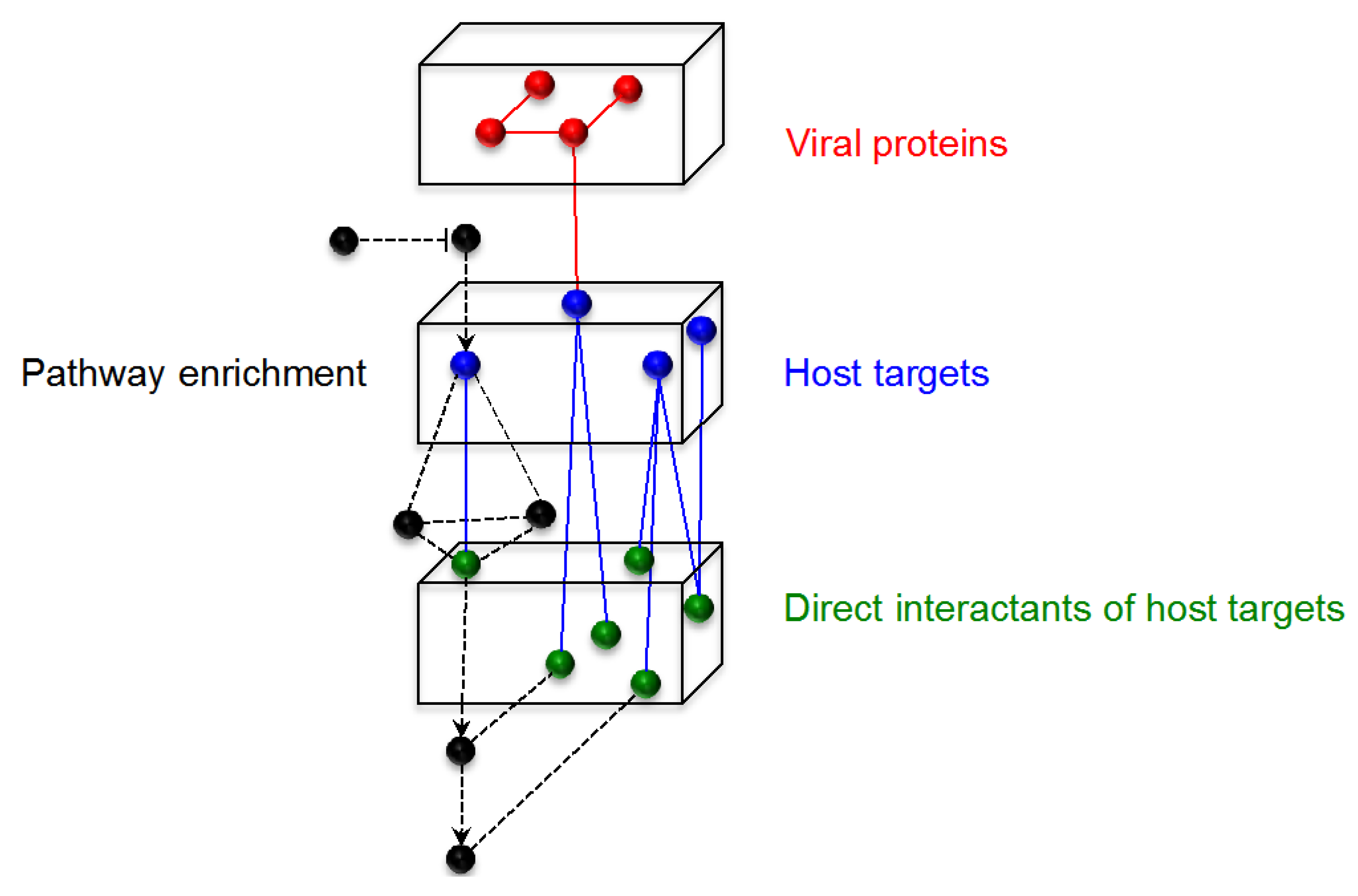
3.2. Programs for Building and Analyzing the Network
3.3. Embedding Information (Attributes) in the Network
3.4. Virus-Specific Elements Encoded within the Network
3.5. Using Filter-Set Subnetworks
3.6. Identifying Potential Viral Targets Through Centrality and Connectivity Values
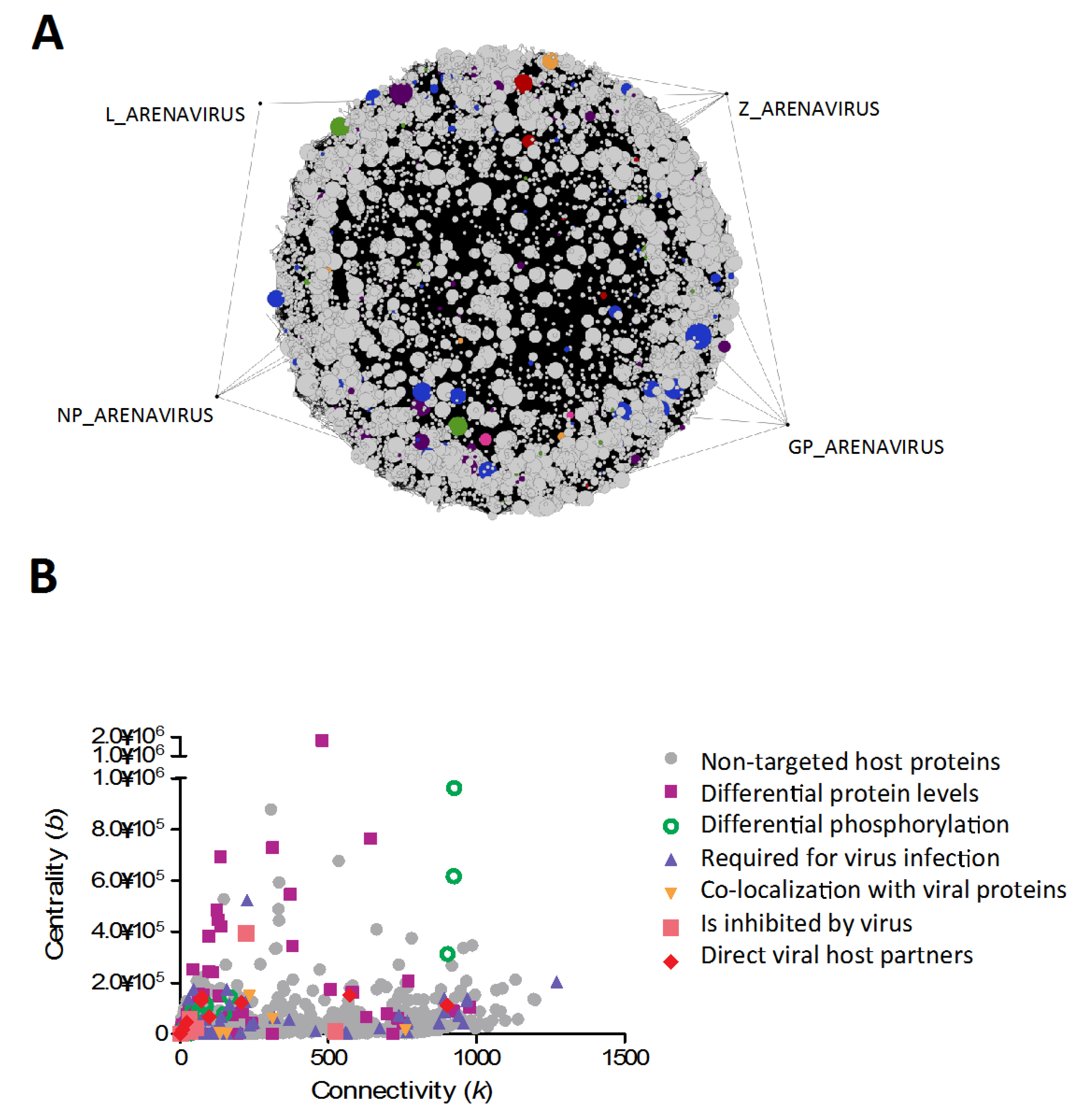
5. Convergence Between Different Virus-Host Networks
6. Systems Biology to Suggest Therapeutic Targets
7. Conclusions
Acknowledgments
Conflict of Interest
Supplementary Files
Supplementary File 1Supplementary File 2References and Notes
- Pawson, T.; Warner, N. Oncogenic re-wiring of cellular signaling pathways. Oncogene 2007, 26, 1268–1275. [Google Scholar] [CrossRef]
- Davey, N.E.; Trave, G.; Gibson, T.J. How viruses hijack cell regulation. Trends Biochem. Sci. 2011, 36, 159–169. [Google Scholar]
- Navratil, V.; de Chassey, B.; Rabourdin-Combe, C.; Lotteau, V. When the human viral infectome and diseasome networks collide: towards a systems biology platform for the aetiology of human diseases. BMC Syst. Biol. 2011, 5, 13. [Google Scholar] [CrossRef]
- Dyer, M.D.; Murali, T.M.; Sobral, B.W. The landscape of human proteins interacting with viruses and other pathogens. PLoSPathog. 2008, 4, e32. [Google Scholar]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar]
- Rasmussen, A.L.; Diamond, D.L.; McDermott, J.E.; Gao, X.; Metz, T.O.; Matzke, M.M.; Carter, V.S.; Belisle, S.E.; Korth, M.J.; Waters, K.M.; et al. Systems virology identifies a mitochondrial fatty acid oxidation enzyme, dodecenoyl coenzyme A delta isomerase, required for hepatitis C virus replication and likely pathogenesis. J. Virol. 2011, 85, 11646–11654. [Google Scholar]
- Garmaroudi, F.S.; Marchant, D.; Si, X.; Khalili, A.; Bashashati, A.; Wong, B.W.; Tabet, A.; Ng, R.T.; Murphy, K.; Luo, H.; et al. Pairwise network mechanisms in the host signaling response to coxsackievirus B3 infection. Proc. Natl. Acad. Sci. USA 2010, 107, 17053–17058. [Google Scholar]
- Borden, K.L.; Campbell Dwyer, E.J.; Salvato, M.S. An arenavirus RING (zinc-binding) protein binds the oncoproteinpromyelocyte leukemia protein (PML) and relocates PML nuclear bodies to the cytoplasm. J. Virol. 1998, 72, 758–766. [Google Scholar]
- Campbell Dwyer, E.J.; Lai, H.; MacDonald, R.C.; Salvato, M.S.; Borden, K.L. The lymphocytic choriomeningitis virus RING protein Z associates with eukaryotic initiation factor 4E and selectively represses translation in a RING-dependent manner. J. Virol. 2000, 74, 3293–3300. [Google Scholar] [CrossRef]
- Kentsis, A.; Dwyer, E.C.; Perez, J.M.; Sharma, M.; Chen, A.; Pan, Z.Q.; Borden, K.L. The RING domains of the promyelocytic leukemia protein PML and the arenaviral protein Z repress translation by directly inhibiting translation initiation factor eIF4E. J. Mol. Biol. 2001, 312, 609–623. [Google Scholar] [CrossRef]
- Borden, K.L.; Campbell-Dwyer, E.J.; Carlile, G.W.; Djavani, M.; Salvato, M.S. Two RING finger proteins, the oncoprotein PML and the arenavirus Z protein, colocalize with the nuclear fraction of the ribosomal P proteins. J. Virol. 1998, 72, 3819–3826. [Google Scholar]
- Bowick, G.C.; Soman, K.V.; Wang, H.; Aronson, J.F.; Luxon, B.A.; Lomas, L.O.; Gorenstein, D.G.; Herzog, N.K. Proteomic analysis of Pichinde virus infection identifies differential expression of prothymosin-alpha. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef]
