Innate Immunity to H5N1 Influenza Viruses in Humans


Abstract
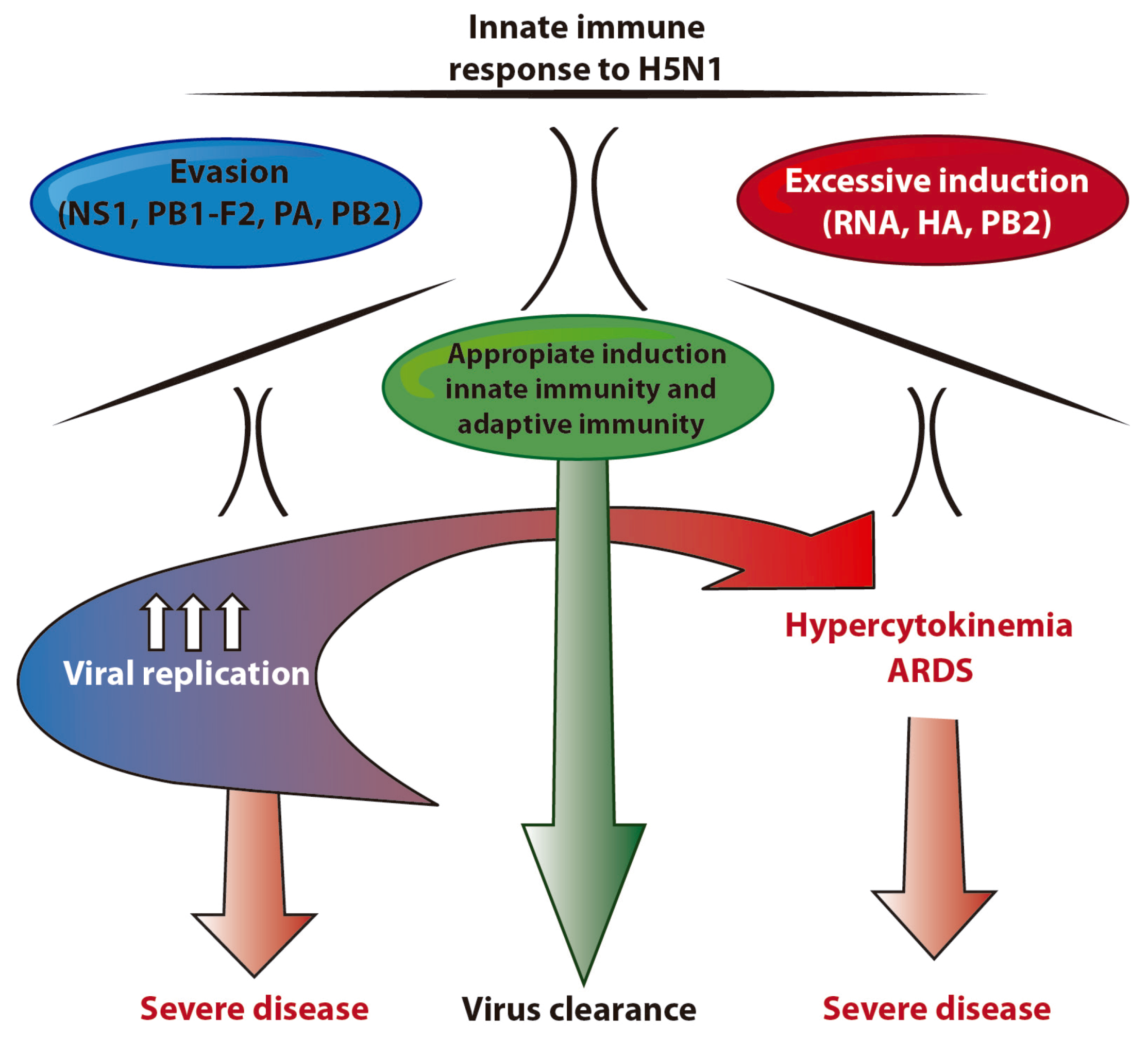
:1. H5N1 viruses and human infection
2. Hypercytokinemia Induction in Humans by H5N1: Insights from ex vivo Experimental Models

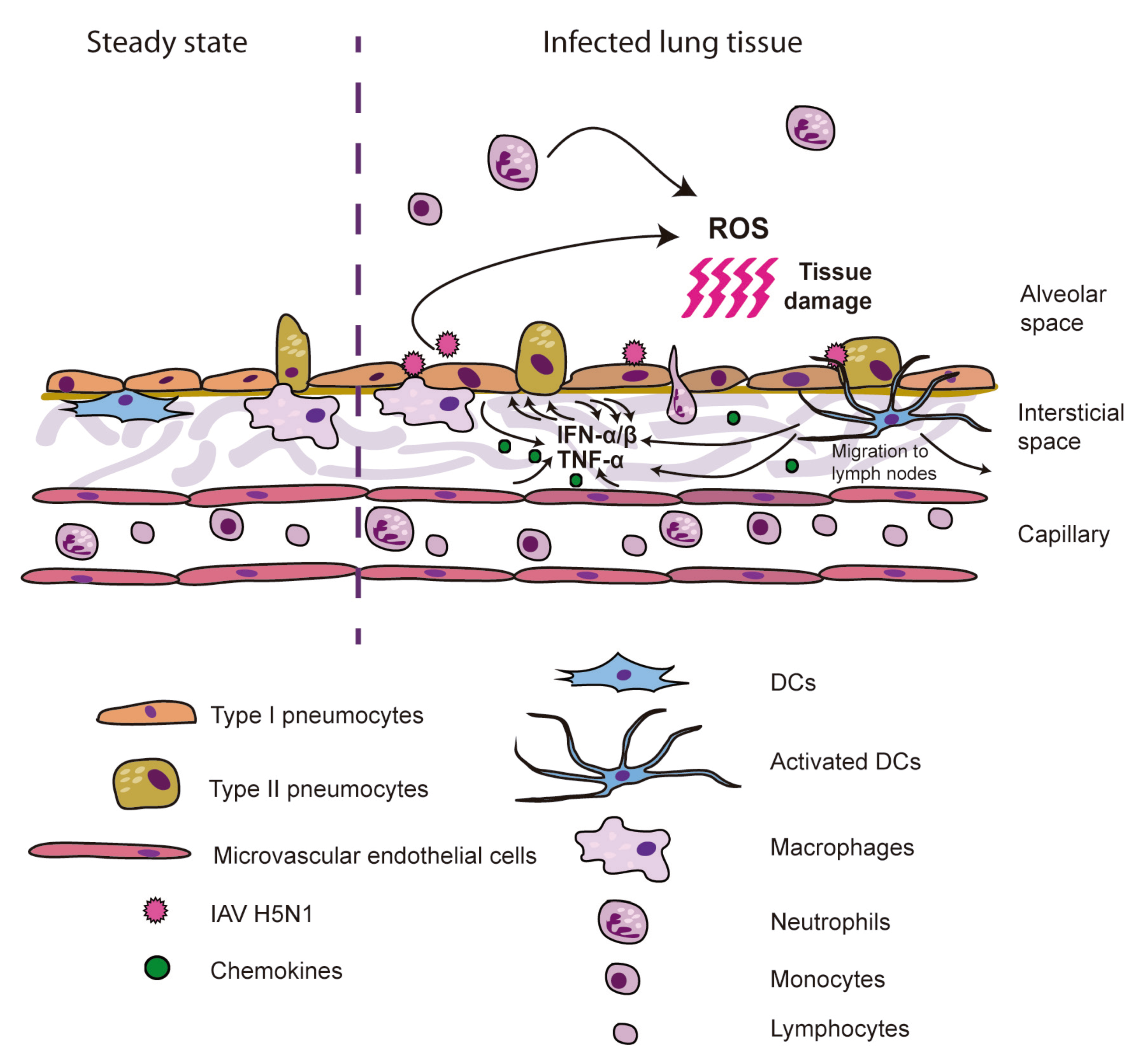{kind=link}
{kind=link}
{kind=link}
| Cell culture system | IAV subtype | Cytokine induction | Genes up-regulated | Refs. | |
|---|---|---|---|---|---|
| Non-immune cells | HTBE (non polarized) | H5N1 vs H1N1 | Higher in H5N1 infected cells | IFN-β, IP-10, RANTES, IL-6, MCP-1, IL-8 | [46] |
| Primary BECs | H5N1 vs H3N2 | Attenuated in H5N1 | IFN, PKR, RIG-I | [45] | |
| Calu-3 | H5N1 vs H3N2 | Attenuated in H5N1 | IFN, PKR, RIG-I | [45] | |
| Polarized HTBE | H5N1 vs H3N2 | Attenuated in H5N1 | IFN-β, ISGs | [51] | |
| Polarized Calu-3 | H5N1 vs H3N2 | Attenuated in H5N1 | IFN-β, ISGs | [51] | |
| Polarized HTBE | H5N1 vs H1N1 | Attenuated in H5N1 | IFN-β | [50] | |
| HTBE (non polarized) | H5N1 vs H1N1 | Higher in H5N1 infected cells | IFN-β | [50] | |
| Primary Type I pneumocytes | H5N1 vs H1N1 | Higher in H5N1 infected cells | IFN, IP-10, RANTES, IL-6 | [49] | |
| Primary Type II pneumocytes | H5N1 vs H1N1 | Higher in H5N1 infected cells | IFN-β, IP-10, RANTES, IL-6 | [46] | |
| HMVEC (Primary endothelial cells) | H5N1 vs H1N1 | Higher in H5N1 infected cells | IFN-β, IL-7, TNF, CCL2 | [52] | |
| HUVEC (Primary endothelial cells) | H5N1 and H1N1 | Higher in H5N1 infected cells | IFN-β, ISGs | [53] | |
| HTBE (non polarized) | H5N1 | Induction of IP-10 | IP-10 | [18] | |
| HMVEC (Primary endothelial cells) | H5N1 | Induction of IP-10 | IP-10 | [18] | |
| Immune cells | hMDMs | H5N1 vs H1N1 | Higher in H5N1 infected cells | TNF-α, MCP-1, RANTES, IP-10, IL-1β, IL-6, IFN-αβ, and TRAIL | [54] |
| hMDMs | H5N1 vs H1N1 | Higher in H5N1 infected cells | RIG-I, MDA5, TLR3, IFN-β, TNF-α, IP-10 | [55] | |
| hMDMs | H5N1 and H1N1 or H3N2 | Higher in H5N1 than H1N1/H3N2 infected cells | NF α, IFN-α/β,IL-1β, MCP-1, MIP-1α, RANTES, IL-12 | [56] | |
| hMDMs | H5N1 and H1N1 or H3N2 | Higher in H5N1/H3N1 than in H1N1 infected cells | TNF α, IL-6, MIP-1α, IP-10 | [57] | |
| hMDMs | H5N1 vs H1N1 | Higher in H1N1 infected cells | TNF-α, IFN-β, IFN-λ1,IFN-α, MCP-1 | [58] | |
| hMDMs | H5N1 vs H1N1 | Higher in H1N1 infected cells | TNF-α, IFN-β, IFN-λ1, IP-10 | [59] | |
| MDDCs, mDCs and pDCs | H5N1 | IFN-α, TNF-α | [60] | ||
| pDCs | H5N1 and H1N1 or H3N2 | Higher in H5N1 than H1N1/H3N2 infected cells | IFN-α, TNF-α | [61] |
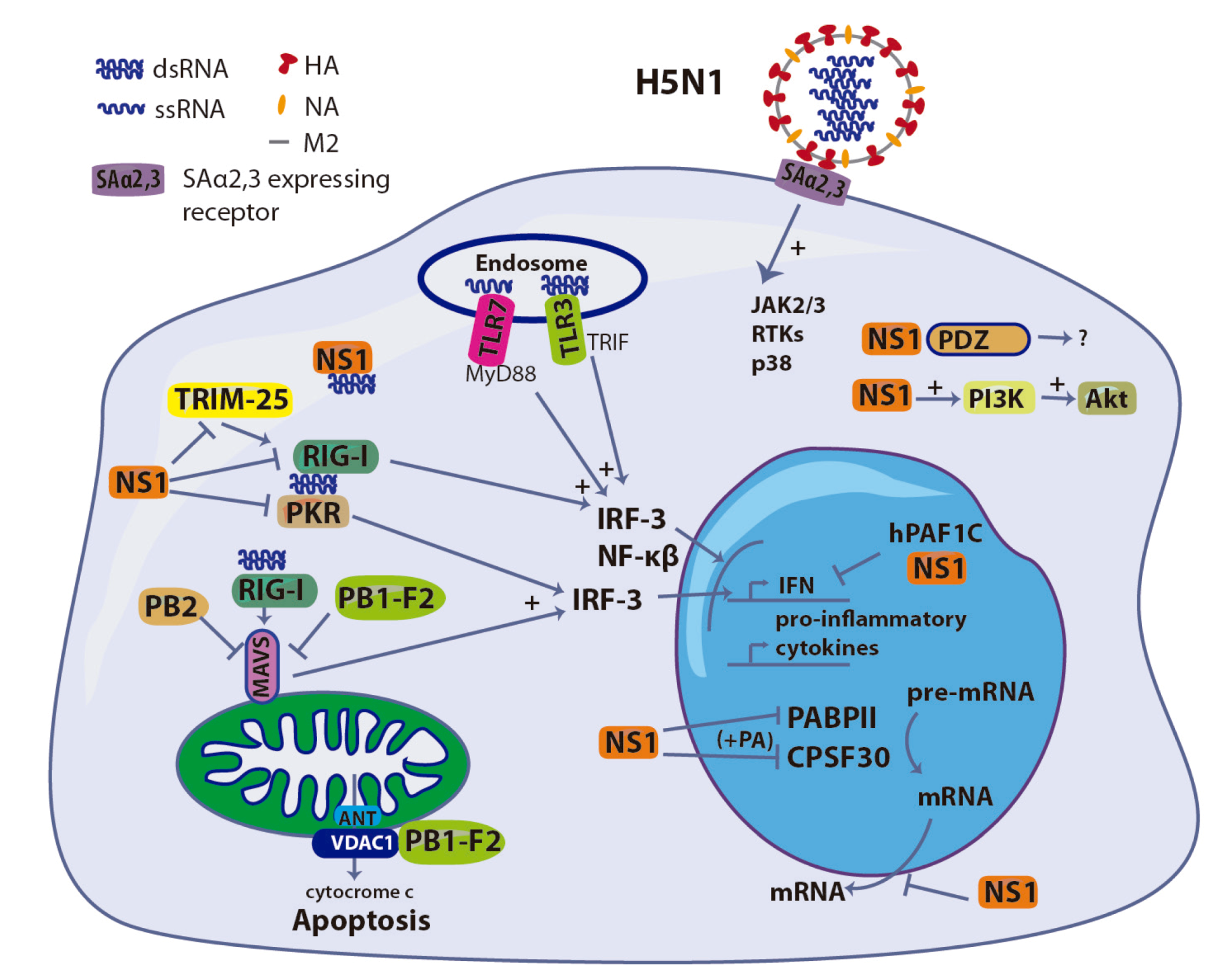
3. HA: Involvement in Entry and Cell Signaling
4. NS1: The Innate Immunity Suppressor
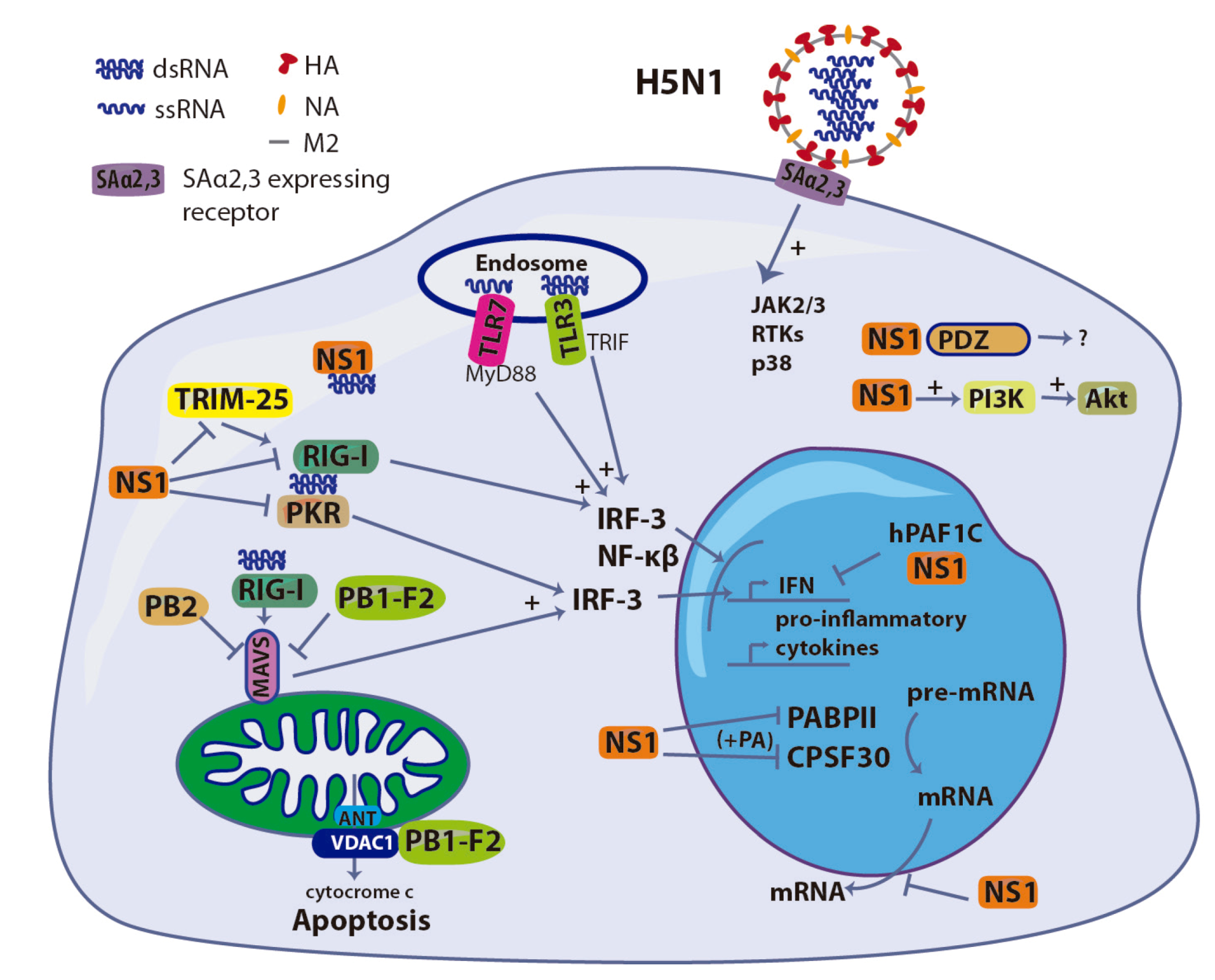
5. PB1-F2: A Double-edged Sword
6. Polymerase
7. Concluding Remarks
Conflict of Interest
Acknowledgements
References
- Palese, P.; Shaw, M.L. Orthomyxoviridae: The viruses and their replication. In Fields virology, 5th; Howley, D.M.K.P.M., Ed.; Lippincott Williams & Wilkins: Philadelphia, PA, 2007; pp. 1647–1689. [Google Scholar]
- Wise, H.M.; Foeglein, A.; Sun, J.; Dalton, R.M.; Patel, S.; Howard, W.; Anderson, E.C.; Barclay, W.S.; Digard, P. A complicated message: Identification of a novel pb1-related protein translated from influenza a virus segment 2 mrna. J. Virol. 2009, 83, 8021–8031. [Google Scholar] [CrossRef]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.A.; Wills, N.M.; Xiao, Y.L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An overlapping protein-coding region in influenza a virus segment 3 modulates the host response. Science 2012, 337, 199–204. [Google Scholar]
- Mubareka, S.; Palese, P. Influenza vaccines for the future. Rino Rappuoli, G.D.G., Ed.; 2011; pp. 3–21. [Google Scholar]
- Tong, S.; Li, Y.; Rivailler, P.; Conrardy, C.; Castillo, D.A.; Chen, L.M.; Recuenco, S.; Ellison, J.A.; Davis, C.T.; York, I.A.; et al. A distinct lineage of influenza a virus from bats. Proc. Nat. Acad. Sci. USA 2012, 109, 4269–4274. [Google Scholar]
- Medina, R.A.; Garcia-Sastre, A. Influenza a viruses: New research developments. Nature Rev. Microbiol. 2011, 9, 590–603. [Google Scholar] [CrossRef]
- Krauss, S.; Webster, R.G. Avian influenza virus surveillance and wild birds: Past and present. Avian. Dis. 2010, 54, 394–398. [Google Scholar] [CrossRef]
- Webster, R.G.; Hulse, D.J. Microbial adaptation and change: Avian influenza. Rev. Sci. Tech. 2004, 23, 453–465. [Google Scholar]
- de Jong, J.C.; Claas, E.C.; Osterhaus, A.D.; Webster, R.G.; Lim, W.L. A pandemic warning? Nature 1997, 389, 554. [Google Scholar]
- Claas, E.C.; de Jong, J.C.; van Beek, R.; Rimmelzwaan, G.F.; Osterhaus, A.D. Human influenza virus a/hongkong/156/97 (h5n1) infection. Vaccine 1998, 16, 977–978. [Google Scholar] [CrossRef]
