Alphaherpesviruses and the Cytoskeleton in Neuronal Infections

{kind=link}
{kind=link}
Abstract
:1. Introduction
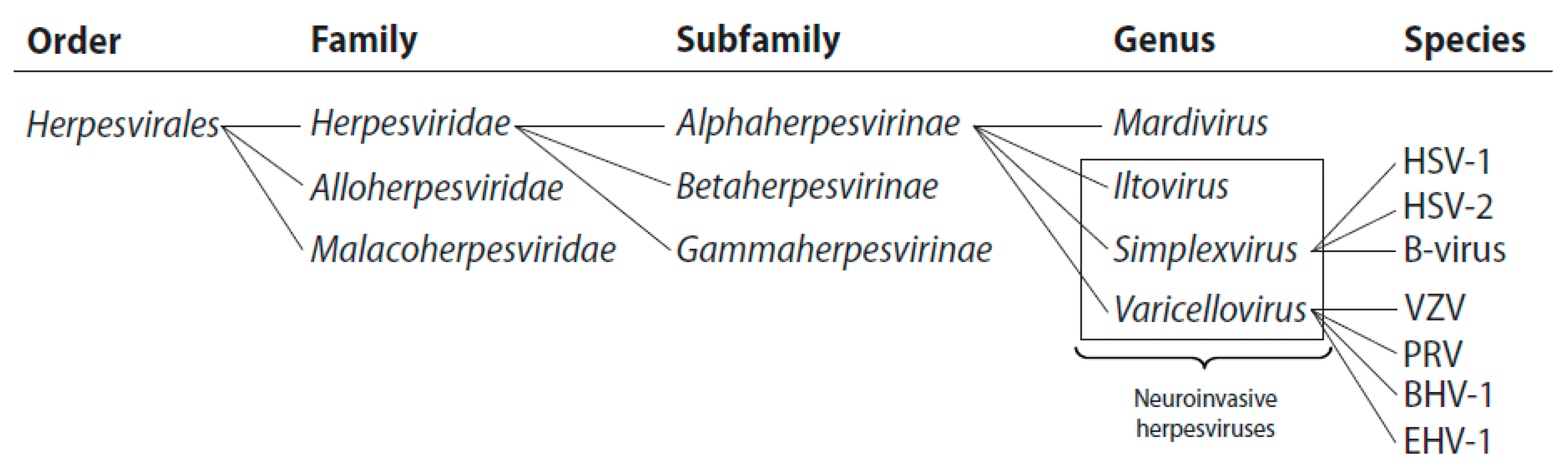
1.1. The Alphaherpesvirinae
1.2. Neuroinvasive Herpesvirus Disease
2. Alphaherpesviruses and the Host Cell Cytoskeleton: Interactions and Virus-Induced Changes
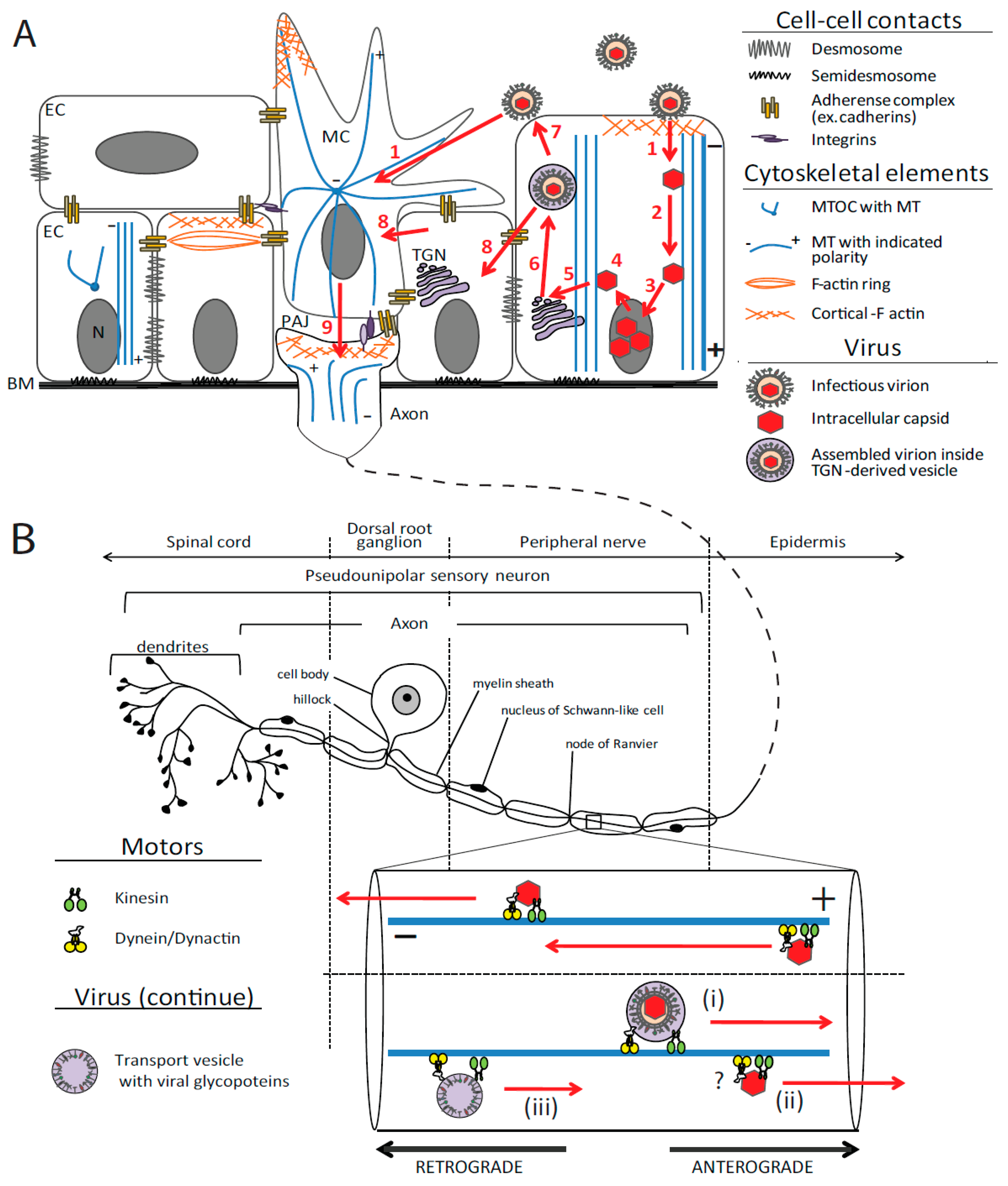
2.1. Cytoskeleton Organization of Polarized Epithelium and Sensory Neurons
2.1.1. Actin
2.1.2. Tubulin and Microtubules
2.1.3. Molecular Motors
2.2. Neuroinvasive Herpesviruses and the Cytoskeleton
2.2.1. Virus Entry: Initiation of Cytoskeleton Rearrangements
2.2.2. Transport of Viral Particles in the Cytoplasm
2.2.2.1. Retrograde Transport: Ingress
2.2.2.2. Assembly, Egress and Cell-to-Cell Spread
2.2.2.2.1. Delivery to TGN and Secondary Envelopment
2.2.2.2.2. TGN to Plasma Membrane Transport in Epithelia
2.2.2.2.3. Anterograde Axon Transport
2.2.3. Viral Protein-Induced Cytoskeleton Rearrangements
3. Concluding Remarks
Acknowledgments
References and Notes
- Savin, K.W.; Cocks, B.G.; Wong, F.; Sawbridge, T.; Cogan, N.; Savage, D.; Warner, S. A neurotropic herpesvirus infecting the gastropod, abalone, shares ancestry with oyster herpesvirus and a herpesvirus associated with the amphioxus genome. Virol. J. 2010, 7, 308. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, C.; Pepin, J.F.; Lapegue, S.; Boudry, P.; Renault, T. Ostreid herpes virus 1 infection in families of the Pacific oyster, Crassostrea gigas, during a summer mortality outbreak: Differences in viral DNA detection and quantification using real-time PCR. Virus Res. 2009, 142, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Vega Thurber, R.L.; Barott, K.L.; Hall, D.; Liu, H.; Rodriguez-Mueller, B.; Desnues, C.; Edwards, R.A.; Haynes, M.; Angly, F.E.; Wegley, L.; et al. Metagenomic analysis indicates that stressors induce production of herpes-like viruses in the coral Porites compressa. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 18413–18418. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Trus, B.L.; Cheng, N.; Steven, A.C.; Watson, M.S.; Cunningham, C.; Le Deuff, R.M.; Renault, T. A novel class of herpesvirus with bivalve hosts. J. Gen. Virol. 2005, 86, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; McGeoch, D.J.; Minson, A.C.; Pellett, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The order Herpesvirales. Arch. Virol. 2009, 154, 171–177. [Google Scholar] [CrossRef]
- Dohner, K.; Nagel, C.H.; Sodeik, B. Viral stop-and-go along microtubules: Taking a ride with dynein and kinesins. Trends Microbiol. 2005, 13, 320–327. [Google Scholar] [CrossRef]
- Radtke, K.; Dohner, K.; Sodeik, B. Viral interactions with the cytoskeleton: A hitchhiker's guide to the cell. Cell. Microbiol. 2006, 8, 387–400. [Google Scholar] [CrossRef]
- Dohner, K.; Sodeik, B. The role of the cytoskeleton during viral infection. Curr. Top. Microbiol. Immunol. 2005, 285, 67–108. [Google Scholar]
- Lyman, M.G.; Enquist, L.W. Herpesvirus interactions with the host cytoskeleton. J. Virol. 2009, 83, 2058–2066. [Google Scholar] [CrossRef]
- Salinas, S.; Schiavo, G.; Kremer, E.J. A hitchhiker's guide to the nervous system: The complex journey of viruses and toxins. Nat. Rev. Microbiol. 2010, 8, 645–655. [Google Scholar] [CrossRef]
- Roizman, B.; Knipe, D.M. Herpes simplex viruses and their replication. In Fields Virology; Fields, B.N., Knipe, D.M., Howley, P.M., Griffin, D.E., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 2, pp. 2399–2459. [Google Scholar]
- Roizman, B.; Baines, J. The diversity and unity of Herpesviridae. Comp. Immunol. Microbiol. Infect. Dis. 1991, 14, 63–79. [Google Scholar] [CrossRef]
- Davison, A.J. Herpesvirus systematics. Vet. Microbiol. 2010, 143, 52–69. [Google Scholar] [CrossRef]
- Roizman, B.; Pellett, P.E. The family Herpesviridae: A brief introduction. In Fields virology; Fields, B.N., Knipe, D.M., Howley, P.M., Griffin, D.E., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 2, pp. 2381–2397. [Google Scholar]
- Roizman, B.; Desrosiers, R.C.; Fleckenstein, B.; Lopez, C.; Minson, A.C.; Studdert, M.J. The family Herpesviridae: An update. The Herpesvirus Study Group of the International Committee on Taxonomy of Viruses. Arch. Virol. 1992, 123, 425–449. [Google Scholar]
- Whitley, R.J. Herpes simplex viruses. In Fields Virology; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Lippincott-Raven Publishers: Philadelphia, PA, USA, 1996; Volume 2, pp. 2297–2342. [Google Scholar]
- Ambinder, R.F.; Cesarman, E. Clinical and pathological aspects of EBV and KSHV infection. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; Chapter 50. [Google Scholar]
- Tischer, B.K.; Osterrieder, N. Herpesviruses—A zoonotic threat? Vet. Microbiol. 2010, 140, 266–270. [Google Scholar] [CrossRef]
- Shek, W.R.; Calnek, B.W.; Schat, K.A.; Chen, C.H. Characterization of Marek's disease virus-infected lymphocytes: Discrimination between cytolytically and latently infected cells. J. Natl. Cancer Inst. 1983, 70, 485–491. [Google Scholar]
- Pepose, J.S.; Stevens, J.G.; Cook, M.L.; Lampert, P.W. Marek's disease as a model for the Landry--Guillain--Barre syndrome: Latent viral infection in nonneuronal cells accompanied by specific immune responses to peripheral nerve and myelin. Am. J. Pathol. 1981, 103, 309–320. [Google Scholar]
- Williams, R.A.; Bennett, M.; Bradbury, J.M.; Gaskell, R.M.; Jones, R.C.; Jordan, F.T. Demonstration of sites of latency of infectious laryngotracheitis virus using the polymerase chain reaction. J. Gen. Virol. 1992, 73, 2415–2420. [Google Scholar] [CrossRef]
- Liesegang, T.J. Herpes simplex virus epidemiology and ocular importance. Cornea 2001, 20, 1–13. [Google Scholar] [CrossRef]
- Sanjuan, N.A.; Lascano, E.F. Autonomic nervous system involvement in experimental genital infection by herpes simplex virus type 2. Arch. Virol. 1986, 91, 329–339. [Google Scholar] [CrossRef]
- Bustos, D.E.; Atherton, S.S. Detection of herpes simplex virus type 1 in human ciliary ganglia. Invest. Ophthalmol. Vis. Sci. 2002, 43, 2244–2249. [Google Scholar]
- Casrouge, A.; Zhang, S.Y.; Eidenschenk, C.; Jouanguy, E.; Puel, A.; Yang, K.; Alcais, A.; Picard, C.; Mahfoufi, N.; Nicolas, N.; et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 2006, 314, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Kimberlin, D.W. Herpes simplex virus infections in neonates and early childhood. Semin. Pediatr. Infect. Dis. 2005, 16, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Reyes, M.; Shaik, N.S.; Graber, J.M.; Nisenbaum, R.; Wetherall, N.T.; Fukuda, K.; Reeves, W.C. Acyclovir-resistant genital herpes among persons attending sexually transmitted disease and human immunodeficiency virus clinics. Arch. Intern. Med. 2003, 163, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Bacon, T.H.; Levin, M.J.; Leary, J.J.; Sarisky, R.T.; Sutton, D. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy. Clin. Microbiol. Rev. 2003, 16, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Ku, C.C.; Besser, J.; Abendroth, A.; Grose, C.; Arvin, A.M. Varicella-Zoster virus pathogenesis and immunobiology: New concepts emerging from investigations with the SCIDhu mouse model. J. Virol. 2005, 79, 2651–2658. [Google Scholar] [CrossRef]
- Josephson, A.; Gombert, M.E. Airborne transmission of nosocomial varicella from localized zoster. J. Infect. Dis. 1988, 158, 238–241. [Google Scholar] [CrossRef]
- Steiner, I.; Kennedy, P.G.; Pachner, A.R. The neurotropic herpes viruses: Herpes simplex and varicella-zoster. Lancet Neurol. 2007, 6, 1015–1028. [Google Scholar] [CrossRef]
- Barnes, D.W.; Whitley, R.J. CNS diseases associated with varicella zoster virus and herpes simplex virus infection. Pathogenesis and current therapy. Neurol. Clin. 1986, 4, 265–283. [Google Scholar] [CrossRef]
- Reed, S.M.; Toribio, R.E. Equine herpesvirus 1 and 4. Vet. Clin. North Am. Equine. Pract. 2004, 20, 631–642. [Google Scholar] [CrossRef]
- Jones, C.A.; Hunt, R.D.; King, N.W. Veterinary Pathology, 6th ed.; Cann, C., Ed.; Lippincott Williams & Wilkins: Baltimore, MD, USA, 1997; pp. 217–234. [Google Scholar]
- Williams, E.S.; Barker, I.K. Infectious Diseases of Wild Mammals, 3rd ed.; Blackwell Publishing: Ames, IA, USA, 2001; pp. 147–179. [Google Scholar]