- Bowick, G.C.; Spratt, H.M.; Hogg, A.E.; Endsley, J.J.; Wiktorowicz, J.E.; Kurosky, A.; Luxon, B.A.; Gorenstein, D.G.; Herzog, N.K. Analysis of the differential host cell nuclear proteome induced by attenuated and virulent hemorrhagic arenavirus infection. J. Virol. 2009, 83, 687–700. [Google Scholar]
- Bowick, G.C.; Fennewald, S.M.; Elsom, B.L.; Aronson, J.F.; Luxon, B.A.; Gorenstein, D.G.; Herzog, N.K. Differential signaling networks induced by mild and lethal hemorrhagic fever virus infections. J. Virol. 2006, 80, 10248–10252. [Google Scholar] [CrossRef]
- Bowick, G.C.; Fennewald, S.M.; Scott, E.P.; Zhang, L.H.; Elsom, B.L.; Aronson, J.F.; Spratt, H.M.; Luxon, B.A.; Gorenstein, D.G.; Herzog, N.K. Identification of differentially activated cell-signaling networks associated with Pichinde virus pathogenesis by using systems kinomics. J. Virol. 2007, 81, 1923–1933. [Google Scholar]
- Djavani, M.; Crasta, O.R.; Zhang, Y.; Zapata, J.C.; Sobral, B.; Lechner, M.G.; Bryant, J.; Davis, H.; Salvato, M.S. Gene expression in primate liver during viral hemorrhagic fever. Virol. J. 2009, 6, 20. [Google Scholar] [CrossRef]
- Djavani, M.M.; Crasta, O.R.; Zapata, J.C.; Fei, Z.; Folkerts, O.; Sobral, B.; Swindells, J.; Bryant, J.; Davis, H.; Pauza, C.D.; et al. Early blood profiles of virus infection in a monkey model for Lassa fever. J. Virol. 2007, 81, 7960–7973. [Google Scholar]
- Muller, S.; Geffers, R.; Gunther, S. Analysis of gene expression in Lassa virus-infected HuH-7 cells. J. Gen. Virol. 2007, 88, 1565–1575. [Google Scholar] [CrossRef]
- Wikoff, W.R.; Kalisak, E.; Trauger, S.; Manchester, M.; Siuzdak, G. Response and recovery in the plasma metabolome tracks the acute LCMV-induced immune response. J. Proteome Res. 2009, 8, 3578–3587. [Google Scholar] [CrossRef]
- Biggs, N.; Lloyd, E.; Wilson, R. Graph Theory 1736–1936; Clarendon Press: Oxford, UK, 1976. [Google Scholar]
- von Bertalanffy, L. An outline of general system theory. Br. J. Philos. Sci. 1950, 1, 139–164. [Google Scholar]
- von Bertalanffy, L. General system theory—A new approach to unity of science. Hum. Biol. 1951, 23, 303–361. [Google Scholar]
- Wagner, A.; Fell, D.A. The small world inside large metabolic networks. Proc. Biol. Sci. 2001, 268, 1803–1810. [Google Scholar] [CrossRef]
- Jeong, H.; Tombor, B.; Albert, R.; Oltvai, Z.N.; Barabasi, A.L. The large-scale organization of metabolic networks. Nature 2000, 407, 651–654. [Google Scholar]
- Maslov, S.; Sneppen, K. Specificity and stability in topology of protein networks. Science 2002, 296, 910–913. [Google Scholar] [CrossRef]
- Spirin, V.; Mirny, L.A. Protein complexes and functional modules in molecular networks. Proc. Natl. Acad. Sci. USA 2003, 100, 12123–12128. [Google Scholar] [CrossRef]
- de la Fuente, A.; Fotia, G.; Maggio, F.; Mancosu, G.; Pieroni, E. Insights into biological information processing: Structural and dynamical analysis of a Human Protein Signaling network. J. Physics A 2008, 41, 224013. [Google Scholar] [CrossRef]
- Joy, M.P.; Brock, A.; Ingber, D.E.; Huang, S. High-betweenness proteins in the yeast protein interaction network. J. Biomed. Biotechnol. 2005, 96–103. [Google Scholar]
- Hahn, M.W.; Kern, A.D. Comparative genomics of centrality and essentiality in three eukaryotic protein-interaction networks. Mol. Biol. Evol. 2005, 22, 803–806. [Google Scholar] [CrossRef]
- Fox, A.D.; Hescott, B.J.; Blumer, A.C.; Slonin, D.K. Connectedness of PPI network neighborhood identifies regulatory hub proteins. Bioinformatics 2011, 27, 1135–1142. [Google Scholar] [CrossRef]