- To, K.F.; Chan, P.K.; Chan, K.F.; Lee, W.K.; Lam, W.Y.; Wong, K.F.; Tang, N.L.; Tsang, D.N.; Sung, R.Y.; Buckley, T.A.; et al. Pathology of fatal human infection associated with avian influenza a h5n1 virus. J. Med. Virol. 2001, 63, 242–246. [Google Scholar] [CrossRef]
- Liem, N.T.; Tung, C.V.; Hien, N.D.; Hien, T.T.; Chau, N.Q.; Long, H.T.; Hien, N.T.; Mai le, Q.; Taylor, W.R.; Wertheim, H.; et al. Clinical features of human influenza a (h5n1) infection in vietnam: 2004-2006. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 2009, 48, 1639–1646. [Google Scholar] [CrossRef]
- WHO. Cumulative number of confirmed human cases of avian influenza a/(h5n1) reported to who. 2012. Available online: Http://www.Who.Int/influenza/human_animal_interface/h5n1_cumulative_table_archives/en/index.Html.
- Wang, T.T.; Parides, M.K.; Palese, P. Seroevidence for h5n1 influenza infections in humans: Meta-analysis. Science 2012, 335, 1463. [Google Scholar] [CrossRef]
- Beare, A.S.; Webster, R.G. Replication of avian influenza viruses in humans. Arch. Virol. 1991, 119, 37–42. [Google Scholar]
- Yang, Y.; Halloran, M.E.; Sugimoto, J.D.; Longini, I.M., Jr. Detecting human-to-human transmission of avian influenza a (h5n1). Emerg. Inf. Dis. 2007, 13, 1348–1353. [Google Scholar]
- de Jong, M.D.; Simmons, C.P.; Thanh, T.T.; Hien, V.M.; Smith, G.J.; Chau, T.N.; Hoang, D.M.; Chau, N.V.; Khanh, T.H.; Dong, V.C.; et al. Fatal outcome of human influenza a (h5n1) is associated with high viral load and hypercytokinemia. Nat. Med. 2006, 12, 1203–1207. [Google Scholar] [CrossRef]
- Thitithanyanont, A.; Engering, A.; Uiprasertkul, M.; Ekchariyawat, P.; Wiboon-Ut, S.; Kraivong, R.; Limsalakpetch, A.; Kum-Arb, U.; Yongvanitchit, K.; Sa-Ard-Iam, N.; et al. Antiviral immune responses in h5n1-infected human lung tissue and possible mechanisms underlying the hyperproduction of interferon-inducible protein ip-10. Biochem. Biophys. Res. Commun. 2010, 398, 752–758. [Google Scholar]
- Ng, W.F.; To, K.F.; Lam, W.W.; Ng, T.K.; Lee, K.C. The comparative pathology of severe acute respiratory syndrome and avian influenza a subtype h5n1--a review. Hum. Pathol. 2006, 37, 381–390. [Google Scholar] [CrossRef]
- Yuen, K.Y.; Chan, P.K.; Peiris, M.; Tsang, D.N.; Que, T.L.; Shortridge, K.F.; Cheung, P.T.; To, W.K.; Ho, E.T.; Sung, R.; et al. Clinical features and rapid viral diagnosis of human disease associated with avian influenza a h5n1 virus. Lancet 1998, 351, 467–471. [Google Scholar]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nature Immun. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef]
- Kanneganti, T.D. Central roles of nlrs and inflammasomes in viral infection. Nat. Rev. Immunol. 2010, 10, 688–698. [Google Scholar]
- Kovach, M.A.; Standiford, T.J. Toll like receptors in diseases of the lung. Int. Immunopharmacol. 2011, 11, 1399–1406. [Google Scholar] [CrossRef]
- Liu, P.; Jamaluddin, M.; Li, K.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Retinoic acid-inducible gene i mediates early antiviral response and toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 2007, 81, 1401–1411. [Google Scholar]
- Matsukura, S.; Kokubu, F.; Kurokawa, M.; Kawaguchi, M.; Ieki, K.; Kuga, H.; Odaka, M.; Suzuki, S.; Watanabe, S.; Takeuchi, H.; et al. Synthetic double-stranded rna induces multiple genes related to inflammation through toll-like receptor 3 depending on nf-kappab and/or irf-3 in airway epithelial cells. Clin. Exp. Allergy 2006, 36, 1049–1062. [Google Scholar]
- Matsukura, S.; Kokubu, F.; Kurokawa, M.; Kawaguchi, M.; Ieki, K.; Kuga, H.; Odaka, M.; Suzuki, S.; Watanabe, S.; Homma, T.; et al. Role of rig-i, mda-5, and pkr on the expression of inflammatory chemokines induced by synthetic dsrna in airway epithelial cells. Int. Arch. Allergy Immunol. 2007, 143 Suppl 1, 80–83. [Google Scholar] [CrossRef]
- Le Goffic, R.; Pothlichet, J.; Vitour, D.; Fujita, T.; Meurs, E.; Chignard, M.; Si-Tahar, M. Cutting edge: Influenza a virus activates tlr3-dependent inflammatory and rig-i-dependent antiviral responses in human lung epithelial cells. J. Immun. 2007, 178, 3368–3372. [Google Scholar]
- Guillot, L.; Le Goffic, R.; Bloch, S.; Escriou, N.; Akira, S.; Chignard, M.; Si-Tahar, M. Involvement of toll-like receptor 3 in the immune response of lung epithelial cells to double-stranded rna and influenza a virus. J. Biol. Chem. 2005, 280, 5571–5580. [Google Scholar]
- Baum, A.; Sachidanandam, R.; Garcia-Sastre, A. Preference of rig-i for short viral rna molecules in infected cells revealed by next-generation sequencing. Proc. Nat. Acad. Sci. USA 2010, 107, 16303–16308. [Google Scholar]
- Pulendran, B.; Palucka, K.; Banchereau, J. Sensing pathogens and tuning immune responses. Science 2001, 293, 253–256. [Google Scholar]
- McGill, J.; Heusel, J.W.; Legge, K.L. Innate immune control and regulation of influenza virus infections. J. Leukoc. Biol. 2009, 86, 803–812. [Google Scholar]