- Glass, C.M.; McLean, R.G.; Katz, J.B.; Maehr, D.S.; Cropp, C.B.; Kirk, L.J.; McKeirnan, A.J.; Evermann, J.F. Isolation of pseudorabies (Aujeszky's disease) virus from a Florida panther. J. Wildl. Dis. 1994, 30, 180–184. [Google Scholar] [CrossRef]
- Weigler, B.J. Biology of B virus in macaque and human hosts: A review. Clin. Infect. Dis. 1992, 14, 555–567. [Google Scholar] [CrossRef]
- Matz-Rensing, K.; Jentsch, K.D.; Rensing, S.; Langenhuyzen, S.; Verschoor, E.; Niphuis, H.; Kaup, F.J. Fatal Herpes simplex infection in a group of common marmosets (Callithrix jacchus). Vet. Pathol. 2003, 40, 405–411. [Google Scholar] [CrossRef]
- Enquist, L.W.; Husak, P.J.; Banfield, B.W.; Smith, G.A. Infection and spread of alphaherpesviruses in the nervous system. Adv. Virus Res. 1998, 51, 237–347. [Google Scholar]
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 2005, 69, 462–500. [Google Scholar] [CrossRef]
- Dotti, C.G.; Simons, K. Polarized sorting of viral glycoproteins to the axon and dendrites of hippocampal neurons in culture. Cell 1990, 62, 63–72. [Google Scholar] [CrossRef]
- Dotti, C.G.; Parton, R.G.; Simons, K. Polarized sorting of glypiated proteins in hippocampal neurons. Nature 1991, 349, 158–161. [Google Scholar] [CrossRef]
- Honda, T.; Sakisaka, T.; Yamada, T.; Kumazawa, N.; Hoshino, T.; Kajita, M.; Kayahara, T.; Ishizaki, H.; Tanaka-Okamoto, M.; Mizoguchi, A.; et al. Involvement of nectins in the formation of puncta adherentia junctions and the mossy fiber trajectory in the mouse hippocampus. Mol. Cell. Neurosci. 2006, 31, 315–325. [Google Scholar] [CrossRef]
- Fannon, A.M.; Colman, D.R. A model for central synaptic junctional complex formation based on the differential adhesive specificities of the cadherins. Neuron 1996, 17, 423–434. [Google Scholar] [CrossRef]
- Mizoguchi, A.; Nakanishi, H.; Kimura, K.; Matsubara, K.; Ozaki-Kuroda, K.; Katata, T.; Honda, T.; Kiyohara, Y.; Heo, K.; Higashi, M.; et al. Nectin: An adhesion molecule involved in formation of synapses. J. Cell Biol. 2002, 156, 555–565. [Google Scholar] [CrossRef]
- Nelson, W.J. Membrane Protein-Cytoskeleton Interactions; Academic Press: London, UK, 1996; Volume 43. [Google Scholar]
- Frixione, E. Recurring views on the structure and function of the cytoskeleton: A 300-year epic. Cell Motil. Cytoskeleton 2000, 46, 73–94. [Google Scholar] [CrossRef]
- Pollard, T.D. Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filaments. J. Cell Biol. 1986, 103, 2747–2754. [Google Scholar] [CrossRef] [PubMed]
- Mullins, R.D.; Heuser, J.A.; Pollard, T.D. The interaction of Arp2/3 complex with actin: Nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 6181–6186. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, E.S.; Higgs, H.N. The many faces of actin: Matching assembly factors with cellular structures. Nat. Cell Biol. 2007, 9, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Luo, L. Actin cytoskeleton regulation in neuronal morphogenesis and structural plasticity. Annu. Rev. Cell Dev. Biol. 2002, 18, 601–635. [Google Scholar] [CrossRef]
- Hall, A. Small GTP-binding proteins and the regulation of the actin cytoskeleton. Annu. Rev. Cell Biol. 1994, 10, 31–54. [Google Scholar] [CrossRef]
- Tapon, N.; Hall, A. Rho, Rac and Cdc42 GTPases regulate the organization of the actin cytoskeleton. Curr. Opin. Cell Biol. 1997, 9, 86–92. [Google Scholar] [CrossRef]
- Zhang, F.; Saha, S.; Shabalina, S.A.; Kashina, A. Differential arginylation of actin isoforms is regulated by coding sequence-dependent degradation. Science 2010, 329, 1534–1537. [Google Scholar] [CrossRef]
- Papakonstanti, E.A.; Stournaras, C. Association of PI-3 kinase with PAK1 leads to actin phosphorylation and cytoskeletal reorganization. Mol. Biol. Cell 2002, 13, 2946–2962. [Google Scholar] [CrossRef]
- Green, K.J.; Getsios, S.; Troyanovsky, S.; Godsel, L.M. Intercellular junction assembly, dynamics, and homeostasis. Cold Spring Harb. Perspect. Biol. 2010, 2, a000125. [Google Scholar] [CrossRef]
- Ivanov, A.I. Actin motors that drive formation and disassembly of epithelial apical junctions. Front. Biosci. 2008, 13, 6662–6681. [Google Scholar] [CrossRef]
- Lowery, L.A.; Van Vactor, D. The trip of the tip: Understanding the growth cone machinery. Nat. Rev. Mol. Cell Biol. 2009, 10, 332–343. [Google Scholar] [CrossRef]
- Braun, N.; Schikorski, T.; Zimmermann, H. Cytoplasmic segregation and cytoskeletal organization in the electric catfish giant electromotoneuron with special reference to the axon hillock region. Neuroscience 1993, 52, 745–756. [Google Scholar] [CrossRef]
- Dillon, C.; Goda, Y. The actin cytoskeleton: Integrating form and function at the synapse. Annu. Rev. Neurosci. 2005, 28, 25–55. [Google Scholar] [CrossRef]
- Wade, R.H. On and around microtubules: An overview. Mol. Biotechnol. 2009, 43, 177–191. [Google Scholar] [CrossRef]
- Luders, J.; Stearns, T. Microtubule-organizing centres: A re-evaluation. Nat. Rev. Mol. Cell Biol. 2007, 8, 161–167. [Google Scholar] [CrossRef]
- Bartolini, F.; Gundersen, G.G. Generation of noncentrosomal microtubule arrays. J. Cell Sci. 2006, 119, 4155–4163. [Google Scholar] [CrossRef]
- Musch, A. Microtubule organization and function in epithelial cells. Traffic 2004, 5, 1–9. [Google Scholar] [CrossRef]
- Meng, W.; Mushika, Y.; Ichii, T.; Takeichi, M. Anchorage of microtubule minus ends to adherens junctions regulates epithelial cell-cell contacts. Cell 2008, 135, 948–959. [Google Scholar] [CrossRef]
- Topp, K.S.; Bisla, K.; Saks, N.D.; Lavail, J.H. Centripetal transport of herpes simplex virus in human retinal pigment epithelial cells in vitro. Neuroscience 1996, 71, 1133–1144. [Google Scholar] [CrossRef]
- Poulain, F.E.; Sobel, A. The microtubule network and neuronal morphogenesis: Dynamic and coordinated orchestration through multiple players. Mol. Cell. Neurosci. 2010, 43, 15–32. [Google Scholar] [CrossRef]
- Topp, K.S.; Meade, L.B.; LaVail, J.H. Microtubule polarity in the peripheral processes of trigeminal ganglion cells: Relevance for the retrograde transport of herpes simplex virus. J. Neurosci. 1994, 14, 318–325. [Google Scholar] [CrossRef] [PubMed]
- O'Connell, C.B.; Tyska, M.J.; Mooseker, M.S. Myosin at work: Motor adaptations for a variety of cellular functions. Biochim. Biophys. Acta 2007, 1773, 615–630. [Google Scholar] [CrossRef] [PubMed]
- De La Cruz, E.M.; Ostap, E.M. Relating biochemistry and function in the myosin superfamily. Curr. Opin. Cell Biol. 2004, 16, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Heidemann, S.R.; Buxbaum, R.E. Cell crawling: First the motor, now the transmission. J. Cell Biol. 1998, 141, 1–4. [Google Scholar] [CrossRef]
- Norstrom, M.F.; Smithback, P.A.; Rock, R.S. Unconventional processive mechanics of non-muscle myosin IIB. J. Biol. Chem. 2010, 285, 26326–26334. [Google Scholar] [CrossRef]
- Bridgman, P.C.; Dave, S.; Asnes, C.F.; Tullio, A.N.; Adelstein, R.S. Myosin IIB is required for growth cone motility. J. Neurosci. 2001, 21, 6159–6169. [Google Scholar] [CrossRef]
- Kovacs, M.; Wang, F.; Hu, A.; Zhang, Y.; Sellers, J.R. Functional divergence of human cytoplasmic myosin II: Kinetic characterization of the non-muscle IIA isoform. J. Biol. Chem. 2003, 278, 38132–38140. [Google Scholar] [CrossRef]
- Even-Ram, S.; Doyle, A.D.; Conti, M.A.; Matsumoto, K.; Adelstein, R.S.; Yamada, K.M. Myosin IIA regulates cell motility and actomyosin-microtubule crosstalk. Nat. Cell Biol. 2007, 9, 299–309. [Google Scholar] [CrossRef]
- Kull, F.J.; Vale, R.D.; Fletterick, R.J. The case for a common ancestor: Kinesin and myosin motor proteins and G proteins. J. Muscle Res. Cell Motil. 1998, 19, 877–886. [Google Scholar] [CrossRef]
- Lawrence, C.J.; Dawe, R.K.; Christie, K.R.; Cleveland, D.W.; Dawson, S.C.; Endow, S.A.; Goldstein, L.S.; Goodson, H.V.; Hirokawa, N.; Howard, J.; et al. A standardized kinesin nomenclature. J. Cell Biol. 2004, 167, 19–22. [Google Scholar] [CrossRef]
- Miki, H.; Okada, Y.; Hirokawa, N. Analysis of the kinesin superfamily: Insights into structure and function. Trends Cell Biol. 2005, 15, 467–476. [Google Scholar] [CrossRef]
- Aizawa, H.; Sekine, Y.; Takemura, R.; Zhang, Z.; Nangaku, M.; Hirokawa, N. Kinesin family in murine central nervous system. J. Cell Biol. 1992, 119, 1287–1296. [Google Scholar] [CrossRef]
- Kanai, Y.; Okada, Y.; Tanaka, Y.; Harada, A.; Terada, S.; Hirokawa, N. KIF5C, a novel neuronal kinesin enriched in motor neurons. J. Neurosci. 2000, 20, 6374–6384. [Google Scholar] [CrossRef]