- Palla, G.; Dereny, I.; Farkas, I.; Vicsek, T. Uncovering the overlapping community structure of complex networks in nature and society. Nature 2005, 435, 814–818. [Google Scholar]
- Kim, W.; Li, M.; Wang, J.; Pan, Y. Biological network motif detection and evaluation. BMC Genom. 2010, 11, S10. [Google Scholar]
- Opgen-Rhein, R.; Strimmer, K. Learning causal networks from systems biology time course data: An effective model selection procedure for the vector autoregressive process. BMC Bioinformatics 2007, 8, S2–S3. [Google Scholar]
- Cui, J.; DeLuca, T.F.; Jung, J.Y.; Wall, D.P. Phylogenetically informed logic relationships improve detection of biological network organization. BMC Bioinformatics 2011, 12, 476. [Google Scholar] [CrossRef]
- Tang, B.; Chen, S.S.; Jin, V.X. Integrative identification of core genetic regulatory modules via a structural model-based clustering method. Int. J. Comput. Biol. Drug Des. 2011, 4, 127–146. [Google Scholar] [CrossRef]
- Marin-Sanguino, A.; Gupta, S.K.; Voit, E.O.; Vera, J. Biochemical pathway modeling tools for drug target detection in cancer and other complex diseases. Methods Enzymol. 2011, 487, 319–369. [Google Scholar]
- Choi, S. Introduction to Systems Biology; Humana press Inc.: New York, NY, USA, 2007. [Google Scholar]
- Alon, U. An Introduction to Systems Biology: Design Principles of Biological Circuits; Chapman and Hall/CRC: Oxford, UK, 2007. [Google Scholar]
- von Mering, D.J.; Krause, R.; Snel, B.; Cornell, M.; Oliver, S.G.; Fields, S.; Bork, P. Comparative assessment of large-scale data sets of protein-protein interactions. Nature 2002, 417, 399–403. [Google Scholar]
- Watts, D.J.; Strogatz, S.H. Collective dynamics of “small-world” networks. Nature 1998, 393, 440–442. [Google Scholar]
- Holland, P.W.; Leinhardt, S. Transitivity in structural models of small groups. Comparative Group Studies 1971, 2, 107–124. [Google Scholar]
- Luce, R.D.; Perry, A.D. A method of matrix analysis of group structure. Psychometrika 1949, 14, 95–116. [Google Scholar] [CrossRef]
- Newman, M.E.J. Detecting community structure in networks. Eur. Phys. J. B 2004, 38, 321–330. [Google Scholar] [CrossRef]
- Girvan, M.; Newman, M.E.J. Community structure in social and biological networks. Proc. Natl. Acad. Sci. USA. 2002, 99, 7821–7826. [Google Scholar] [CrossRef]
- Radicchi, F.; Castellano, C.; Cecconi, F.; Loreto, V.; Parisi, D. Defining and identifying communities in networks. Proc. Natl. Acad. Sci. USA. 2004, 101, 2658–2663. [Google Scholar] [CrossRef]
- Shtanko, O.; Watanabe, S.; Jasenosky, L.D.; Watanabe, T.; Kawaoka, Y. ALIX/AIP1 is required for NP incorporation into Mopeia virus Z-induced virus-like particles. J. Virol. 2011, 85, 3631–3641. [Google Scholar] [CrossRef]
- Volpon, L.; Osborne, M.J.; Capul, A.A.; de la Torre, J.C.; Borden, K.L. Structural characterization of the Z RING-eIF4E complex reveals a distinct mode of control for eIF4E. Proc. Natl. Acad. Sci. USA. 2010, 107, 5441–5446. [Google Scholar]
- Labudova, M.; Tomaskova, J.; Skultety, L.; Pastorek, J.; Pastorekova, S. The nucleoprotein of lymphocytic choriomeningitis virus facilitates spread of persistent infection through stabilization of the keratin network. J. Virol. 2009, 83, 7842–7849. [Google Scholar] [CrossRef]
- Pythoud, C.; Rodrigo, W.W.; Pasqual, G.; Rothenberger, S.; Martínez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKK(varepsilon). J. Virol. 2012, 86, 7728–7738. [Google Scholar]