- Demedts, I.K.; Brusselle, G.G.; Vermaelen, K.Y.; Pauwels, R.A. Identification and characterization of human pulmonary dendritic cells. Am. J. Respir. Cell Mol. Biol. 2005, 32, 177–184. [Google Scholar] [CrossRef]
- van Haarst, J.M.; de Wit, H.J.; Drexhage, H.A.; Hoogsteden, H.C. Distribution and immunophenotype of mononuclear phagocytes and dendritic cells in the human lung. Am. J. Respir. Cell Mol. Biol. 1994, 10, 487–492. [Google Scholar]
- Sertl, K.; Takemura, T.; Tschachler, E.; Ferrans, V.J.; Kaliner, M.A.; Shevach, E.M. Dendritic cells with antigen-presenting capability reside in airway epithelium, lung parenchyma, and visceral pleura. J. Exp. Med. 1986, 163, 436–451. [Google Scholar] [CrossRef]
- Cochand, L.; Isler, P.; Songeon, F.; Nicod, L.P. Human lung dendritic cells have an immature phenotype with efficient mannose receptors. Am. J. Respir. Cell Mol. Biol. 1999, 21, 547–554. [Google Scholar]
- Perrot, I.; Deauvieau, F.; Massacrier, C.; Hughes, N.; Garrone, P.; Durand, I.; Demaria, O.; Viaud, N.; Gauthier, L.; Blery, M.; et al. Tlr3 and rig-like receptor on myeloid dendritic cells and rig-like receptor on human nk cells are both mandatory for production of ifn-gamma in response to double-stranded rna. J. Immun. 2010, 185, 2080–2088. [Google Scholar] [CrossRef]
- Matsumoto, M.; Funami, K.; Tanabe, M.; Oshiumi, H.; Shingai, M.; Seto, Y.; Yamamoto, A.; Seya, T. Subcellular localization of toll-like receptor 3 in human dendritic cells. J. Immun. 2003, 171, 3154–3162. [Google Scholar]
- Jarrossay, D.; Napolitani, G.; Colonna, M.; Sallusto, F.; Lanzavecchia, A. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur. J. Immunol. 2001, 31, 3388–3393. [Google Scholar] [CrossRef]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of tlr7-mediated recognition of single-stranded rna. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded rna viruses by toll-like receptor 7. Proc. Nat. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar]
- Maris, N.A.; Dessing, M.C.; de Vos, A.F.; Bresser, P.; van der Zee, J.S.; Jansen, H.M.; Spek, C.A.; van der Poll, T. Toll-like receptor mrna levels in alveolar macrophages after inhalation of endotoxin. Eur. Respir. J. 2006, 28, 622–626. [Google Scholar] [CrossRef]
- Striz, I.; Wang, Y.M.; Svarcova, I.; Trnka, L.; Sorg, C.; Costabel, U. The phenotype of alveolar macrophages and its correlation with immune cells in bronchoalveolar lavage. Eur. Respir. J. 1993, 6, 1287–1294. [Google Scholar]
- Ohman, T.; Rintahaka, J.; Kalkkinen, N.; Matikainen, S.; Nyman, T.A. Actin and rig-i/mavs signaling components translocate to mitochondria upon influenza a virus infection of human primary macrophages. J. Immun. 2009, 182, 5682–5692. [Google Scholar] [CrossRef]
- Hsu, A.C.; Parsons, K.; Barr, I.; Lowther, S.; Middleton, D.; Hansbro, P.M.; Wark, P.A. Critical role of constitutive type i interferon response in bronchial epithelial cell to influenza infection. PLoS One 2012, 7, e32947. [Google Scholar]
- Chan, M.C.; Cheung, C.Y.; Chui, W.H.; Tsao, S.W.; Nicholls, J.M.; Chan, Y.O.; Chan, R.W.; Long, H.T.; Poon, L.L.; Guan, Y.; et al. Proinflammatory cytokine responses induced by influenza a (h5n1) viruses in primary human alveolar and bronchial epithelial cells. Respir. Res. 2005, 6, 135. [Google Scholar]
- Gray, T.E.; Guzman, K.; Davis, C.W.; Abdullah, L.H.; Nettesheim, P. Mucociliary differentiation of serially passaged normal human tracheobronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 1996, 14, 104–112. [Google Scholar]
- Matrosovich, M.N.; Matrosovich, T.Y.; Gray, T.; Roberts, N.A.; Klenk, H.D. Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc. Nat. Acad. Sci. USA 2004, 101, 4620–4624. [Google Scholar]
- Chan, M.C.; Chan, R.W.; Yu, W.C.; Ho, C.C.; Chui, W.H.; Lo, C.K.; Yuen, K.M.; Guan, Y.I.; Nicholls, J.M.; Peiris, J.S. Influenza h5n1 virus infection of polarized human alveolar epithelial cells and lung microvascular endothelial cells. Resp. Res. 2009, 10, 102. [Google Scholar]
- Chan, R.W.; Yuen, K.M.; Yu, W.C.; Ho, C.C.; Nicholls, J.M.; Peiris, J.S.; Chan, M.C. Influenza h5n1 and h1n1 virus replication and innate immune responses in bronchial epithelial cells are influenced by the state of differentiation. PLoS One 2010, 5, e8713. [Google Scholar]
- Zeng, H.; Goldsmith, C.; Thawatsupha, P.; Chittaganpitch, M.; Waicharoen, S.; Zaki, S.; Tumpey, T.M.; Katz, J.M. Highly pathogenic avian influenza h5n1 viruses elicit an attenuated type i interferon response in polarized human bronchial epithelial cells. J. Virol. 2007, 81, 12439–12449. [Google Scholar] [CrossRef]
- Zeng, H.; Pappas, C.; Belser, J.A.; Houser, K.V.; Zhong, W.; Wadford, D.A.; Stevens, T.; Balczon, R.; Katz, J.M.; Tumpey, T.M. Human pulmonary microvascular endothelial cells support productive replication of highly pathogenic avian influenza viruses: Possible involvement in the pathogenesis of human h5n1 virus infection. J. Virol. 2012, 86, 667–678. [Google Scholar]