- Lee, J.R.; Shin, H.; Ko, J.; Choi, J.; Lee, H.; Kim, E. Characterization of the movement of the kinesin motor KIF1A in living cultured neurons. J. Biol. Chem. 2003, 278, 2624–2629. [Google Scholar] [CrossRef]
- Niwa, S.; Tanaka, Y.; Hirokawa, N. KIF1Bbeta- and KIF1A-mediated axonal transport of presynaptic regulator Rab3 occurs in a GTP-dependent manner through DENN/MADD. Nat. Cell Biol. 2008, 10, 1269–1279. [Google Scholar] [CrossRef]
- Saito, N.; Okada, Y.; Noda, Y.; Kinoshita, Y.; Kondo, S.; Hirokawa, N. KIFC2 is a novel neuron-specific C-terminal type kinesin superfamily motor for dendritic transport of multivesicular body-like organelles. Neuron 1997, 18, 425–438. [Google Scholar] [CrossRef]
- Yang, Z.; Xia, C.; Roberts, E.A.; Bush, K.; Nigam, S.K.; Goldstein, L.S. Molecular cloning and functional analysis of mouse C-terminal kinesin motor KifC3. Mol. Cell Biol. 2001, 21, 765–770. [Google Scholar] [CrossRef]
- Hanlon, D.W.; Yang, Z.; Goldstein, L.S. Characterization of KIFC2, a neuronal kinesin superfamily member in mouse. Neuron 1997, 18, 439–451. [Google Scholar] [CrossRef]
- Dhar, S.S.; Wong-Riley, M.T. The kinesin superfamily protein KIF17 is regulated by the same transcription factor (NRF-1) as its cargo NR2B in neurons. Biochim. Biophys. Acta 2011, 1813, 403–411. [Google Scholar] [CrossRef]
- Davidovic, L.; Jaglin, X.H.; Lepagnol-Bestel, A.M.; Tremblay, S.; Simonneau, M.; Bardoni, B.; Khandjian, E.W. The fragile X mental retardation protein is a molecular adaptor between the neurospecific KIF3C kinesin and dendritic RNA granules. Hum. Mol. Genet. 2007, 16, 3047–3058. [Google Scholar] [CrossRef]
- Muresan, V.; Abramson, T.; Lyass, A.; Winter, D.; Porro, E.; Hong, F.; Chamberlin, N.L.; Schnapp, B.J. KIF3C and KIF3A form a novel neuronal heteromeric kinesin that associates with membrane vesicles. Mol. Biol. Cell 1998, 9, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.J.; Allan, V.J. Cargo selection by specific kinesin light chain 1 isoforms. EMBO J. 2006, 25, 5457–5468. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.W.; Chuang, J.Z.; Sung, C.H. Cytoplasmic dynein regulation by subunit heterogeneity and its role in apical transport. J. Cell Biol. 2001, 153, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- King, S.J.; Schroer, T.A. Dynactin increases the processivity of the cytoplasmic dynein motor. Nat. Cell Biol. 2000, 2, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.A.; Knight, P.J. Is the dynein motor a winch? Curr. Opin. Struct. Biol. 2004, 14, 138–146. [Google Scholar] [CrossRef]
- Schroer, T.A. Dynactin. Annu. Rev. Cell Dev. Biol. 2004, 20, 759–779. [Google Scholar] [CrossRef]
- Rodriguez, O.C.; Cheney, R.E. A new direction for myosin. Trends Cell Biol. 2000, 10, 307–311. [Google Scholar] [CrossRef]
- Chibalina, M.V.; Puri, C.; Kendrick-Jones, J.; Buss, F. Potential roles of myosin VI in cell motility. Biochem. Soc. Trans. 2009, 37, 966–970. [Google Scholar] [CrossRef]
- Caviston, J.P.; Holzbaur, E.L. Microtubule motors at the intersection of trafficking and transport. Trends Cell Biol. 2006, 16, 530–537. [Google Scholar] [CrossRef]
- Parks, L.D.; Zalups, R.K.; Barfuss, D.W. Luminal and basolateral membrane transport of glutathione in isolated perfused S(1), S(2), and S(3) segments of the rabbit proximal tubule. J. Am. Soc. Nephrol. 2000, 11, 1008–1015. [Google Scholar] [CrossRef]
- Bi, G.Q.; Morris, R.L.; Liao, G.; Alderton, J.M.; Scholey, J.M.; Steinhardt, R.A. Kinesin- and myosin-driven steps of vesicle recruitment for Ca2+-regulated exocytosis. J. Cell Biol. 1997, 138, 999–1008. [Google Scholar] [CrossRef]
- Park, C.S.; Leem, C.H.; Jang, Y.J.; Shim, Y.H. Vesicular transport as a new paradigm in short-term regulation of transepithelial transport. J. Korean Med. Sci. 2000, 15, 123–132. [Google Scholar] [CrossRef]
- Vilfan, A.; Frey, E.; Schwabl, F.; Thormahlen, M.; Song, Y.H.; Mandelkow, E. Dynamics and cooperativity of microtubule decoration by the motor protein kinesin. J. Mol. Biol. 2001, 312, 1011–1026. [Google Scholar] [CrossRef]
- Kural, C.; Kim, H.; Syed, S.; Goshima, G.; Gelfand, V.I.; Selvin, P.R. Kinesin and dynein move a peroxisome in vivo: a tug-of-war or coordinated movement? Science 2005, 308, 1469–1472. [Google Scholar] [CrossRef]
- Muto, E.; Sakai, H.; Kaseda, K. Long-range cooperative binding of kinesin to a microtubule in the presence of ATP. J. Cell Biol. 2005, 168, 691–696. [Google Scholar] [CrossRef]
- Song, H.; Endow, S.A. Binding sites on microtubules of kinesin motors of the same or opposite polarity. Biochemistry 1996, 35, 11203–11209. [Google Scholar] [CrossRef]
- Gross, S.P.; Tuma, M.C.; Deacon, S.W.; Serpinskaya, A.S.; Reilein, A.R.; Gelfand, V.I. Interactions and regulation of molecular motors in Xenopus melanophores. J. Cell Biol. 2002, 156, 855–865. [Google Scholar] [CrossRef]
- Gross, S.P.; Welte, M.A.; Block, S.M.; Wieschaus, E.F. Coordination of opposite-polarity microtubule motors. J. Cell Biol. 2002, 156, 715–724. [Google Scholar] [CrossRef]
- Muresan, V.; Stankewich, M.C.; Steffen, W.; Morrow, J.S.; Holzbaur, E.L.; Schnapp, B.J. Dynactin-dependent, dynein-driven vesicle transport in the absence of membrane proteins: A role for spectrin and acidic phospholipids. Mol. Cell 2001, 7, 173–183. [Google Scholar] [CrossRef]
- Haghnia, M.; Cavalli, V.; Shah, S.B.; Schimmelpfeng, K.; Brusch, R.; Yang, G.; Herrera, C.; Pilling, A.; Goldstein, L.S. Dynactin is required for coordinated bidirectional motility, but not for dynein membrane attachment. Mol. Biol. Cell 2007, 18, 2081–2089. [Google Scholar] [CrossRef]
- Gross, S.P. Dynactin: coordinating motors with opposite inclinations. Curr. Biol. 2003, 13, R320–R322. [Google Scholar] [CrossRef] [PubMed]
- Deacon, S.W.; Serpinskaya, A.S.; Vaughan, P.S.; Lopez Fanarraga, M.; Vernos, I.; Vaughan, K.T.; Gelfand, V.I. Dynactin is required for bidirectional organelle transport. J. Cell Biol. 2003, 160, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Splinter, D.; Tanenbaum, M.E.; Lindqvist, A.; Jaarsma, D.; Flotho, A.; Yu, K.L.; Grigoriev, I.; Engelsma, D.; Haasdijk, E.D.; Keijzer, N.; et al. Bicaudal D2, dynein, and kinesin-1 associate with nuclear pore complexes and regulate centrosome and nuclear positioning during mitotic entry. PLoS Biol. 2010, 8, e1000350. [Google Scholar] [CrossRef] [PubMed]
- Ligon, L.A.; Tokito, M.; Finklestein, J.M.; Grossman, F.E.; Holzbaur, E.L. A direct interaction between cytoplasmic dynein and kinesin I may coordinate motor activity. J. Biol. Chem. 2004, 279, 19201–19208. [Google Scholar] [CrossRef] [PubMed]
- Akhmanova, A.; Hammer, J.A., 3rd. Linking molecular motors to membrane cargo. Curr. Opin. Cell Biol. 2010, 22, 479–487. [Google Scholar] [CrossRef]
- Verhey, K.J.; Hammond, J.W. Traffic control: Regulation of kinesin motors. Nat. Rev. Mol. Cell Biol. 2009, 10, 765–777. [Google Scholar] [CrossRef]
- Prekeris, R.; Terrian, D.M. Brain myosin V is a synaptic vesicle-associated motor protein: Evidence for a Ca2+-dependent interaction with the synaptobrevin-synaptophysin complex. J. Cell Biol. 1997, 137, 1589–1601. [Google Scholar] [CrossRef]
- Fielitz, J.; Kim, M.S.; Shelton, J.M.; Latif, S.; Spencer, J.A.; Glass, D.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Myosin accumulation and striated muscle myopathy result from the loss of muscle RING finger 1 and 3. J. Clin. Invest. 2007, 117, 2486–2495. [Google Scholar] [CrossRef]
- Hoppe, T.; Cassata, G.; Barral, J.M.; Springer, W.; Hutagalung, A.H.; Epstein, H.F.; Baumeister, R. Regulation of the myosin-directed chaperone UNC-45 by a novel E3/E4-multiubiquitylation complex in C. elegans. Cell 2004, 118, 337–349. [Google Scholar] [CrossRef]
- Kohta, R.; Kotake, Y.; Hosoya, T.; Hiramatsu, T.; Otsubo, Y.; Koyama, H.; Hirokane, Y.; Yokoyama, Y.; Ikeshoji, H.; Oofusa, K.; et al. 1-Benzyl-1,2,3,4-tetrahydroisoquinoline binds with tubulin beta, a substrate of parkin, and reduces its polyubiquitination. J. Neurochem. 2010, 114, 1291–1301. [Google Scholar]
- Goode, B.L.; Drubin, D.G.; Barnes, G. Functional cooperation between the microtubule and actin cytoskeletons. Curr. Opin. Cell Biol. 2000, 12, 63–71. [Google Scholar] [CrossRef]
- Herold, B.C.; Visalli, R.J.; Susmarski, N.; Brandt, C.R.; Spear, P.G. Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulphate and glycoprotein B. J. Gen. Virol. 1994, 75, 1211–1222. [Google Scholar] [CrossRef]