- Radoshitzky, S.R.; Abraham, J.; Spiropoulou, J.F.; Kuhn, J.H.; Nguyen, D.; Li, W.; Nagel, J.; Schmidt, P.J.; Nunberg, J.H.; Andrews, N.C.; et al. Transferrin receptor 1 is a cellular receptor for New World hemorrhagic fever arenaviruses. Nature 2007, 446, 92–96. [Google Scholar]
- Flanagan, M.L; Oldenburg, J.; Reignier, T.; Holt, N.; Hamilton, G.A.; Martin, V.K.; Cannon, P.M. New world clade B arenaviruses can use transferrin receptor 1 (TfR1)-dependent and -independent entry pathways, and glycoproteins from human pathogenic strains are associated with the use of TfR1. J. Virol. 2008, 82, 938–948. [Google Scholar]
- Reignier, T.; Oldenburg, J.; Flanagan, M.L.; Hamilton, G.A.; Martin, V.K.; Cannon, P.M. Receptor use by the Whitewater Arroyo virus glycoprotein. Virology 2008, 371, 439–446. [Google Scholar] [CrossRef]
- Droniou-Bonzom, M.E.; Reignier, T.; Oldenburg, J.E.; Cox, A.U.; Exline, C.M.; Rathbun, J.Y.; Cannon, P.M. Substitutions in the glycoprotein (GP) of the Candid#1 vaccine strain of Junin virus increase dependence on human transferrin receptor 1 for entry and destabilize the metastable conformation of GP. J. Virol. 2011, 85, 13457–13462. [Google Scholar]
- Cao, W.; Henry, M.D.; Borrow, P.; Yamada, H.; Elder, J.H.; Ravkov, E.V; Nichol, S.T.; Compans, R.W.; Campbell, K.P.; Oldstone, M.B. Identification of alpha-dystroglycan as a receptor for lymphocytic chriomeningitis virus and Lassa fever virus. Science 1998, 282, 2079–2081. [Google Scholar]
- Spiropoulou, C.F.; Kunz, S.; Rollin, P.E.; Campbell, K.P.; Oldstone, M.B. New World arenavirusclade C, but not clade A and B viruses, utilizes α-dystroglycan as its major receptor. J. Virol. 2002, 76, 5140–5146. [Google Scholar] [CrossRef]
- Abraham, J.; Kwong, J.A.; Albariño, C.G.; Lu, J.G.; Radoshitzky, S.R.; Salazar-Bravo, J.; Farzan, M.; Spiropoulou, C.F.; Choe, H. Host-species transferrin receptor 1 orthologs are cellular receptors for non-pathogenic new World clade B arenaviruses. PLoS Pathog. 2009, 5, e1000358. [Google Scholar]
- Rojek, J.M.; Spiropoulou, C.F.; Kunz, S. Characterization of the cellular receptors for the South American hemorrhagic fever viruses Junin, Guanarito and Machupo. Virology 2006, 349, 476–491. [Google Scholar] [CrossRef]
- Shimojima, M.; Kawaoka, Y. Cell surface molecules involved in infection mediated by Lymphocytic choriomeningitis virus glycoprotein. J. Vet. Med. Sci. 2012, 10, 1363–1366. [Google Scholar] [CrossRef]
- Shimojima, M.; Ströher, U.; Ebihara, H.; Feldmann, H.; Kawaoka, Y. Identification of cell surface molecules involved in dystroglycan-independent Lassa virus cell entry. J. Virol. 2012, 86, 2067–2078. [Google Scholar]
- Martínez, M.G.; Prado Acosta, M.; Candurra, N.A.; Ruzal, S.M. S-layer proteins of Lactobacillus acidophilus inhibits JUNV infection. Biochem. Biophys. Res. Commun. 2012, 422, 590–595. [Google Scholar] [CrossRef]
- Fan, L.; Briese, T.; Lipkin, W.I. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J. Virol. 2010, 84, 1785–1791. [Google Scholar] [CrossRef]
- Zhou, S.; Cerny, A.M.; Zacharia, A.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Finberg, R.W. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J. Virol. 2010, 84, 9452–9462. [Google Scholar] [CrossRef]
- Perez, M.; Craven, R.C.; de la Torre, J.C. The small RING finger protein Z drives arenavirus budding: Implications for antiviral strategies. Proc. Natl. Acad. Sci. USA 2003, 100, 12978–12983. [Google Scholar]
- Urata, S.; Noda, T.; Kawaoka, Y.; Yokosawa, H.; Yasuda, J. Cellular factors required for Lassa virus budding. J. Virol. 2006, 80, 4191–4195. [Google Scholar] [CrossRef]