- Schmolke, M.; Viemann, D.; Roth, J.; Ludwig, S. Essential impact of nf-kappab signaling on the h5n1 influenza a virus-induced transcriptome. J. Immun. 2009, 183, 5180–5189. [Google Scholar] [CrossRef]
- Hui, K.P.; Lee, S.M.; Cheung, C.Y.; Ng, I.H.; Poon, L.L.; Guan, Y.; Ip, N.Y.; Lau, A.S.; Peiris, J.S. Induction of proinflammatory cytokines in primary human macrophages by influenza a virus (h5n1) is selectively regulated by ifn regulatory factor 3 and p38 mapk. J. Immun. 2009, 182, 1088–1098. [Google Scholar]
- Lee, S.M.; Cheung, C.Y.; Nicholls, J.M.; Hui, K.P.; Leung, C.Y.; Uiprasertkul, M.; Tipoe, G.L.; Lau, Y.L.; Poon, L.L.; Ip, N.Y.; et al. Hyperinduction of cyclooxygenase-2-mediated proinflammatory cascade: A mechanism for the pathogenesis of avian influenza h5n1 infection. J. Infect. Dis. 2008, 198, 525–535. [Google Scholar]
- Cheung, C.Y.; Poon, L.L.; Lau, A.S.; Luk, W.; Lau, Y.L.; Shortridge, K.F.; Gordon, S.; Guan, Y.; Peiris, J.S. Induction of proinflammatory cytokines in human macrophages by influenza a (h5n1) viruses: A mechanism for the unusual severity of human disease? Lancet 2002, 360, 1831–1837. [Google Scholar]
- Geiler, J.; Michaelis, M.; Sithisarn, P.; Cinatl, J., Jr. Comparison of pro-inflammatory cytokine expression and cellular signal transduction in human macrophages infected with different influenza a viruses. Med. Microbiol. Immunol. 2011, 200, 53–60. [Google Scholar] [CrossRef]
- Lee, D.C.; Law, A.H.; Hui, K.; Tam, A.H.; Peiris, J.S.; Lau, A.S. Interferon dysregulation and virus-induced cell death in avian influenza h5n1 virus infections. Hong Kong Med. J. 2012, 18 Suppl 2, 12–16. [Google Scholar]
- Hui, K.P.; Lee, S.M.; Cheung, C.Y.; Mao, H.; Lai, A.K.; Chan, R.W.; Chan, M.C.; Tu, W.; Guan, Y.; Lau, Y.L.; et al. H5n1 influenza virus-induced mediators upregulate rig-i in uninfected cells by paracrine effects contributing to amplified cytokine cascades. J. Infect. Dis. 2011, 204, 1866–1878. [Google Scholar] [CrossRef]
- Thitithanyanont, A.; Engering, A.; Ekchariyawat, P.; Wiboon-ut, S.; Limsalakpetch, A.; Yongvanitchit, K.; Kum-Arb, U.; Kanchongkittiphon, W.; Utaisincharoen, P.; Sirisinha, S.; et al. High susceptibility of human dendritic cells to avian influenza h5n1 virus infection and protection by ifn-alpha and tlr ligands. J. Immun. 2007, 179, 5220–5227. [Google Scholar]
- Sandbulte, M.R.; Boon, A.C.; Webby, R.J.; Riberdy, J.M. Analysis of cytokine secretion from human plasmacytoid dendritic cells infected with h5n1 or low-pathogenicity influenza viruses. Virology 2008, 381, 22–28. [Google Scholar]
- Viemann, D.; Schmolke, M.; Lueken, A.; Boergeling, Y.; Friesenhagen, J.; Wittkowski, H.; Ludwig, S.; Roth, J. H5n1 virus activates signaling pathways in human endothelial cells resulting in a specific imbalanced inflammatory response. J. Immun. 2011, 186, 164–173. [Google Scholar] [CrossRef]
- Peschke, T.; Bender, A.; Nain, M.; Gemsa, D. Role of macrophage cytokines in influenza a virus infections. Immunobiol. 1993, 189, 340–355. [Google Scholar] [CrossRef]
- Hofmann, P.; Sprenger, H.; Kaufmann, A.; Bender, A.; Hasse, C.; Nain, M.; Gemsa, D. Susceptibility of mononuclear phagocytes to influenza a virus infection and possible role in the antiviral response. J. Leukoc. Biol. 1997, 61, 408–414. [Google Scholar]
- Lehmann, C.; Sprenger, H.; Nain, M.; Bacher, M.; Gemsa, D. Infection of macrophages by influenza a virus: Characteristics of tumour necrosis factor-alpha (tnf alpha) gene expression. Res. Virol. 1996, 147, 123–130. [Google Scholar]
- Ramos, I.; Bernal-Rubio, D.; Durham, N.; Belicha-Villanueva, A.; Lowen, A.C.; Steel, J.; Fernandez-Sesma, A. Effects of receptor binding specificity of avian influenza virus on the human innate immune response. J. Virol. 2011, 85, 4421–4431. [Google Scholar] [CrossRef]
- Wang, J.; Nikrad, M.P.; Travanty, E.A.; Zhou, B.; Phang, T.; Gao, B.; Alford, T.; Ito, Y.; Nahreini, P.; Hartshorn, K.; et al. Innate immune response of human alveolar macrophages during influenza a infection. PLoS One 2012, 7, e29879. [Google Scholar]
- Sakabe, S.; Iwatsuki-Horimoto, K.; Takano, R.; Nidom, C.A.; Le, M.; Nagamura-Inoue, T.; Horimoto, T.; Yamashita, N.; Kawaoka, Y. Cytokine production by primary human macrophages infected with highly pathogenic h5n1 or pandemic h1n1 2009 influenza viruses. J. Virol. 2011, 92, 1428–1434. [Google Scholar] [CrossRef]
- Lee, S.M.; Gardy, J.L.; Cheung, C.Y.; Cheung, T.K.; Hui, K.P.; Ip, N.Y.; Guan, Y.; Hancock, R.E.; Peiris, J.S. Systems-level comparison of host-responses elicited by avian h5n1 and seasonal h1n1 influenza viruses in primary human macrophages. PLoS One 2009, 4, e8072. [Google Scholar]