- Herold, B.C.; WuDunn, D.; Soltys, N.; Spear, P.G. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J. Virol. 1991, 65, 1090–1098. [Google Scholar] [CrossRef]
- Svennerholm, B.; Jeansson, S.; Vahlne, A.; Lycke, E. Involvement of glycoprotein C (gC) in adsorption of herpes simplex virus type 1 (HSV-1) to the cell. Arch. Virol. 1991, 120, 273–279. [Google Scholar] [CrossRef]
- Meckes, D.G., Jr.; Wills, J.W. Structural rearrangement within an enveloped virus upon binding to the host cell. J. Virol. 2008, 82, 10429–10435. [Google Scholar] [CrossRef]
- Rue, C.A.; Ryan, P. A role for glycoprotein C in pseudorabies virus entry that is independent of virus attachment to heparan sulfate and which involves the actin cytoskeleton. Virology 2003, 307, 12–21. [Google Scholar] [CrossRef]
- Martinho, R.G.; Castel, S.; Urena, J.; Fernandez-Borja, M.; Makiya, R.; Olivecrona, G.; Reina, M.; Alonso, A.; Vilaro, S. Ligand binding to heparan sulfate proteoglycans induces their aggregation and distribution along actin cytoskeleton. Mol. Biol. Cell 1996, 7, 1771–1788. [Google Scholar] [CrossRef]
- Oh, M.J.; Akhtar, J.; Desai, P.; Shukla, D. A role for heparan sulfate in viral surfing. Biochem. Biophys. Res. Commun. 2010, 391, 176–181. [Google Scholar] [CrossRef]
- Lehmann, M.J.; Sherer, N.M.; Marks, C.B.; Pypaert, M.; Mothes, W. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell Biol. 2005, 170, 317–325. [Google Scholar] [CrossRef]
- Nobes, C.D.; Hall, A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef]
- Tiwari, V.; Shukla, D. Phosphoinositide 3 kinase signalling may affect multiple steps during herpes simplex virus type-1 entry. J. Gen. Virol. 2010, 91, 3002–3009. [Google Scholar] [CrossRef] [PubMed]
- Kawakatsu, T.; Shimizu, K.; Honda, T.; Fukuhara, T.; Hoshino, T.; Takai, Y. Trans-interactions of nectins induce formation of filopodia and Lamellipodia through the respective activation of Cdc42 and Rac small G proteins. J. Biol. Chem. 2002, 277, 50749–50755. [Google Scholar] [CrossRef] [PubMed]
- Mandai, K.; Nakanishi, H.; Satoh, A.; Obaishi, H.; Wada, M.; Nishioka, H.; Itoh, M.; Mizoguchi, A.; Aoki, T.; Fujimoto, T.; et al. Afadin: A novel actin filament-binding protein with one PDZ domain localized at cadherin-based cell-to-cell adherens junction. J. Cell Biol. 1997, 139, 517–528. [Google Scholar] [CrossRef]
- Nicola, A.V.; Straus, S.E. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J. Virol. 2004, 78, 7508–7517. [Google Scholar] [CrossRef] [PubMed]
- Heine, J.W.; Spear, P.G.; Roizman, B. Proteins specified by herpes simplex virus. VI. Viral proteins in the plasma membrane. J. Virol. 1972, 9, 431–439. [Google Scholar] [CrossRef]
- Mothes, W.; Sherer, N.M.; Jin, J.; Zhong, P. Virus cell-to-cell transmission. J. Virol. 2010, 84, 8360–8368. [Google Scholar] [CrossRef]
- Aubert, M.; Yoon, M.; Sloan, D.D.; Spear, P.G.; Jerome, K.R. The virological synapse facilitates herpes simplex virus entry into T cells. J. Virol. 2009, 83, 6171–6183. [Google Scholar] [CrossRef]
- De Regge, N.; Nauwynck, H.J.; Geenen, K.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Mettenleiter, T.C.; Favoreel, H.W. Alpha-herpesvirus glycoprotein D interaction with sensory neurons triggers formation of varicosities that serve as virus exit sites. J. Cell Biol. 2006, 174, 267–275. [Google Scholar] [CrossRef]
- Robbins, A.K.; Whealy, M.E.; Watson, R.J.; Enquist, L.W. Pseudorabies virus gene encoding glycoprotein gIII is not essential for growth in tissue culture. J. Virol. 1986, 59, 635–645. [Google Scholar] [CrossRef]
- Nalwanga, D.; Rempel, S.; Roizman, B.; Baines, J.D. The UL 16 gene product of herpes simplex virus 1 is a virion protein that colocalizes with intranuclear capsid proteins. Virology 1996, 226, 236–242. [Google Scholar] [CrossRef]
- Laquerre, S.; Argnani, R.; Anderson, D.B.; Zucchini, S.; Manservigi, R.; Glorioso, J.C. Heparan sulfate proteoglycan binding by herpes simplex virus type 1 glycoproteins B and C, which differ in their contributions to virus attachment, penetration, and cell-to-cell spread. J. Virol. 1998, 72, 6119–6130. [Google Scholar] [CrossRef]
- Holland, T.C.; Marlin, S.D.; Levine, M.; Glorioso, J. Antigenic variants of herpes simplex virus selected with glycoprotein-specific monoclonal antibodies. J. Virol. 1983, 45, 672–682. [Google Scholar] [CrossRef]
- Ruyechan, W.T.; Morse, L.S.; Knipe, D.M.; Roizman, B. Molecular genetics of herpes simplex virus. II. Mapping of the major viral glycoproteins and of the genetic loci specifying the social behavior of infected cells. J. Virol. 1979, 29, 677–697. [Google Scholar] [CrossRef]
- Baines, J.D.; Roizman, B. The open reading frames UL3, UL4, UL10, and UL16 are dispensable for the replication of herpes simplex virus 1 in cell culture. J. Virol. 1991, 65, 938–944. [Google Scholar] [CrossRef]
- Rosenthal, K.S.; Perez, R.; Hodnichak, C. Inhibition of herpes simplex virus type 1 penetration by cytochalasins B and D. J. Gen. Virol. 1985, 66, 1601–1605. [Google Scholar] [CrossRef]
- Sodeik, B.; Ebersold, M.W.; Helenius, A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 1997, 136, 1007–1021. [Google Scholar] [CrossRef]
- Nicola, A.V.; Hou, J.; Major, E.O.; Straus, S.E. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 2005, 79, 7609–7616. [Google Scholar] [CrossRef]
- Lycke, E.; Hamark, B.; Johansson, M.; Krotochwil, A.; Lycke, J.; Svennerholm, B. Herpes simplex virus infection of the human sensory neuron. An electron microscopy study. Arch. Virol. 1988, 101, 87–104. [Google Scholar] [CrossRef]
- Maurer, U.E.; Sodeik, B.; Grunewald, K. Native 3D intermediates of membrane fusion in herpes simplex virus 1 entry. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 10559–10564. [Google Scholar] [CrossRef]
- Milne, R.S.; Nicola, A.V.; Whitbeck, J.C.; Eisenberg, R.J.; Cohen, G.H. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J. Virol. 2005, 79, 6655–6663. [Google Scholar] [CrossRef]
- Nicola, A.V.; McEvoy, A.M.; Straus, S.E. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 2003, 77, 5324–5332. [Google Scholar] [CrossRef]
- Peeters, B.; Pol, J.; Gielkens, A.; Moormann, R. Envelope glycoprotein gp50 of pseudorabies virus is essential for virus entry but is not required for viral spread in mice. J. Virol. 1993, 67, 170–177. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.J. Evolutionary relationships of virion glycoprotein genes in the S regions of alphaherpesvirus genomes. J. Gen. Virol. 1990, 71, 2361–2367. [Google Scholar] [CrossRef] [PubMed]
- Heffner, S.; Kovacs, F.; Klupp, B.G.; Mettenleiter, T.C. Glycoprotein gp50-negative pseudorabies virus: A novel approach toward a nonspreading live herpesvirus vaccine. J. Virol. 1993, 67, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Hanssens, F.P.; Nauwynck, H.J.; Mettenlieter, T.C. Role of glycoprotein gD in the adhesion of pseudorabies virus infected cells and subsequent cell-associated virus spread. Arch. Virol. 1995, 140, 1855–1862. [Google Scholar] [CrossRef]
- Satoh, T.; Arase, H. HSV-1 infection through inhibitory receptor, PILRalpha. Uirusu 2008, 58, 27–36. [Google Scholar] [CrossRef]
- Satoh, T.; Arii, J.; Suenaga, T.; Wang, J.; Kogure, A.; Uehori, J.; Arase, N.; Shiratori, I.; Tanaka, S.; Kawaguchi, Y.; et al. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 2008, 132, 935–944. [Google Scholar] [CrossRef]
- Arii, J.; Goto, H.; Suenaga, T.; Oyama, M.; Kozuka-Hata, H.; Imai, T.; Minowa, A.; Akashi, H.; Arase, H.; Kawaoka, Y.; et al. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 2010, 467, 859–862. [Google Scholar] [CrossRef]
- Fournier, N.; Chalus, L.; Durand, I.; Garcia, E.; Pin, J.J.; Churakova, T.; Patel, S.; Zlot, C.; Gorman, D.; Zurawski, S.; et al. FDF03, a novel inhibitory receptor of the immunoglobulin superfamily, is expressed by human dendritic and myeloid cells. J. Immunol. 2000, 165, 1197–1209. [Google Scholar] [CrossRef]
- Wilson, M.D.; Cheung, J.; Martindale, D.W.; Scherer, S.W.; Koop, B.F. Comparative analysis of the paired immunoglobulin-like receptor (PILR) locus in six mammalian genomes: Duplication, conversion, and the birth of new genes. Physiol. Genom. 2006, 27, 201–218. [Google Scholar] [CrossRef]