- Urata, S.; Yasuda, J.; de la Torre, J.C. The Z protein of the New World arenavirustacaribe virus has bonafide budding activity that does not depend on known late domain motifs. J. Virol. 2009, 83, 12651–12655. [Google Scholar] [CrossRef]
- Urata, S.; Ngo, N.; de la Torre, J.C. The PI3K/Akt pathway contributes to arenavirus budding. J. Virol. 2012, 86, 4578–4585. [Google Scholar] [CrossRef]
- Pasqual, G.; Rojek, J.M.; Masin, M.; Chatton, J.Y.; Kunz, S. Old World arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoSPathog. 2011, 7, e1002232. [Google Scholar]
- Linero, F.N.; Scolaro, L.A. Participation of the phosphatidylinositol 3-kinase/Akt pathway in Junin virus replication in vitro. Virus Res. 2009, 145, 166–170. [Google Scholar] [CrossRef]
- Panda, D.; Das, A.; Dinh, P.X.; Subramaniam, S.; Nayak, D.; Barrows, N.J.; Pearson, J.L.; Thompson, J.; Kelly, D.L.; Ladunga, I.; et al. RNAi screening reveals requirement for host cell secretory pathway in infection by diverse families of negative-strand RNA viruses. Proc. Natl. Acad. of Sci. USA 2011, 47, 19036–19041. [Google Scholar]
- Candurra, N.A.; Lago, M.J.; Maskin, L.; Damonte, E.B. Involvement of the cytoskeleton in Junin virus multiplication. J. Gen. Virol. 1999, 80, 147–156. [Google Scholar]
- Cordo, S.M.; Candurra, N.A. Intermediate filament integrity is required for Junin virus replication. Virus Res. 2003, 97, 47–55. [Google Scholar] [CrossRef]
- Martinez, M.G.; Cordo, S.M.; Candurra, N.A. Involvement of cytoskeleton in Junin virus entry. Virus Res. 2008, 138, 17–25. [Google Scholar] [CrossRef]
- Martinez, M.G.; Cordo, S.M.; Candurra, N.A. Characterization of Juninarenavirus cell entry. J. Gen. Virol. 2007, 88, 1776–1784. [Google Scholar] [CrossRef]
- Martinez, M.G.; Forlenza, M.B.; Candurra, N.A. Involvement of cellular proteins in Junin arenavirus entry. Biotechnology J. 2009, 4, 866–870. [Google Scholar]
- Borrow, P.; Oldstone, M.B. Mechanism of lymphocytic choriomeningitis virus entry into cells. Virology 1994, 198, 1–9. [Google Scholar] [CrossRef]
- Rojek, J.M.; Perez, M.; Kunz, S. Cellular entry of lymphocytic choriomeningitis virus. J. Virol. 2008, 82, 1505–1517. [Google Scholar] [CrossRef]
- Rojek, J.M.; Sanchez, A.B.; Nguyen, N.T.; de la Torre, J.C.; Kunz, S. Different mechanisms of cell entry by human pathogenic Old World and New World arenaviruses. J. Virol. 2008, 82, 7677–7687. [Google Scholar] [CrossRef]
- Castilla, V.; Palermo, L.M.; Coto, C.E. Involvement of vacuolar proton ATPase in Junin virus multiplication. Arch. Virol. 2001, 146, 251–263. [Google Scholar] [CrossRef]
- Lenz, O.; ter Meulen, J.; Klenk, H.D.; Seidah, N.G.; Garten, W. The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc. Natl. Acad. Sci. USA 2001, 98, 12701–12705. [Google Scholar] [CrossRef]
- Beyer, W.R.; Pöpplau, D.; Garten, W.; von Laer, D.; Lenz, O. Endoproteolytic processing of the lymphocytic choriomeningitis virus glycoprotein by the subtilase SKI-1/S1P. J. Virol. 2003, 77, 2866–2872. [Google Scholar] [CrossRef]
- Kunz, S.; Edelmann, K.H.; de la Torre, J.C.; Gorney, R.; Oldstone, M.B. Mechanisms for lymphocytic choriomeningitis virus glycoprotein cleavage, transport, and incorporation into virions. Virology 2003, 314, 168–178. [Google Scholar] [CrossRef]
- Rojek, J.M.; Lee, A.M.; Nguyen, N.; Spiropoulou, C.F.; Kunz, S. Site 1 protease is required for proteolytic processing of the glycoproteins of the South American hemorrhagic fever viruses Junin, Machupo and Guanarito. J. Virol. 2008, 82, 6045–6051. [Google Scholar]