- Cheung, C.Y.; Chan, E.Y.; Krasnoselsky, A.; Purdy, D.; Navare, A.T.; Bryan, J.T.; Leung, C.K.; Hui, K.P.; Peiris, J.S.; Katze, M.G. H5N1 virus causes significant perturbations in host proteome very early in influenza virus-infected primary human monocyte-derived macrophages. J. Infect. Dis. 2012, 206, 640–645. [Google Scholar]
- Zhou, J.; Law, H.K.; Cheung, C.Y.; Ng, I.H.; Peiris, J.S.; Lau, Y.L. Differential expression of chemokines and their receptors in adult and neonatal macrophages infected with human or avian influenza viruses. J. Infect. Dis. 2006, 194, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Ekchariyawat, P.; Thitithanyanont, A.; Sirisinha, S.; Utaisincharoen, P. Apoptosis induced by avian h5n1 virus in human monocyte-derived macrophages involves trail-inducing caspase-10 activation. Innate Immun. 2012, 18, 390–397. [Google Scholar] [CrossRef]
- Mok, C.K.; Lee, D.C.; Cheung, C.Y.; Peiris, M.; Lau, A.S. Differential onset of apoptosis in influenza a virus h5n1- and h1n1-infected human blood macrophages. J. Gen. Virol. 2007, 88, 1275–1280. [Google Scholar] [CrossRef]
- Haye, K.; Burmakina, S.; Moran, T.; Garcia-Sastre, A.; Fernandez-Sesma, A. The ns1 protein of a human influenza virus inhibits type i interferon production and the induction of antiviral responses in primary human dendritic and respiratory epithelial cells. J. Virol. 2009, 83, 6849–6862. [Google Scholar]
- Weinheimer, V.K.; Becher, A.; Tonnies, M.; Holland, G.; Knepper, J.; Bauer, T.T.; Schneider, P.; Neudecker, J.; Ruckert, J.C.; Szymanski, K.; et al. Influenza a viruses target type ii pneumocytes in the human lung. J. Infect. Dis. 2012, 206, 1685–1694. [Google Scholar] [CrossRef]
- Chabot, F.; Mitchell, J.A.; Gutteridge, J.M.; Evans, T.W. Reactive oxygen species in acute lung injury. Eur. Respir. J. 1998, 11, 745–757. [Google Scholar]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef]
- Smits, S.L.; van den Brand, J.M.; de Lang, A.; Leijten, L.M.; van Ijcken, W.F.; van Amerongen, G.; Osterhaus, A.D.; Andeweg, A.C.; Haagmans, B.L. Distinct severe acute respiratory syndrome coronavirus-induced acute lung injury pathways in two different nonhuman primate species. J. Virol. 2011, 85, 4234–4245. [Google Scholar]
- Chow, C.W.; Herrera Abreu, M.T.; Suzuki, T.; Downey, G.P. Oxidative stress and acute lung injury. Am. J. Respir. Cell Mol. Biol. 2003, 29, 427–431. [Google Scholar]
- Weis, W.; Brown, J.H.; Cusack, S.; Paulson, J.C.; Skehel, J.J.; Wiley, D.C. Structure of the influenza virus haemagglutinin complexed with its receptor, sialic acid. Nature 1988, 333, 426–431. [Google Scholar] [CrossRef]
- Wiley, D.C.; Skehel, J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Ann. Rev. Biochem. 1987, 56, 365–394. [Google Scholar]
- Shinya, K.; Ebina, M.; Yamada, S.; Ono, M.; Kasai, N.; Kawaoka, Y. Avian flu: Influenza virus receptors in the human airway. Nature 2006, 440, 435–436. [Google Scholar]
- van Riel, D.; Munster, V.J.; de Wit, E.; Rimmelzwaan, G.F.; Fouchier, R.A.; Osterhaus, A.D.; Kuiken, T. H5n1 virus attachment to lower respiratory tract. Science 2006, 312, 399. [Google Scholar] [CrossRef]
- Nicholls, J.M.; Chan, M.C.; Chan, W.Y.; Wong, H.K.; Cheung, C.Y.; Kwong, D.L.; Wong, M.P.; Chui, W.H.; Poon, L.L.; Tsao, S.W.; et al. Tropism of avian influenza a (h5n1) in the upper and lower respiratory tract. Nat. Med. 2007, 13, 147–149. [Google Scholar]
- Rogers, G.N.; Paulson, J.C. Receptor determinants of human and animal influenza virus isolates: Differences in receptor specificity of the h3 hemagglutinin based on species of origin. Virology 1983, 127, 361–373. [Google Scholar] [CrossRef]
- Herfst, S.; Schrauwen, E.J.; Linster, M.; Chutinimitkul, S.; de Wit, E.; Munster, V.J.; Sorrell, E.M.; Bestebroer, T.M.; Burke, D.F.; Smith, D.J.; et al. Airborne transmission of influenza a/h5n1 virus between ferrets. Science 2012, 336, 1534–1541. [Google Scholar]
- Imai, M.; Watanabe, T.; Hatta, M.; Das, S.C.; Ozawa, M.; Shinya, K.; Zhong, G.; Hanson, A.; Katsura, H.; Watanabe, S.; et al. Experimental adaptation of an influenza h5 ha confers respiratory droplet transmission to a reassortant h5 ha/h1n1 virus in ferrets. Nature 2012, 486, 420–428. [Google Scholar]
- Imai, M.; Kawaoka, Y. The role of receptor binding specificity in interspecies transmission of influenza viruses. Curr. Opin. Virol. 2012, 2, 160–167. [Google Scholar]
- Steel, J.; Lowen, A.C.; Mubareka, S.; Palese, P. Transmission of influenza virus in a mammalian host is increased by pb2 amino acids 627k or 627e/701n. PLoS Pathog. 2009, 5, e1000252. [Google Scholar] [CrossRef]
- Van Hoeven, N.; Pappas, C.; Belser, J.A.; Maines, T.R.; Zeng, H.; Garcia-Sastre, A.; Sasisekharan, R.; Katz, J.M.; Tumpey, T.M. Human ha and polymerase subunit pb2 proteins confer transmission of an avian influenza virus through the air. Proc. Nat. Acad. Sci. USA 2009, 106, 3366–3371. [Google Scholar]