- Shukla, S.Y.; Singh, Y.K.; Shukla, D. Role of nectin-1, HVEM, and PILR-alpha in HSV-2 entry into human retinal pigment epithelial cells. Invest. Ophthalmol. Vis. Sci. 2009, 50, 2878–2887. [Google Scholar] [CrossRef]
- Schelhaas, M.; Ewers, H.; Rajamaki, M.L.; Day, P.M.; Schiller, J.T.; Helenius, A. Human papillomavirus type 16 entry: Retrograde cell surface transport along actin-rich protrusions. PLoS Pathog. 2008, 4, e1000148. [Google Scholar] [CrossRef]
- Suenaga, T.; Satoh, T.; Somboonthum, P.; Kawaguchi, Y.; Mori, Y.; Arase, H. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 866–871. [Google Scholar] [CrossRef]
- Goh, E.L.; Young, J.K.; Kuwako, K.; Tessier-Lavigne, M.; He, Z.; Griffin, J.W.; Ming, G.L. beta1-integrin mediates myelin-associated glycoprotein signaling in neuronal growth cones. Mol. Brain 2008, 1, 10. [Google Scholar] [CrossRef]
- Leung, T.; Chen, X.Q.; Manser, E.; Lim, L. The p160 RhoA-binding kinase ROK alpha is a member of a kinase family and is involved in the reorganization of the cytoskeleton. Mol. Cell Biol. 1996, 16, 5313–5327. [Google Scholar] [CrossRef]
- Imamura, F.; Mukai, M.; Ayaki, M.; Akedo, H. Y-27632, an inhibitor of rho-associated protein kinase, suppresses tumor cell invasion via regulation of focal adhesion and focal adhesion kinase. Jpn. J. Cancer Res. 2000, 91, 811–816. [Google Scholar] [CrossRef]
- Cheshenko, N.; Liu, W.; Satlin, L.M.; Herold, B.C. Focal adhesion kinase plays a pivotal role in herpes simplex virus entry. J. Biol. Chem. 2005, 280, 31116–31125. [Google Scholar] [CrossRef]
- Frampton, A.R., Jr.; Stolz, D.B.; Uchida, H.; Goins, W.F.; Cohen, J.B.; Glorioso, J.C. Equine herpesvirus 1 enters cells by two different pathways, and infection requires the activation of the cellular kinase ROCK1. J. Virol. 2007, 81, 10879–10889. [Google Scholar] [CrossRef]
- Hasebe, R.; Sasaki, M.; Sawa, H.; Wada, R.; Umemura, T.; Kimura, T. Infectious entry of equine herpesvirus-1 into host cells through different endocytic pathways. Virology 2009, 393, 198–209. [Google Scholar] [CrossRef]
- Mundy, D.I.; Machleidt, T.; Ying, Y.S.; Anderson, R.G.; Bloom, G.S. Dual control of caveolar membrane traffic by microtubules and the actin cytoskeleton. J. Cell Sci. 2002, 115, 4327–4339. [Google Scholar] [CrossRef]
- Watanabe, T.; Noritake, J.; Kaibuchi, K. Regulation of microtubules in cell migration. Trends Cell Biol. 2005, 15, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Antinone, S.E.; Smith, G.A. Two modes of herpesvirus trafficking in neurons: Membrane acquisition directs motion. J. Virol. 2006, 80, 11235–11240. [Google Scholar] [CrossRef] [PubMed]
- Fuller, A.O.; Santos, R.E.; Spear, P.G. Neutralizing antibodies specific for glycoprotein H of herpes simplex virus permit viral attachment to cells but prevent penetration. J. Virol. 1989, 63, 3435–3443. [Google Scholar] [CrossRef] [PubMed]
- Fuller, A.O.; Spear, P.G. Anti-glycoprotein D antibodies that permit adsorption but block infection by herpes simplex virus 1 prevent virion-cell fusion at the cell surface. Proc. Natl. Acad. Sci. U. S. A. 1987, 84, 5454–5458. [Google Scholar] [CrossRef] [PubMed]
- Lycke, E.; Kristensson, K.; Svennerholm, B.; Vahlne, A.; Ziegler, R. Uptake and transport of herpes simplex virus in neurites of rat dorsal root ganglia cells in culture. J. Gen. Virol. 1984, 65, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Morgan, C.; Rose, H.M.; Mednis, B. Electron microscopy of herpes simplex virus. I. Entry. J. Virol. 1968, 2, 507–516. [Google Scholar] [CrossRef]
- Smith, J.D.; de Harven, E. Herpes simplex virus and human cytomegalovirus replication in WI-38 cells. II. An ultrastructural study of viral penetration. J. Virol. 1974, 14, 945–956. [Google Scholar] [CrossRef]
- Granzow, H.; Klupp, B.G.; Mettenleiter, T.C. Entry of pseudorabies virus: An immunogold-labeling study. J. Virol. 2005, 79, 3200–3205. [Google Scholar] [CrossRef]
- Luxton, G.W.; Haverlock, S.; Coller, K.E.; Antinone, S.E.; Pincetic, A.; Smith, G.A. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 5832–5837. [Google Scholar] [CrossRef]
- Antinone, S.E.; Smith, G.A. Retrograde axon transport of herpes simplex virus and pseudorabies virus: a live-cell comparative analysis. J. Virol. 2010, 84, 1504–1512. [Google Scholar] [CrossRef]
- Coller, K.E.; Smith, G.A. Two viral kinases are required for sustained long distance axon transport of a neuroinvasive herpesvirus. Traffic 2008, 9, 1458–1470. [Google Scholar] [CrossRef]
- Kristensson, K.; Lycke, E.; Roytta, M.; Svennerholm, B.; Vahlne, A. Neuritic transport of herpes simplex virus in rat sensory neurons in vitro. Effects of substances interacting with microtubular function and axonal flow [nocodazole, taxol and erythro-9–3-(2-hydroxynonyl)adenine]. J. Gen. Virol. 1986, 67, 2023–2028. [Google Scholar] [CrossRef]
- Bacallao, R.; Antony, C.; Dotti, C.; Karsenti, E.; Stelzer, E.H.; Simons, K. The subcellular organization of Madin-Darby canine kidney cells during the formation of a polarized epithelium. J. Cell Biol. 1989, 109, 2817–2832. [Google Scholar] [CrossRef]
- Troutt, L.L.; Burnside, B. The unusual microtubule polarity in teleost retinal pigment epithelial cells. J. Cell Biol. 1988, 107, 1461–1464. [Google Scholar] [CrossRef]
- Mogensen, M.M.; Tucker, J.B.; Stebbings, H. Microtubule polarities indicate that nucleation and capture of microtubules occurs at cell surfaces in Drosophila. J. Cell Biol. 1989, 108, 1445–1452. [Google Scholar] [CrossRef]
- Marozin, S.; Prank, U.; Sodeik, B. Herpes simplex virus type 1 infection of polarized epithelial cells requires microtubules and access to receptors present at cell-cell contact sites. J. Gen. Virol. 2004, 85, 775–786. [Google Scholar] [CrossRef]
- Hara, Y.; Hasebe, R.; Sunden, Y.; Ochiai, K.; Honda, E.; Sakoda, Y.; Umemura, T. Propagation of swine hemagglutinating encephalomyelitis virus and pseudorabies virus in dorsal root ganglia cells. J. Vet. Med. Sci. 2009, 71, 595–601. [Google Scholar] [CrossRef]
- Dohner, K.; Wolfstein, A.; Prank, U.; Echeverri, C.; Dujardin, D.; Vallee, R.; Sodeik, B. Function of dynein and dynactin in herpes simplex virus capsid transport. Mol. Biol. Cell 2002, 13, 2795–2809. [Google Scholar] [CrossRef]
- Smith, G.A.; Pomeranz, L.; Gross, S.P.; Enquist, L.W. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 16034–16039. [Google Scholar] [CrossRef]
- Radtke, K.; Kieneke, D.; Wolfstein, A.; Michael, K.; Steffen, W.; Scholz, T.; Karger, A.; Sodeik, B. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 2010, 6, e1000991. [Google Scholar] [CrossRef]
- Strunze, S.; Trotman, L.C.; Boucke, K.; Greber, U.F. Nuclear targeting of adenovirus type 2 requires CRM1-mediated nuclear export. Mol. Biol. Cell 2005, 16, 2999–3009. [Google Scholar] [CrossRef] [PubMed]
- Koshizuka, T.; Kawaguchi, Y.; Nishiyama, Y. Herpes simplex virus type 2 membrane protein UL56 associates with the kinesin motor protein KIF1A. J. Gen. Virol. 2005, 86, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.W.; Diefenbach, R.J.; Homa, F.L.; Miranda-Saksena, M.; Rixon, F.J.; Vittone, V.; Byth, K.; Cunningham, A.L. Herpes simplex virus type 1 capsid protein VP26 interacts with dynein light chains RP3 and Tctex1 and plays a role in retrograde cellular transport. J. Biol. Chem. 2004, 279, 28522–28530. [Google Scholar] [CrossRef] [PubMed]
- Ye, G.J.; Vaughan, K.T.; Vallee, R.B.; Roizman, B. The herpes simplex virus 1 U(L)34 protein interacts with a cytoplasmic dynein intermediate chain and targets nuclear membrane. J. Virol. 2000, 74, 1355–1363. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Miranda-Saksena, M.; Diefenbach, E.; Holland, D.J.; Boadle, R.A.; Armati, P.J.; Cunningham, A.L. Herpes simplex virus tegument protein US11 interacts with conventional kinesin heavy chain. J. Virol. 2002, 76, 3282–3291. [Google Scholar] [CrossRef]
- Trus, B.L.; Homa, F.L.; Booy, F.P.; Newcomb, W.W.; Thomsen, D.R.; Cheng, N.; Brown, J.C.; Steven, A.C. Herpes simplex virus capsids assembled in insect cells infected with recombinant baculoviruses: structural authenticity and localization of VP26. J. Virol. 1995, 69, 7362–7366. [Google Scholar] [CrossRef]
- Zhou, Z.H.; He, J.; Jakana, J.; Tatman, J.D.; Rixon, F.J.; Chiu, W. Assembly of VP26 in herpes simplex virus-1 inferred from structures of wild-type and recombinant capsids. Nat. Struct. Biol. 1995, 2, 1026–1030. [Google Scholar] [CrossRef]