- Pasquato, A.; Burri, D.J.; Traba, E.G.; Hanna-El-Daher, L.; Seidah, N.G.; Kunz, S. Arenavirus envelope glycoproteins mimic autoprocessing sites of the cellular proproteinconvertasesubtilisinkexin isozyme-1/ site-1 protease. Virology 2011, 417, 18–26. [Google Scholar]
- Martinez-Sobrido, L.; Zúñiga, E.I.; Rosario, D.; Garcia-Sastre, A.; de la Torre, J.C. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2006, 80, 9192–9199. [Google Scholar] [CrossRef]
- Martinez-Sobrido, L.; Giannakas, P.; Cubitt, B; Garcia-Sastre, A.; de la Torre, J.C. Differential inhibition of type I interferon induction by arenavirus nucleoproteins. J. Virol. 2007, 81, 12696–12703. [Google Scholar] [CrossRef]
- Rodrigo, W.W.; Ortiz-Riaño, E.; Pythoud, C.; Kunz, S.; de la Torre, J.C.; Martínez-Sobrido, L. Arenavirus nucleoproteins prevent activation of nuclear factor kappa B. J. Virol. 2012, 86, 8185–8197. [Google Scholar]
- Gomez, R.M.; Pozner, R.G.; Lazzari, M.A.; D’Atri, L.P; Negrotto, S.; Chudzinski-Tavassi, A.M.; Berria, M.I.; Schattner, M. Endothelial cell function alteration after Junin virus infection. Thromb. Haemost. 2003, 90, 326–333. [Google Scholar]
- Molinas, F.C.; de Bracco, M.M.; Maitzegui, J.I. Hemostasis and the complement system in Argentine hemorrhagic fever. Rev. Infect. Dis. 1989, 11, S762–S770. [Google Scholar]
- Bowick, G.C.; Fennewald, S.M.; Zhang, L.; Yang, X.; Aronson, J.F.; Shope, R.E.; Luxon, B.A.; Gorenstein, D.G.; Herzog, N.K. Attenuated and lethal variants of Pichinde virus induce differential patterns of NF-kappaB activation suggesting a potential target for novel therapeutics. Viral Immunol. 2009, 22, 457–462. [Google Scholar] [CrossRef]
- Fennewald, S.M.; Aronson, J.F.; Zhang, L.; Herzog, N.K. Alterations in NK-kappaB and RBP-Jkappa by arenavirus infection of macrophages in vitro and in vivo. J. Virol. 2002, 76, 1154–1162. [Google Scholar] [CrossRef]
- Hayes, M.W.; Carrion, R., Jr.; Nunneley, J.; Medvedev, A.E.; Salvato, M.S.; Lukashevich, I.S. Pathogenic Old World arenaviruses inhibit TLR2/Mal-dependent proinflammatory cytokines in vitro. J. Virol. 2012, 86, 7216–7226. [Google Scholar] [CrossRef]
- Rojek, J.M.; Campbell, K.P.; Oldstone, M.B.; Kunz, S. Old World arenavirus infection interferes with the expression of functional alpha-dystroglycan in the host cell. Mol. Biol. Cell 2007, 18, 4493–4507. [Google Scholar] [CrossRef]
- Aronson, J.F.; Herzog, N.K.; Jerrells, T.R. Tumor-necrosis-factor and the pathogenesis of Pichinde virus infection in guinea pigs. Am. J. Trop. Med. Hyg. 1995, 52, 262–269. [Google Scholar]
- Marta, R.F.; Montero, V.S.; Hack, C.E.; Sturk, A.; Maiztegui, J.I.; Molinas, F.C. Proinflammatory cytokines and elastase-α-1-antitrypsin in Argentine hemorrhagic fever. Am. J. Trop. Med. Hyg. 1999, 60, 85–89. [Google Scholar]
- Djavani, M.; Topisirovic, I.; Zapata, J.C.; Sadowska, M.; Yang, Y.; Rodas, J.; Lukashevich, I.S.; Bogue, C.W.; Pauza, C.D.; Borden, K.L.; et al. The proline-rich homeodomain (PRH/HEX) protein is down-regulated in liver during infection with lymphocytic choriomeningitis virus. J. Virol. 2005, 79, 2461–2473. [Google Scholar]
- Topcu, Z.; Mack, D.L; Hromas, R.A.; Borden, K.L.B. The promyelocytic leukemia protein PML interacts with the proline-rich homeodomain protein PRH: a RING may link hematopoiesis and growth control. Oncogene 1999, 18, 7091–7100. [Google Scholar] [CrossRef]
- Jaquenod De Giusti, C.; Alberdi, L.; Frik, J.; Ferrer, M.F.; Scharrig, E.; Schattner, M.; Gomwez, R.M. Galectin-3 is upregulated in activated glia during Junin virus-induced murine encephalitis. Neurosci. Lett. 2011, 501, 163–166. [Google Scholar] [CrossRef]