- Ocana-Macchi, M.; Bel, M.; Guzylack-Piriou, L.; Ruggli, N.; Liniger, M.; McCullough, K.C.; Sakoda, Y.; Isoda, N.; Matrosovich, M.; Summerfield, A. Hemagglutinin-dependent tropism of h5n1 avian influenza virus for human endothelial cells. J. Virol. 2009, 83, 12947–12955. [Google Scholar]
- Ramos, I.; Fernandez-Sesma, A. Cell receptors for influenza a viruses and the innate immune response. Front. Microbiol. 2012, 3, 117. [Google Scholar]
- Xu, W.; Chen, M.; Ge, N.; Xu, J. Hemagglutinin from the h5n1 virus activates janus kinase 3 to dysregulate innate immunity. PLoS One 2012, 7, e31721. [Google Scholar]
- Liu, W.C.; Lin, S.C.; Yu, Y.L.; Chu, C.L.; Wu, S.C. Dendritic cell activation by recombinant hemagglutinin proteins of h1n1 and h5n1 influenza a viruses. J. Vvirol. 2010, 84, 12011–12017. [Google Scholar] [CrossRef]
- Eierhoff, T.; Hrincius, E.R.; Rescher, U.; Ludwig, S.; Ehrhardt, C. The epidermal growth factor receptor (egfr) promotes uptake of influenza a viruses (iav) into host cells. PLoS Pathog. 2010, 6, e1001099. [Google Scholar] [CrossRef]
- Marchant, D.; Singhera, G.K.; Utokaparch, S.; Hackett, T.L.; Boyd, J.H.; Luo, Z.; Si, X.; Dorscheid, D.R.; McManus, B.M.; Hegele, R.G. Toll-like receptor 4-mediated activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. J. Virol. 2010, 84, 11359–11373. [Google Scholar]
- Suguitan, A.L., Jr.; Matsuoka, Y.; Lau, Y.F.; Santos, C.P.; Vogel, L.; Cheng, L.I.; Orandle, M.; Subbarao, K. The multibasic cleavage site of the hemagglutinin of highly pathogenic a/vietnam/1203/2004 (h5n1) avian influenza virus acts as a virulence factor in a host-specific manner in mammals. J. Virol. 2012, 86, 2706–2714. [Google Scholar]
- Horimoto, T.; Nakayama, K.; Smeekens, S.P.; Kawaoka, Y. Proprotein-processing endoproteases pc6 and furin both activate hemagglutinin of virulent avian influenza viruses. J. Virol. 1994, 68, 6074–6078. [Google Scholar]
- Horimoto, T.; Kawaoka, Y. Reverse genetics provides direct evidence for a correlation of hemagglutinin cleavability and virulence of an avian influenza a virus. J. Virol. 1994, 68, 3120–3128. [Google Scholar]
- Stieneke-Grober, A.; Vey, M.; Angliker, H.; Shaw, E.; Thomas, G.; Roberts, C.; Klenk, H.D.; Garten, W. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J. 1992, 11, 2407–2414. [Google Scholar]
- Barbey-Morel, C.L.; Oeltmann, T.N.; Edwards, K.M.; Wright, P.F. Role of respiratory tract proteases in infectivity of influenza a virus. J. Infect. Dis. 1987, 155, 667–672. [Google Scholar]
- Schrauwen, E.J.; Bestebroer, T.M.; Munster, V.J.; de Wit, E.; Herfst, S.; Rimmelzwaan, G.F.; Osterhaus, A.D.; Fouchier, R.A. Insertion of a multibasic cleavage site in the haemagglutinin of human influenza h3n2 virus does not increase pathogenicity in ferrets. J. Gen. Virol. 2011, 92, 1410–1415. [Google Scholar] [CrossRef]
- Schrauwen, E.J.; Herfst, S.; Leijten, L.M.; van Run, P.; Bestebroer, T.M.; Linster, M.; Bodewes, R.; Kreijtz, J.H.; Rimmelzwaan, G.F.; Osterhaus, A.D.; et al. The multibasic cleavage site in h5n1 virus is critical for systemic spread along the olfactory and hematogenous routes in ferrets. J. Virol. 2012, 86, 3975–3984. [Google Scholar]
- Brandstadter, J.D.; Yang, Y. Natural killer cell responses to viral infection. J. Innate Immun. 2011, 3, 274–279. [Google Scholar]
- Arnon, T.I.; Lev, M.; Katz, G.; Chernobrov, Y.; Porgador, A.; Mandelboim, O. Recognition of viral hemagglutinins by nkp44 but not by nkp30. Eur. J. Immunol. 2001, 31, 2680–2689. [Google Scholar] [CrossRef]
- Mandelboim, O.; Lieberman, N.; Lev, M.; Paul, L.; Arnon, T.I.; Bushkin, Y.; Davis, D.M.; Strominger, J.L.; Yewdell, J.W.; Porgador, A. Recognition of haemagglutinins on virus-infected cells by nkp46 activates lysis by human nk cells. Nature 2001, 409, 1055–1060. [Google Scholar]
- Ho, J.W.; Hershkovitz, O.; Peiris, M.; Zilka, A.; Bar-Ilan, A.; Nal, B.; Chu, K.; Kudelko, M.; Kam, Y.W.; Achdout, H.; et al. H5-type influenza virus hemagglutinin is functionally recognized by the natural killer-activating receptor nkp44. J. Virol. 2008, 82, 2028–2032. [Google Scholar] [CrossRef]
- Achdout, H.; Meningher, T.; Hirsh, S.; Glasner, A.; Bar-On, Y.; Gur, C.; Porgador, A.; Mendelson, M.; Mandelboim, M.; Mandelboim, O. Killing of avian and swine influenza virus by natural killer cells. J. Virol. 2010, 84, 3993–4001. [Google Scholar]
- Du, N.; Zhou, J.; Lin, X.; Zhang, Y.; Yang, X.; Wang, Y.; Shu, Y. Differential activation of nk cells by influenza a pseudotype h5n1 and 1918 and 2009 pandemic h1n1 viruses. J. Virol. 2010, 84, 7822–7831. [Google Scholar]
- Stevens, J.; Blixt, O.; Paulson, J.C.; Wilson, I.A. Glycan microarray technologies: Tools to survey host specificity of influenza viruses. Nat. Rev. Microbiol. 2006, 4, 857–864. [Google Scholar] [CrossRef]