- Wolfstein, A.; Nagel, C.H.; Radtke, K.; Dohner, K.; Allan, V.J.; Sodeik, B. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 2006, 7, 227–237. [Google Scholar] [CrossRef]
- Desai, P.; DeLuca, N.A.; Person, S. Herpes simplex virus type 1 VP26 is not essential for replication in cell culture but influences production of infectious virus in the nervous system of infected mice. Virology 1998, 247, 115–124. [Google Scholar] [CrossRef]
- Antinone, S.E.; Shubeita, G.T.; Coller, K.E.; Lee, J.I.; Haverlock-Moyns, S.; Gross, S.P.; Smith, G.A. The Herpesvirus capsid surface protein, VP26, and the majority of the tegument proteins are dispensable for capsid transport toward the nucleus. J. Virol. 2006, 80, 5494–5498. [Google Scholar] [CrossRef]
- Dohner, K.; Radtke, K.; Schmidt, S.; Sodeik, B. Eclipse phase of herpes simplex virus type 1 infection: Efficient dynein-mediated capsid transport without the small capsid protein VP26. J. Virol. 2006, 80, 8211–8224. [Google Scholar] [CrossRef]
- Batterson, W.; Furlong, D.; Roizman, B. Molecular genetics of herpes simplex virus. VIII. further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J. Virol. 1983, 45, 397–407. [Google Scholar] [CrossRef]
- Jovasevic, V.; Liang, L.; Roizman, B. Proteolytic cleavage of VP1–2 is required for release of herpes simplex virus 1 DNA into the nucleus. J. Virol. 2008, 82, 3311–3319. [Google Scholar] [CrossRef]
- Desai, P.J. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 2000, 74, 11608–11618. [Google Scholar] [CrossRef]
- Fuchs, W.; Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J. Virol. 2004, 78, 11879–11889. [Google Scholar] [CrossRef]
- Abaitua, F.; Souto, R.N.; Browne, H.; Daikoku, T.; O'Hare, P. Characterization of the herpes simplex virus (HSV)-1 tegument protein VP1-2 during infection with the HSV temperature-sensitive mutant tsB7. J. Gen. Virol. 2009, 90, 2353–2363. [Google Scholar] [CrossRef]
- Uetz, P.; Dong, Y.A.; Zeretzke, C.; Atzler, C.; Baiker, A.; Berger, B.; Rajagopala, S.V.; Roupelieva, M.; Rose, D.; Fossum, E.; et al. Herpesviral protein networks and their interaction with the human proteome. Science 2006, 311, 239–242. [Google Scholar] [CrossRef]
- Coller, K.E.; Lee, J.I.; Ueda, A.; Smith, G.A. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J. Virol. 2007, 81, 11790–11797. [Google Scholar] [CrossRef]
- Pasdeloup, D.; Blondel, D.; Isidro, A.L.; Rixon, F.J. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J. Virol. 2009, 83, 6610–6623. [Google Scholar] [CrossRef]
- Vittone, V.; Diefenbach, E.; Triffett, D.; Douglas, M.W.; Cunningham, A.L.; Diefenbach, R.J. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 2005, 79, 9566–9571. [Google Scholar] [CrossRef]
- Klupp, B.G.; Fuchs, W.; Granzow, H.; Nixdorf, R.; Mettenleiter, T.C. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 2002, 76, 3065–3071. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.H.; Cunningham, A.L.; Diefenbach, R.J. The major determinant for addition of tegument protein pUL48 (VP16) to capsids in herpes simplex virus type 1 is the presence of the major tegument protein pUL36 (VP1/2). J. Virol. 2010, 84, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- McNabb, D.S.; Courtney, R.J. Characterization of the large tegument protein (ICP1/2) of herpes simplex virus type 1. Virology 1992, 190, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.H.; Chen, D.H.; Jakana, J.; Rixon, F.J.; Chiu, W. Visualization of tegument-capsid interactions and DNA in intact herpes simplex virus type 1 virions. J. Virol. 1999, 73, 3210–3218. [Google Scholar] [CrossRef]
- Gibson, W.; Roizman, B. Proteins specified by herpes simplex virus. 8. Characterization and composition of multiple capsid forms of subtypes 1 and 2. J. Virol. 1972, 10, 1044–1052. [Google Scholar] [CrossRef]
- Vernon, S.K.; Lawrence, W.C.; Long, C.A.; Rubin, B.A.; Sheffield, J.B. Morphological components of herpesvirus. IV. Ultrastructural features of the envelope and tegument. J. Ultrastruct. Res. 1982, 81, 163–171. [Google Scholar] [CrossRef]
- Leege, T.; Granzow, H.; Fuchs, W.; Klupp, B.G.; Mettenleiter, T.C. Phenotypic similarities and differences between UL37-deleted pseudorabies virus and herpes simplex virus type 1. J. Gen. Virol. 2009, 90, 1560–1568. [Google Scholar] [CrossRef]
- Klupp, B.G.; Granzow, H.; Mundt, E.; Mettenleiter, T.C. Pseudorabies virus UL37 gene product is involved in secondary envelopment. J. Virol. 2001, 75, 8927–8936. [Google Scholar] [CrossRef]
- Luxton, G.W.; Lee, J.I.; Haverlock-Moyns, S.; Schober, J.M.; Smith, G.A. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 2006, 80, 201–209. [Google Scholar] [CrossRef]
- Desai, P.; Sexton, G.L.; McCaffery, J.M.; Person, S. A null mutation in the gene encoding the herpes simplex virus type 1 UL37 polypeptide abrogates virus maturation. J. Virol. 2001, 75, 10259–10271. [Google Scholar] [CrossRef]
- Krautwald, M.; Fuchs, W.; Klupp, B.G.; Mettenleiter, T.C. Translocation of incoming pseudorabies virus capsids to the cell nucleus is delayed in the absence of tegument protein pUL37. J. Virol. 2009, 83, 3389–3396. [Google Scholar] [CrossRef]
- Roberts, A.P.; Abaitua, F.; O'Hare, P.; McNab, D.; Rixon, F.J.; Pasdeloup, D. Differing roles of inner tegument proteins pUL36 and pUL37 during entry of herpes simplex virus type 1. J. Virol. 2009, 83, 105–116. [Google Scholar] [CrossRef]
- Forest, T.; Barnard, S.; Baines, J.D. Active intranuclear movement of herpesvirus capsids. Nat. Cell Biol. 2005, 7, 429–431. [Google Scholar] [CrossRef]
- Baines, J.D.; Hsieh, C.E.; Wills, E.; Mannella, C.; Marko, M. Electron tomography of nascent herpes simplex virus virions. J. Virol. 2007, 81, 2726–2735. [Google Scholar] [CrossRef]
- Leuzinger, H.; Ziegler, U.; Schraner, E.M.; Fraefel, C.; Glauser, D.L.; Heid, I.; Ackermann, M.; Mueller, M.; Wild, P. Herpes simplex virus 1 envelopment follows two diverse pathways. J. Virol. 2005, 79, 13047–13059. [Google Scholar] [CrossRef]
- Mettenleiter, T.C.; Minson, T. Egress of alphaherpesviruses. J. Virol. 2006, 80, 1610–1611; author reply 1611–1612. [Google Scholar] [CrossRef]
- Campadelli-Fiume, G.; Roizman, B. The egress of herpesviruses from cells: The unanswered questions. J. Virol. 2006, 80, 6716–6717; author replies 6717–6719. [Google Scholar] [CrossRef]
- Granzow, H.; Klupp, B.G.; Fuchs, W.; Veits, J.; Osterrieder, N.; Mettenleiter, T.C. Egress of alphaherpesviruses: comparative ultrastructural study. J. Virol. 2001, 75, 3675–3684. [Google Scholar] [CrossRef]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: An update. Virus Res. 2009, 143, 222–234. [Google Scholar] [CrossRef]
- Fuchs, W.; Granzow, H.; Klupp, B.G.; Kopp, M.; Mettenleiter, T.C. The UL48 tegument protein of pseudorabies virus is critical for intracytoplasmic assembly of infectious virions. J. Virol. 2002, 76, 6729–6742. [Google Scholar] [CrossRef]
- Lee, J.H.; Vittone, V.; Diefenbach, E.; Cunningham, A.L.; Diefenbach, R.J. Identification of structural protein-protein interactions of herpes simplex virus type 1. Virology 2008, 378, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Loomis, J.S.; Courtney, R.J.; Wills, J.W. Binding partners for the UL11 tegument protein of herpes simplex virus type 1. J. Virol. 2003, 77, 11417–11424. [Google Scholar] [CrossRef] [PubMed]
- de Wind, N.; Wagenaar, F.; Pol, J.; Kimman, T.; Berns, A. The pseudorabies virus homology of the herpes simplex virus UL21 gene product is a capsid protein which is involved in capsid maturation. J. Virol. 1992, 66, 7096–7103. [Google Scholar] [CrossRef] [PubMed]
- Klupp, B.G.; Bottcher, S.; Granzow, H.; Kopp, M.; Mettenleiter, T.C. Complex formation between the UL16 and UL21 tegument proteins of pseudorabies virus. J. Virol. 2005, 79, 1510–1522. [Google Scholar] [CrossRef]
- Wang, L.; Liu, L.; Che, Y.; Jiang, L.; Dong, C.; Zhang, Y.; Li, Q. Egress of HSV-1 capsid requires the interaction of VP26 and a cellular tetraspanin membrane protein. Virol. J. 2010, 7, 156. [Google Scholar] [CrossRef]
- Charrin, S.; le Naour, F.; Silvie, O.; Milhiet, P.E.; Boucheix, C.; Rubinstein, E. Lateral organization of membrane proteins: tetraspanins spin their web. Biochem. J. 2009, 420, 133–154. [Google Scholar] [CrossRef]
- Desai, P.; Sexton, G.L.; Huang, E.; Person, S. Localization of herpes simplex virus type 1 UL37 in the Golgi complex requires UL36 but not capsid structures. J. Virol. 2008, 82, 11354–11361. [Google Scholar] [CrossRef]