- Huang, C.; Kolokoltsova, O.A.; Yun, N.E.; Seregin, A.V.; Poussard, A.L.; Walker, A.G.; Brasier, A.R.; Zhao, Y.; Tian, B.; de la Torre, J.C.; et al. Junin virus infection activates the type I interferon pathway in a RIG-I-dependent manner. PLoS Negl. Trop. Dis. 2012, 6, e1659. [Google Scholar] [CrossRef]
- Cuevas, C.D.; Lavanaya, M.; Wnag, E.; Ross, S.R. Junin virus infects mouse cells and induces innate immune responses. J. Virol. 2011, 85, 11058–11068. [Google Scholar] [CrossRef]
- Dejean, C.B.; Oubina, J.R.; Carballal, G.; Teyssie, A.R. Circulating interferon in the guinea pig infected with the XJ prototype Junin virus strain. J. Med. Virol. 1988, 24, 97–99. [Google Scholar]
- Dejean, C.B.; Ayerra, B.L.; Teyssie, A.R. Interferon response in the guinea pig infected with Junin virus. J. Med. Virol. 1987, 23, 83–91. [Google Scholar] [CrossRef]
- Levis, S.C.; Saavedra, M.C.; Ceccoli, C.; Falcoff, E.; Feuillade, M.R.; Enria, D.A.; Maitzegui, J.I.; Falcoff, R. Endogenous interferon in Argentine hemorrhagic fever. J. Infect. Dis. 1984, 149, 428–433. [Google Scholar] [CrossRef]
- Groseth, A.; Hoenen, T.; Weber, M.; Wolff, S.; Herwig, A.; Kaufmann, A.; Becker, S. Tacaribe virus but not junin virus infection induces cytokine release from primary human monocytes and macrophages. PLoSNegl. Trop. Dis. 2011, 5, e1137. [Google Scholar] [CrossRef]
- Marta, R.F.; Enria, D.; Molinas, F.C. Relationship between hematopoietic growth factors levels and hematological parameters in Argentine hemorrhagic fever. Am. J. Hematol. 2000, 64, 1–6. [Google Scholar] [CrossRef]
- Mahanty, S.; Bausch, D.G.; Thomas, R.L.; Goba, A.; Bah, A.; Peters, C.J.; Rollin, P.E. Low levels of interleukin-8 and interferon-inducible protein-10 in serum are associated with fatal infections in acute Lassa fever. J. Infect. Dis. 2001, 183, 1713–1721. [Google Scholar]
- Baize, S.; Marianneau, P.; Loth, P.; Reynard, S.; Journeaux, A.; Chevallier, M.; Tordo, N.; Deubel, V.; Contamin, H. Early and strong immune responses are associated with control of viral replication and recovery in lassa-virus-infected cynomolgus monkeys. J. Virol. 2009, 83, 5890–5903. [Google Scholar]
- Zhou, S.; Halle, A.; Kurt-Jones, E.A.; Cerny, A.M.; Porpiglia, E.; Rogers, M.; Golenbock, D.T.; Finberg, R.W. Lymphocytic choriomeningitis virus (LCMV) infection of CNS glial cells results in TLR2-MyD88/Mal-dependent inflammatory responses. J. Neuroimmunol. 2008, 194, 70–82. [Google Scholar] [CrossRef]
- Baird, N.L.; York, J.; Nunberg, J.H. Arenavirus infection induces discrete cytosolic structures for RNA replication. J. Virol. 2012, 86, 11301–11310. [Google Scholar] [CrossRef]
- Radoshitzky, S.R.; Dong, L.; Chi, X.; Clester, J.C.; Retterer, C.; Spurgers, K.; Kuhn, J.H.; Sandwick, S.; Ruthel, G.; Kota, K.; et al. Infectious Lassa virus, but not filoviruses, is restricted by BST-2/ tetherin. J. Virol. 2010, 84, 10569–10580. [Google Scholar]
- Sakuma, T.; Noda, T.; Urata, S.; Kawaoka, Y.; Yasuda, J. Inhibition of Lassa and Marburg virus production by tetherin. J. Virol. 2009, 83, 2382–2385. [Google Scholar] [CrossRef]
- Cerami, E.G.; Gross, B.E.; Demir, E.; Rodchenkov, I.; Babur, O.; Anwar, N.; Schultz, N.; Bader, G.D.; Sander, C. Pathway Commons, a web ressource for biological pathway data. Nucleic. Acids Res. 2011, 39, D685–D690. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef]
- Cytoscape. Available online: www.cytoscape.org (accessed on 25 July 2012).