- Stevens, J.; Blixt, O.; Tumpey, T.M.; Taubenberger, J.K.; Paulson, J.C.; Wilson, I.A. Structure and receptor specificity of the hemagglutinin from an h5n1 influenza virus. Science 2006, 312, 404–410. [Google Scholar] [CrossRef]
- Wang, X.; Li, M.; Zheng, H.; Muster, T.; Palese, P.; Beg, A.A.; Garcia-Sastre, A. Influenza a virus ns1 protein prevents activation of nf-kappab and induction of alpha/beta interferon. J. Virol. 2000, 74, 11566–11573. [Google Scholar]
- Garcia-Sastre, A. Induction and evasion of type i interferon responses by influenza viruses. Virus Res. 2011, 162, 12–18. [Google Scholar]
- Hale, B.G.; Randall, R.E.; Ortin, J.; Jackson, D. The multifunctional ns1 protein of influenza a viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef]
- Lu, Y.; Wambach, M.; Katze, M.G.; Krug, R.M. Binding of the influenza virus ns1 protein to double-stranded rna inhibits the activation of the protein kinase that phosphorylates the elf-2 translation initiation factor. Virology 1995, 214, 222–228. [Google Scholar] [CrossRef]
- Min, J.Y.; Krug, R.M. The primary function of rna binding by the influenza a virus ns1 protein in infected cells: Inhibiting the 2'-5' oligo (a) synthetase/rnase l pathway. Proc. Nat. Acad. Sci. USA 2006, 103, 7100–7105. [Google Scholar]
- Bergmann, M.; Garcia-Sastre, A.; Carnero, E.; Pehamberger, H.; Wolff, K.; Palese, P.; Muster, T. Influenza virus ns1 protein counteracts pkr-mediated inhibition of replication. J. Virol. 2000, 74, 6203–6206. [Google Scholar] [CrossRef]
- Tan, S.L.; Katze, M.G. Biochemical and genetic evidence for complex formation between the influenza a virus ns1 protein and the interferon-induced pkr protein kinase. J. Interferon. Cytokine Res. 1998, 18, 757–766. [Google Scholar] [CrossRef]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; Garcia-Sastre, A. Influenza a virus ns1 targets the ubiquitin ligase trim25 to evade recognition by the host viral rna sensor rig-i. Cell Host Microbe 2009, 5, 439–449. [Google Scholar]
- Mibayashi, M.; Martinez-Sobrido, L.; Loo, Y.M.; Cardenas, W.B.; Gale, M., Jr.; Garcia-Sastre, A. Inhibition of retinoic acid-inducible gene i-mediated induction of beta interferon by the ns1 protein of influenza a virus. J. Virol. 2007, 81, 514–524. [Google Scholar] [CrossRef]
- Nemeroff, M.E.; Barabino, S.M.; Li, Y.; Keller, W.; Krug, R.M. Influenza virus ns1 protein interacts with the cellular 30 kda subunit of cpsf and inhibits 3'end formation of cellular pre-mrnas. Mol. Cell 1998, 1, 991–1000. [Google Scholar]
- Noah, D.L.; Twu, K.Y.; Krug, R.M. Cellular antiviral responses against influenza a virus are countered at the posttranscriptional level by the viral ns1a protein via its binding to a cellular protein required for the 3' end processing of cellular pre-mrnas. Virology 2003, 307, 386–395. [Google Scholar] [CrossRef]
- Chen, Z.; Li, Y.; Krug, R.M. Influenza a virus ns1 protein targets poly(a)-binding protein ii of the cellular 3'-end processing machinery. EMBO J. 1999, 18, 2273–2283. [Google Scholar]
- Gao, S.; Song, L.; Li, J.; Zhang, Z.; Peng, H.; Jiang, W.; Wang, Q.; Kang, T.; Chen, S.; Huang, W. Influenza a virus-encoded ns1 virulence factor protein inhibits innate immune response by targeting ikk. Cell Microbiol. 2012, 14, 1849–1866. [Google Scholar] [CrossRef]
- Burgui, I.; Aragon, T.; Ortin, J.; Nieto, A. Pabp1 and eif4gi associate with influenza virus ns1 protein in viral mrna translation initiation complexes. J. Gen. Virol. 2003, 84, 3263–3274. [Google Scholar]
- Aragon, T.; de la Luna, S.; Novoa, I.; Carrasco, L.; Ortin, J.; Nieto, A. Eukaryotic translation initiation factor 4gi is a cellular target for ns1 protein, a translational activator of influenza virus. Mol. Cell. Biol. 2000, 20, 6259–6268. [Google Scholar] [CrossRef]
- Hale, B.G.; Jackson, D.; Chen, Y.H.; Lamb, R.A.; Randall, R.E. Influenza a virus ns1 protein binds p85beta and activates phosphatidylinositol-3-kinase signaling. Proc. Nat. Acad. Sci. USA 2006, 103, 14194–14199. [Google Scholar]
- Marazzi, I.; Ho, J.S.; Kim, J.; Manicassamy, B.; Dewell, S.; Albrecht, R.A.; Seibert, C.W.; Schaefer, U.; Jeffrey, K.L.; Prinjha, R.K.; et al. Suppression of the antiviral response by an influenza histone mimic. Nature 2012, 483, 428–433. [Google Scholar] [CrossRef]
- Kainov, D.E.; Muller, K.H.; Theisen, L.L.; Anastasina, M.; Kaloinen, M.; Muller, C.P. Differential effects of ns1 proteins of human pandemic h1n1/2009, avian highly pathogenic h5n1, and low pathogenic h5n2 influenza a viruses on cellular pre-mrna polyadenylation and mrna translation. J. Biol. Chem. 2011, 286, 7239–7247. [Google Scholar]
- Kochs, G.; Garcia-Sastre, A.; Martinez-Sobrido, L. Multiple anti-interferon actions of the influenza a virus ns1 protein. J. Virol. 2007.