- Johnson, D.C.; Webb, M.; Wisner, T.W.; Brunetti, C. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 2001, 75, 821–833. [Google Scholar] [CrossRef]
- McMillan, T.N.; Johnson, D.C. Cytoplasmic domain of herpes simplex virus gE causes accumulation in the trans-Golgi network, a site of virus envelopment and sorting of virions to cell junctions. J. Virol. 2001, 75, 1928–1940. [Google Scholar] [CrossRef]
- Sugimoto, K.; Uema, M.; Sagara, H.; Tanaka, M.; Sata, T.; Hashimoto, Y.; Kawaguchi, Y. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J. Virol. 2008, 82, 5198–5211. [Google Scholar] [CrossRef]
- Avitabile, E.; Di Gaeta, S.; Torrisi, M.R.; Ward, P.L.; Roizman, B.; Campadelli-Fiume, G. Redistribution of microtubules and Golgi apparatus in herpes simplex virus-infected cells and their role in viral exocytosis. J. Virol. 1995, 69, 7472–7482. [Google Scholar] [CrossRef]
- Campadelli, G.; Brandimarti, R.; Di Lazzaro, C.; Ward, P.L.; Roizman, B.; Torrisi, M.R. Fragmentation and dispersal of Golgi proteins and redistribution of glycoproteins and glycolipids processed through the Golgi apparatus after infection with herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 2798–2802. [Google Scholar] [CrossRef]
- McGraw, H.M.; Friedman, H.M. Herpes simplex virus type 1 glycoprotein E mediates retrograde spread from epithelial cells to neurites. J. Virol. 2009, 83, 4791–4799. [Google Scholar] [CrossRef]
- Remillard-Labrosse, G.; Mihai, C.; Duron, J.; Guay, G.; Lippe, R. Protein kinase D-dependent trafficking of the large Herpes simplex virus type 1 capsids from the TGN to plasma membrane. Traffic 2009, 10, 1074–1083. [Google Scholar] [CrossRef]
- Liljedahl, M.; Maeda, Y.; Colanzi, A.; Ayala, I.; Van Lint, J.; Malhotra, V. Protein kinase D regulates the fission of cell surface destined transport carriers from the trans-Golgi network. Cell 2001, 104, 409–420. [Google Scholar] [CrossRef]
- Yeaman, C.; Ayala, M.I.; Wright, J.R.; Bard, F.; Bossard, C.; Ang, A.; Maeda, Y.; Seufferlein, T.; Mellman, I.; Nelson, W.J.; et al. Protein kinase D regulates basolateral membrane protein exit from trans-Golgi network. Nat. Cell Biol. 2004, 6, 106–112. [Google Scholar] [CrossRef]
- Lee, J.I.; Sollars, P.J.; Baver, S.B.; Pickard, G.E.; Leelawong, M.; Smith, G.A. A herpesvirus encoded deubiquitinase is a novel neuroinvasive determinant. PLoS Pathog. 2009, 5, e1000387. [Google Scholar] [CrossRef]
- Lee, G.E.; Murray, J.W.; Wolkoff, A.W.; Wilson, D.W. Reconstitution of herpes simplex virus microtubule-dependent trafficking in vitro. J. Virol. 2006, 80, 4264–4275. [Google Scholar] [CrossRef]
- Shanda, S.K.; Wilson, D.W. UL36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J. Virol. 2008, 82, 7388–7394. [Google Scholar] [CrossRef]
- Szilagyi, J.F.; Cunningham, C. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J. Gen. Virol. 1991, 72, 661–668. [Google Scholar] [CrossRef]
- Zaichick, S.V.; Smith, G.A. Northwestern University, Evanston, IL, USA. To be submitted for publication, 2011.
- Bisbal, M.; Conde, C.; Donoso, M.; Bollati, F.; Sesma, J.; Quiroga, S.; Diaz Anel, A.; Malhotra, V.; Marzolo, M.P.; Caceres, A. Protein kinase d regulates trafficking of dendritic membrane proteins in developing neurons. J. Neurosci. 2008, 28, 9297–9308. [Google Scholar] [CrossRef] [PubMed]
- Benboudjema, L.; Mulvey, M.; Gao, Y.; Pimplikar, S.W.; Mohr, I. Association of the herpes simplex virus type 1 Us11 gene product with the cellular kinesin light-chain-related protein PAT1 results in the redistribution of both polypeptides. J. Virol. 2003, 77, 9192–9203. [Google Scholar] [CrossRef] [PubMed]
- Koshizuka, T.; Goshima, F.; Takakuwa, H.; Nozawa, N.; Daikoku, T.; Koiwai, O.; Nishiyama, Y. Identification and characterization of the UL56 gene product of herpes simplex virus type 2. J. Virol. 2002, 76, 6718–6728. [Google Scholar] [CrossRef] [PubMed]
- Rosen-Wolff, A.; Lamade, W.; Berkowitz, C.; Becker, Y.; Darai, G. Elimination of UL56 gene by insertion of LacZ cassette between nucleotide position 116030 to 121753 of the herpes simplex virus type 1 genome abrogates intraperitoneal pathogenicity in tree shrews and mice. Virus Res. 1991, 20, 205–221. [Google Scholar] [CrossRef]
- van Leeuwen, H.; Elliott, G.; O'Hare, P. Evidence of a role for nonmuscle myosin II in herpes simplex virus type 1 egress. J. Virol. 2002, 76, 3471–3481. [Google Scholar] [CrossRef]
- Roberts, K.L.; Baines, J.D. Myosin Va enhances secretion of herpes simplex virus 1 virions and cell surface expression of viral glycoproteins. J. Virol. 2010, 84, 9889–9896. [Google Scholar] [CrossRef]
- Bonanomi, D.; Benfenati, F.; Valtorta, F. Protein sorting in the synaptic vesicle life cycle. Prog. Neurobiol. 2006, 80, 177–217. [Google Scholar] [CrossRef]
- Tomishima, M.J.; Enquist, L.W. A conserved alpha-herpesvirus protein necessary for axonal localization of viral membrane proteins. J. Cell Biol. 2001, 154, 741–752. [Google Scholar] [CrossRef]
- Lyman, M.G.; Feierbach, B.; Curanovic, D.; Bisher, M.; Enquist, L.W. Pseudorabies virus Us9 directs axonal sorting of viral capsids. J. Virol. 2007, 81, 11363–11371. [Google Scholar] [CrossRef]
- Lyman, M.G.; Kemp, C.D.; Taylor, M.P.; Enquist, L.W. Comparison of the pseudorabies virus Us9 protein with homologs from other veterinary and human alphaherpesviruses. J. Virol. 2009, 83, 6978–6986. [Google Scholar] [CrossRef]
- Koshizuka, T.; Kawaguchi, Y.; Nozawa, N.; Mori, I.; Nishiyama, Y. Herpes simplex virus protein UL11 but not UL51 is associated with lipid rafts. Virus Genes 2007, 35, 571–575. [Google Scholar] [CrossRef]
- Lyman, M.G.; Curanovic, D.; Enquist, L.W. Targeting of pseudorabies virus structural proteins to axons requires association of the viral Us9 protein with lipid rafts. PLoS Pathog. 2008, 4, e1000065. [Google Scholar] [CrossRef]
- Cook, M.L.; Stevens, J.G. Pathogenesis of herpetic neuritis and ganglionitis in mice: evidence for intra-axonal transport of infection. Infect. Immun. 1973, 7, 272–288. [Google Scholar] [CrossRef]
- Hill, T.J.; Field, H.J.; Roome, A.P. Intra-axonal location of herpes simplex virus particles. J. Gen. Virol. 1972, 15, 233–235. [Google Scholar] [CrossRef]
- Kristensson, K.; Ghetti, B.; Wisniewski, H.M. Study on the propagation of Herpes simplex virus (type 2) into the brain after intraocular injection. Brain Res. 1974, 69, 189–201. [Google Scholar] [CrossRef]
- LaVail, J.H.; Topp, K.S.; Giblin, P.A.; Garner, J.A. Factors that contribute to the transneuronal spread of herpes simplex virus. J. Neurosci. Res. 1997, 49, 485–496. [Google Scholar] [CrossRef]
- Penfold, M.E.; Armati, P.; Cunningham, A.L. Axonal transport of herpes simplex virions to epidermal cells: evidence for a specialized mode of virus transport and assembly. Proc. Natl. Acad. Sci. U. S. A. 1994, 91, 6529–6533. [Google Scholar] [CrossRef]
- Holland, D.J.; Miranda-Saksena, M.; Boadle, R.A.; Armati, P.; Cunningham, A.L. Anterograde transport of herpes simplex virus proteins in axons of peripheral human fetal neurons: An immunoelectron microscopy study. J. Virol. 1999, 73, 8503–8511. [Google Scholar] [CrossRef]
- Negatsch, A.; Granzow, H.; Maresch, C.; Klupp, B.G.; Fuchs, W.; Teifke, J.P.; Mettenleiter, T.C. Ultrastructural analysis of virion formation and intraaxonal transport of herpes simplex virus type 1 in primary rat neurons. J. Virol. 2010, 84, 13031–13035. [Google Scholar] [CrossRef]
- Huang, J.; Lazear, H.M.; Friedman, H.M. Completely assembled virus particles detected by transmission electron microscopy in proximal and mid-axons of neurons infected with herpes simplex virus type 1, herpes simplex virus type 2 and pseudorabies virus. Virology 2011, 409, 12–16. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Miranda-Saksena, M.; Douglas, M.W.; Cunningham, A.L. Transport and egress of herpes simplex virus in neurons. Rev. Med. Virol. 2008, 18, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Curanovic, D.; Enquist, L. Directional transneuronal spread of alpha-herpesvirus infection. Future Virol. 2009, 4, 591. [Google Scholar] [CrossRef] [PubMed]
- Wisner, T.W.; Sugimoto, K.; Howard, P.W.; Kawaguchi, Y.; Johnson, D.C. Anterograde transport of herpes simplex virus capsids in neurons by both Separate and Married mechanisms. J. Virol. 2011, 85, 5919–5928. [Google Scholar] [CrossRef] [PubMed]