- Cerami, E.G.; Bader, G.D.; Gross, B.E.; Sander, C. cPath: Open source software for collecting, storing, and querying biological pathways. BMC Bioinformatics 2006, 7, 497. [Google Scholar] [CrossRef]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal 2006, 1695. [Google Scholar]
- Jahrling, P.B.; Hesse, R.A.; Rhoderick, J.B.; Elwell, M.A.; Moe, J.B. Pathogenesis of a Pichinde virus strain adapted to produce lethal infections in guinea pigs. Infect. Immunol. 1981, 32, 872–880. [Google Scholar]
- Aronson, J.F.; Herzog, N.K.; Jerrels, T.R. Pathological and virological features of arenavirus disease in guinea pigs. Comparison of two Pichinde virus strains. Am. J. Pathol. 1994, 145, 228–235. [Google Scholar]
- Walker, D.H.; McCormick, J.B.; Johnson, K.M.; Webb, P.A.; Komba-Kono, G.; Elliott, L.H.; Gardner, J.J. Pathologic and virologic study of fatal Lassa fever in man. Am. J. Pathol. 1982, 107, 349–356. [Google Scholar]
- Moraz, M.L.; Kunz, S. Pathogenesis of arenavirus hemorrhagic fevers. Expert Rev. Anti. Infect. Ther. 2011, 9, 49–59. [Google Scholar]
- Bowick, G.C.; McAuley, A.J. Meta-analysis of high-throughput datasets reveals cellular responses following hemorrhagic fever virus infection. Viruses 2011, 3, 613–619. [Google Scholar] [CrossRef]
- Hartman, A.L.; Ling, L.; Nichol, S.T.; Hibberd, M.L. Whole-genome expression profiling reveals that inhibition of host innate immune response pathways by Ebola virus can be reversed by a single amino acid change in the VP35 protein. J. Virol. 2008, 82, 5348–5358. [Google Scholar] [CrossRef]
- do Valle, T.Z.; Billecocq, A.; Guillemot, L.; Alberts, R.; Gommet, C.; Geffers, R.; Calabrese, K.; Schughart, K.; Bouloy, M.; Montagutelli, X.; et al. A new mouse model reveals a critical role for host innate immunity in resistance to Rift Valley fever. J. Immunol. 2010, 185, 6146–6156. [Google Scholar] [CrossRef]
- Wu, W.L.; Ho, L.J.; Chang, D.M.; Chen, C.H.; Lai, J.H. Triggering of DC migration by dengue virus stimulation of COX-2-dependent signaling cascades in vitro highlights the significance of these cascades beyond inflammation. Europ. J. Immunol. 2009, 39, 3413–3422. [Google Scholar] [CrossRef]
- O’Banion, M.K. Cyclooxygenase-2: Molecular biology, pharmacology, and neurobiology. Crit. Rev. Neurobiol. 1999, 13, 45–82. [Google Scholar]
- Pichlmair, A.; Kandasamy, K.; Alvisi, G.; Mulhern, O.; Sacco, R.; Habjan, M.; Binder, M.; Stefanovic, A.; Eberle, C.A.; Goncalves, A.; et al. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 2012, 26, 486–490. [Google Scholar]
- Holt, N.; Wang, J.; Kim, K.; Friedman, G.; Wang, X.; Taupin, V.; Crooks, G.M.; Kohn, D.B.; Gregory, P.D.; Holmes, M.C.; et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 2010, 28, 839–847. [Google Scholar] [CrossRef]
- Spiro, Z.; Kovacs, I.A.; Csermely, P. Drug-therapy networks and the prediction of novel drug targets. J. Biol. 2008, 7, 20. [Google Scholar] [CrossRef]
- Faustino, R.S.; Terzic, A. Bioinformatic networks: molecular reticles for pinpointing pharmalogical target selection. Clin. Pharmacol. Ther. 2008, 5, 543–545. [Google Scholar] [CrossRef]
- Campillos, M.; Kuhn, M.; Gavin, A.C.; Jensen, L.J.; Bork, P. Drug target identification using side-effect similarity. Science 2008, 321, 263–266. [Google Scholar] [CrossRef]
- Navratil, V.; de Chassey, B.; Meyniel, L.; Delmotte, S.; Gautier, C.; Andre, P.; Lotteau, V.; Rabourdin-Combe, C. VirHostNet: A knowledge base for the management and the analysis of proteome-wide virus-host interaction networks. Nucleic Acids Res. 2009, 37, D661–D668. [Google Scholar]
- Chatr-aryamontri, A.; Ceol, A.; Peluso, D.; Nardozza, A.; Panni, S.; Sacco, F.; Tinti, M.; Smolyar, A.; Castagnoli, L.; Vidal, M.; et al. VirusMINT: a viral protein interaction database. Nucleic Acids Res. 2009, 37, D669–D673. [Google Scholar] [CrossRef]
- Kwofie, S.K.; Schaefer, U.; Sundararajan, V.S.; Bajic, V.B.; Christoffels, A. HCVpro: Hepatitis C virus protein interaction database. Infect. Genet. Evol. 2011, 11, 1971–1977. [Google Scholar] [CrossRef]
- Shoemaker, J.E.; Fukuyama, S.; Eisfeld, A.J.; Muramoto, Y.; Watanabe, S.; Watanabe, T.; Matsuoka, Y.; Kitano, H.; Kawaoka, Y. Integrated network analysis reveals a novel role for the cell cycle in 2009 pandemic influenza virus-induced inflammation in macaque lungs. BMC Syst. Biol. 2012, 6, 117. [Google Scholar] [CrossRef]
- McDermott, J.E.; Diamond, D.L.; Corley, C.; Rasmussen, A.L.; Katze, M.G.; Waters, K.M. Topological analysis of protein co-abundance networks identified novel host targets important for HCV infection and pathogenesis. BMC Syst. Biol. 2012, 6, 28. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Droniou-Bonzom, M.E.; Cannon, P.M. A Systems Biology Starter Kit for Arenaviruses. Viruses 2012, 4, 3625-3646. https://doi.org/10.3390/v4123625
Droniou-Bonzom ME, Cannon PM. A Systems Biology Starter Kit for Arenaviruses. Viruses. 2012; 4(12):3625-3646. https://doi.org/10.3390/v4123625
Chicago/Turabian StyleDroniou-Bonzom, Magali E., and Paula M. Cannon. 2012. "A Systems Biology Starter Kit for Arenaviruses" Viruses 4, no. 12: 3625-3646. https://doi.org/10.3390/v4123625
APA StyleDroniou-Bonzom, M. E., & Cannon, P. M. (2012). A Systems Biology Starter Kit for Arenaviruses. Viruses, 4(12), 3625-3646. https://doi.org/10.3390/v4123625