- Das, K.; Ma, L.C.; Xiao, R.; Radvansky, B.; Aramini, J.; Zhao, L.; Marklund, J.; Kuo, R.L.; Twu, K.Y.; Arnold, E.; et al. Structural basis for suppression of a host antiviral response by influenza a virus. Proc. Nat. Acad. Sci. USA 2008, 105, 13093–13098. [Google Scholar]
- Kuo, R.L.; Krug, R.M. Influenza a virus polymerase is an integral component of the cpsf30-ns1a protein complex in infected cells. J. Virol. 2009, 83, 1611–1616. [Google Scholar] [CrossRef]
- Twu, K.Y.; Noah, D.L.; Rao, P.; Kuo, R.L.; Krug, R.M. The cpsf30 binding site on the ns1a protein of influenza a virus is a potential antiviral target. J. Virol. 2006, 80, 3957–3965. [Google Scholar]
- Spesock, A.; Malur, M.; Hossain, M.J.; Chen, L.M.; Njaa, B.L.; Davis, C.T.; Lipatov, A.S.; York, I.A.; Krug, R.M.; Donis, R.O. The virulence of 1997 h5n1 influenza viruses in the mouse model is increased by correcting a defect in their ns1 proteins. J. Virol. 2011, 85, 7048–7058. [Google Scholar]
- Twu, K.Y.; Kuo, R.L.; Marklund, J.; Krug, R.M. The h5n1 influenza virus ns genes selected after 1998 enhance virus replication in mammalian cells. J. Virol. 2007, 81, 8112–8121. [Google Scholar]
- Obenauer, J.C.; Denson, J.; Mehta, P.K.; Su, X.; Mukatira, S.; Finkelstein, D.B.; Xu, X.; Wang, J.; Ma, J.; Fan, Y.; et al. Large-scale sequence analysis of avian influenza isolates. Science 2006, 311, 1576–1580. [Google Scholar] [CrossRef]
- Zielecki, F.; Semmler, I.; Kalthoff, D.; Voss, D.; Mauel, S.; Gruber, A.D.; Beer, M.; Wolff, T. Virulence determinants of avian h5n1 influenza a virus in mammalian and avian hosts: Role of the c-terminal esev motif in the viral ns1 protein. J. Virol. 2010, 84, 10708–10718. [Google Scholar] [CrossRef]
- Jackson, D.; Hossain, M.J.; Hickman, D.; Perez, D.R.; Lamb, R.A. A new influenza virus virulence determinant: The ns1 protein four c-terminal residues modulate pathogenicity. Proc. Natl. Acad. Sci. USA 2008, 105, 4381–4386. [Google Scholar]
- Sheng, M.; Sala, C. Pdz domains and the organization of supramolecular complexes. Annu Rev Neurosci. 2001, 24, 1–29. [Google Scholar]
- Zhang, H.; Li, W.; Wang, G.; Su, Y.; Zhang, C.; Chen, X.; Xu, Y.; Li, K. The distinct binding properties between avian/human influenza a virus ns1 and postsynaptic density protein-95 (psd-95), and inhibition of nitric oxide production. J. Virol. 2011, 8, 298. [Google Scholar] [CrossRef]
- Bavagnoli, L.; Dundon, W.G.; Garbelli, A.; Zecchin, B.; Milani, A.; Parakkal, G.; Baldanti, F.; Paolucci, S.; Volmer, R.; Tu, Y.; et al. The pdz-ligand and src-homology type 3 domains of epidemic avian influenza virus ns1 protein modulate human src kinase activity during viral infection. PLoS One 2011, 6, e27789. [Google Scholar]
- Golebiewski, L.; Liu, H.; Javier, R.T.; Rice, A.P. The avian influenza virus ns1 esev pdz binding motif associates with dlg1 and scribble to disrupt cellular tight junctions. J. Virol. 2011, 85, 10639–10648. [Google Scholar]
- Yu, J.; Li, X.; Wang, Y.; Li, B.; Li, H.; Li, Y.; Zhou, W.; Zhang, C.; Rao, Z.; Bartlam, M.; et al. Pdlim2 selectively interacts with the pdz binding motif of highly pathogenic avian h5n1 influenza a virus ns1. PLoS One 2011, 6, e19511. [Google Scholar]
- Liu, H.; Golebiewski, L.; Dow, E.C.; Krug, R.M.; Javier, R.T.; Rice, A.P. The esev pdz-binding motif of the avian influenza a virus ns1 protein protects infected cells from apoptosis by directly targeting scribble. J. Virol. 2010, 84, 11164–11174. [Google Scholar]
- Chen, W.; Calvo, P.A.; Malide, D.; Gibbs, J.; Schubert, U.; Bacik, I.; Basta, S.; O'Neill, R.; Schickli, J.; Palese, P.; et al. A novel influenza a virus mitochondrial protein that induces cell death. Nat. Med. 2001, 7, 1306–1312. [Google Scholar] [CrossRef]
- Gibbs, J.S.; Malide, D.; Hornung, F.; Bennink, J.R.; Yewdell, J.W. The influenza a virus pb1-f2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J. Virol. 2003, 77, 7214–7224. [Google Scholar] [CrossRef]
- Zamarin, D.; Garcia-Sastre, A.; Xiao, X.; Wang, R.; Palese, P. Influenza virus pb1-f2 protein induces cell death through mitochondrial ant3 and vdac1. PLoS Pathog 2005, 1, e4. [Google Scholar]
- Zamarin, D.; Ortigoza, M.B.; Palese, P. Influenza a virus pb1-f2 protein contributes to viral pathogenesis in mice. J. Virol. 2006, 80, 7976–7983. [Google Scholar] [CrossRef]
- McAuley, J.L.; Hornung, F.; Boyd, K.L.; Smith, A.M.; McKeon, R.; Bennink, J.; Yewdell, J.W.; McCullers, J.A. Expression of the 1918 influenza a virus pb1-f2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell. Host. Microbe. 2007, 2, 240–249. [Google Scholar] [CrossRef]
- Conenello, G.M.; Zamarin, D.; Perrone, L.A.; Tumpey, T.; Palese, P. A single mutation in the pb1-f2 of h5n1 (hk/97) and 1918 influenza a viruses contributes to increased virulence. PLoS Pathog. 2007, 3, 1414–1421. [Google Scholar]
- Zell, R.; Krumbholz, A.; Eitner, A.; Krieg, R.; Halbhuber, K.J.; Wutzler, P. Prevalence of pb1-f2 of influenza a viruses. J. Gen. Virol. 2007, 88, 536–546. [Google Scholar]
- Hai, R.; Schmolke, M.; Varga, Z.T.; Manicassamy, B.; Wang, T.T.; Belser, J.A.; Pearce, M.B.; Garcia-Sastre, A.; Tumpey, T.M.; Palese, P. Pb1-f2 expression by the 2009 pandemic h1n1 influenza virus has minimal impact on virulence in animal models. J. Virol. 2010, 84, 4442–4450. [Google Scholar]
- Bruns, K.; Studtrucker, N.; Sharma, A.; Fossen, T.; Mitzner, D.; Eissmann, A.; Tessmer, U.; Roder, R.; Henklein, P.; Wray, V.; et al. Structural characterization and oligomerization of pb1-f2, a proapoptotic influenza a virus protein. J. Biol. Chem. 2007, 282, 353–363. [Google Scholar]
- Varga, Z.T.; Ramos, I.; Hai, R.; Schmolke, M.; Garcia-Sastre, A.; Fernandez-Sesma, A.; Palese, P. The influenza virus protein pb1-f2 inhibits the induction of type i interferon at the level of the mavs adaptor protein. PLoS Pathog. 2011, 7, e1002067. [Google Scholar] [CrossRef]
- Varga, Z.T.; Grant, A.; Manicassamy, B.; Palese, P. Influenza virus protein pb1-f2 inhibits the induction of type i interferon by binding to mavs and decreasing mitochondrial membrane potential. J. Virol. 2012, 86, 8359–8366. [Google Scholar]
- Varga, Z.T.; Palese, P. The influenza a virus protein pb1-f2: Killing two birds with one stone? Virulence 2011, 2, 542–546. [Google Scholar] [CrossRef]
- Hatta, M.; Gao, P.; Halfmann, P.; Kawaoka, Y. Molecular basis for high virulence of hong kong h5n1 influenza a viruses. Science 2001, 293, 1840–1842. [Google Scholar]
- Mok, K.P.; Wong, C.H.; Cheung, C.Y.; Chan, M.C.; Lee, S.M.; Nicholls, J.M.; Guan, Y.; Peiris, J.S. Viral genetic determinants of h5n1 influenza viruses that contribute to cytokine dysregulation. J. Infect. Dis. 2009, 200, 1104–1112. [Google Scholar] [CrossRef] [Green Version]
- Iwai, A.; Shiozaki, T.; Kawai, T.; Akira, S.; Kawaoka, Y.; Takada, A.; Kida, H.; Miyazaki, T. Influenza a virus polymerase inhibits type i interferon induction by binding to interferon beta promoter stimulator 1. J. Biol. Chem. 2010, 285, 32064–32074. [Google Scholar]
- Graef, K.M.; Vreede, F.T.; Lau, Y.F.; McCall, A.W.; Carr, S.M.; Subbarao, K.; Fodor, E. The pb2 subunit of the influenza virus rna polymerase affects virulence by interacting with the mitochondrial antiviral signaling protein and inhibiting expression of beta interferon. J. Virol. 2010, 84, 8433–8445. [Google Scholar] [CrossRef]
- Vreede, F.T.; Chan, A.Y.; Sharps, J.; Fodor, E. Mechanisms and functional implications of the degradation of host rna polymerase ii in influenza virus infected cells. Virology 2010, 396, 125–134. [Google Scholar]
- Vreede, F.T.; Fodor, E. The role of the influenza virus rna polymerase in host shut-off. Virulence 2010, 1, 436–439. [Google Scholar] [CrossRef]
- Carter, M.J. A rationale for using steroids in the treatment of severe cases of h5n1 avian influenza. J. Med. Microbiol. 2007, 56, 875–883. [Google Scholar]
- Chan, P.K.; Lee, N.; Zaman, M.; Adisasmito, W.; Coker, R.; Hanshaoworakul, W.; Gasimov, V.; Oner, A.F.; Dogan, N.; Tsang, O.; et al. Determinants of antiviral effectiveness in h5n1 avian influenza. J. Iinfect. Dis. 2012, 206, 1359–1366. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ramos, I.; Fernandez-Sesma, A. Innate Immunity to H5N1 Influenza Viruses in Humans. Viruses 2012, 4, 3363-3388. https://doi.org/10.3390/v4123363
Ramos I, Fernandez-Sesma A. Innate Immunity to H5N1 Influenza Viruses in Humans. Viruses. 2012; 4(12):3363-3388. https://doi.org/10.3390/v4123363
Chicago/Turabian StyleRamos, Irene, and Ana Fernandez-Sesma. 2012. "Innate Immunity to H5N1 Influenza Viruses in Humans" Viruses 4, no. 12: 3363-3388. https://doi.org/10.3390/v4123363