- Antinone, S.E.; Zaichick, S.V.; Smith, G.A. Resolving the assembly state of herpes simplex virus during axon transport by live-cell imaging. J. Virol. 2010, 84, 13019–13030. [Google Scholar] [CrossRef]
- Miranda-Saksena, M.; Boadle, R.A.; Aggarwal, A.; Tijono, B.; Rixon, F.J.; Diefenbach, R.J.; Cunningham, A.L. Herpes simplex virus utilizes the large secretory vesicle pathway for anterograde transport of tegument and envelope proteins and for viral exocytosis from growth cones of human fetal axons. J. Virol. 2009, 83, 3187–3199. [Google Scholar] [CrossRef]
- Smith, G.A.; Gross, S.P.; Enquist, L.W. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 3466–3470. [Google Scholar] [CrossRef]
- Tannous, R.; Grose, C. Calculation of the anterograde velocity of varicella-zoster virions in a human sciatic nerve during shingles. J. Infect. Dis. 2011, 203, 324–326. [Google Scholar] [CrossRef]
- Laing, J.M.; Smith, C.C.; Aurelian, L. Multi-targeted neuroprotection by the HSV-2 gene ICP10PK includes robust bystander activity through PI3-K/Akt and/or MEK/ERK-dependent neuronal release of vascular endothelial growth factor and fractalkine. J. Neurochem. 2010, 112, 662–676. [Google Scholar] [CrossRef]
- Goold, R.G.; Gordon-Weeks, P.R. Glycogen synthase kinase 3beta and the regulation of axon growth. Biochem. Soc. Trans. 2004, 32, 809–811. [Google Scholar] [CrossRef]
- Ada-Nguema, A.S.; Xenias, H.; Hofman, J.M.; Wiggins, C.H.; Sheetz, M.P.; Keely, P.J. The small GTPase R-Ras regulates organization of actin and drives membrane protrusions through the activity of PLCepsilon. J. Cell Sci. 2006, 119, 1307–1319. [Google Scholar] [CrossRef]
- Van den Broeke, C.; Deruelle, M.; Nauwynck, H.J.; Coller, K.E.; Smith, G.A.; Van Doorsselaere, J.; Favoreel, H.W. The kinase activity of pseudorabies virus US3 is required for modulation of the actin cytoskeleton. Virology 2009, 385, 155–160. [Google Scholar] [CrossRef]
- Van Minnebruggen, G.; Favoreel, H.W.; Jacobs, L.; Nauwynck, H.J. Pseudorabies virus US3 protein kinase mediates actin stress fiber breakdown. J. Virol. 2003, 77, 9074–9080. [Google Scholar] [CrossRef]
- Finnen, R.L.; Banfield, B.W. Subcellular localization of the alphaherpesvirus serine/threonine kinase Us3 as a determinant of Us3 function. Virulence 2010, 1, 291–294. [Google Scholar] [CrossRef]
- Finnen, R.L.; Roy, B.B.; Zhang, H.; Banfield, B.W. Analysis of filamentous process induction and nuclear localization properties of the HSV-2 serine/threonine kinase Us3. Virology 2010, 397, 23–33. [Google Scholar] [CrossRef]
- Murata, T.; Goshima, F.; Daikoku, T.; Takakuwa, H.; Nishiyama, Y. Expression of herpes simplex virus type 2 US3 affects the Cdc42/Rac pathway and attenuates c-Jun N-terminal kinase activation. Genes Cells 2000, 5, 1017–1027. [Google Scholar] [CrossRef]
- Brzozowska, A.; Rychlowski, M.; Lipinska, A.D.; Bienkowska-Szewczyk, K. Point mutations in BHV-1 Us3 gene abolish its ability to induce cytoskeletal changes in various cell types. Vet. Microbiol. 2010, 143, 8–13. [Google Scholar] [CrossRef]
- Favoreel, H.W.; Van Minnebruggen, G.; Adriaensen, D.; Nauwynck, H.J. Cytoskeletal rearrangements and cell extensions induced by the US3 kinase of an alphaherpesvirus are associated with enhanced spread. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 8990–8995. [Google Scholar] [CrossRef]
- Kimman, T.G.; De Wind, N.; De Bruin, T.; de Visser, Y.; Voermans, J. Inactivation of glycoprotein gE and thymidine kinase or the US3-encoded protein kinase synergistically decreases in vivo replication of pseudorabies virus and the induction of protective immunity. Virology 1994, 205, 511–518. [Google Scholar] [CrossRef]
- Kimman, T.G.; de Wind, N.; Oei-Lie, N.; Pol, J.M.; Berns, A.J.; Gielkens, A.L. Contribution of single genes within the unique short region of Aujeszky's disease virus (suid herpesvirus type 1) to virulence, pathogenesis and immunogenicity. J. Gen. Virol. 1992, 73, 243–251. [Google Scholar] [CrossRef]
- Olsen, L.M.; Ch'ng, T.H.; Card, J.P.; Enquist, L.W. Role of pseudorabies virus Us3 protein kinase during neuronal infection. J. Virol. 2006, 80, 6387–6398. [Google Scholar] [CrossRef]
- Mori, I.; Goshima, F.; Koshizuka, T.; Koide, N.; Sugiyama, T.; Yoshida, T.; Yokochi, T.; Kimura, Y.; Nishiyama, Y. The US3 protein kinase of herpes simplex virus attenuates the activation of the c-Jun N-terminal protein kinase signal transduction pathway in infected piriform cortex neurons of C57BL/6 mice. Neurosci. Lett. 2003, 351, 201–205. [Google Scholar] [CrossRef] [PubMed]
- Mori, I.; Goshima, F.; Watanabe, D.; Ito, H.; Koide, N.; Yoshida, T.; Liu, B.; Kimura, Y.; Yokochi, T.; Nishiyama, Y. Herpes simplex virus US3 protein kinase regulates virus-induced apoptosis in olfactory and vomeronasal chemosensory neurons in vivo. Microbes Infect. 2006, 8, 1806–1812. [Google Scholar] [CrossRef] [PubMed]
- Kurachi, R.; Daikoku, T.; Tsurumi, T.; Maeno, K.; Nishiyama, Y.; Kurata, T. The pathogenicity of a US3 protein kinase-deficient mutant of herpes simplex virus type 2 in mice. Arch. Virol. 1993, 133, 259–273. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Yamada, Y.; Kurachi, R.; Daikoku, T. Construction of a US3 lacZ insertion mutant of herpes simplex virus type 2 and characterization of its phenotype in vitro and in vivo. Virology 1992, 190, 256–268. [Google Scholar] [CrossRef] [PubMed]
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef]
- Daire, V.; Giustiniani, J.; Leroy-Gori, I.; Quesnoit, M.; Drevensek, S.; Dimitrov, A.; Perez, F.; Pous, C. Kinesin-1 regulates microtubule dynamics via a c-Jun N-terminal kinase-dependent mechanism. J. Biol. Chem. 2009, 284, 31992–32001. [Google Scholar] [CrossRef]
- Van den Broeke, C.; Radu, M.; Nauwynck, H.J.; Chernoff, J.; Favoreel, H.W. Role of group A p21-activated kinases in the anti-apoptotic activity of the pseudorabies virus US3 protein kinase. Virus Res. 2011, 155, 376–380. [Google Scholar] [CrossRef]
- Geiss, B.J.; Tavis, J.E.; Metzger, L.M.; Leib, D.A.; Morrison, L.A. Temporal regulation of herpes simplex virus type 2 VP22 expression and phosphorylation. J. Virol. 2001, 75, 10721–10729. [Google Scholar] [CrossRef]
- Labiuk, S.L.; Lobanov, V.; Lawman, Z.; Snider, M.; Babiuk, L.A.; van Drunen Littel-van den Hurk, S. Bovine herpesvirus-1 US3 protein kinase: Critical residues and involvement in the phosphorylation of VP22. J. Gen. Virol. 2010, 91, 1117–1126. [Google Scholar] [CrossRef]
- Elliott, G.; O'Reilly, D.; O'Hare, P. Phosphorylation of the herpes simplex virus type 1 tegument protein VP22. Virology 1996, 226, 140–145. [Google Scholar] [CrossRef]
- Elliott, G.; O'Hare, P. Herpes simplex virus type 1 tegument protein VP22 induces the stabilization and hyperacetylation of microtubules. J. Virol. 1998, 72, 6448–6455. [Google Scholar] [CrossRef]
- Martin, A.; O'Hare, P.; McLauchlan, J.; Elliott, G. Herpes simplex virus tegument protein VP22 contains overlapping domains for cytoplasmic localization, microtubule interaction, and chromatin binding. J. Virol. 2002, 76, 4961–4970. [Google Scholar] [CrossRef]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef]
- Vaughan, E.E.; Geiger, R.C.; Miller, A.M.; Loh-Marley, P.L.; Suzuki, T.; Miyata, N.; Dean, D.A. Microtubule acetylation through HDAC6 inhibition results in increased transfection efficiency. Mol. Ther. 2008, 16, 1841–1847. [Google Scholar] [CrossRef]
- Harms, J.S.; Ren, X.; Oliveira, S.C.; Splitter, G.A. Distinctions between bovine herpesvirus 1 and herpes simplex virus type 1 VP22 tegument protein subcellular associations. J. Virol. 2000, 74, 3301–3312. [Google Scholar] [CrossRef]
- Brewis, N.; Phelan, A.; Webb, J.; Drew, J.; Elliott, G.; O'Hare, P. Evaluation of VP22 spread in tissue culture. J. Virol. 2000, 74, 1051–1056. [Google Scholar] [CrossRef]
- Aints, A.; Dilber, M.S.; Smith, C.I. Intercellular spread of GFP-VP22. J. Gene Med. 1999, 1, 275–279. [Google Scholar] [CrossRef]
- Dilber, M.S.; Phelan, A.; Aints, A.; Mohamed, A.J.; Elliott, G.; Smith, C.I.; O'Hare, P. Intercellular delivery of thymidine kinase prodrug activating enzyme by the herpes simplex virus protein, VP22. Gene Ther. 1999, 6, 12–21. [Google Scholar] [CrossRef]
- Elliott, G.; O'Hare, P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell 1997, 88, 223–233. [Google Scholar] [CrossRef]
- Sciortino, M.T.; Taddeo, B.; Poon, A.P.; Mastino, A.; Roizman, B. Of the three tegument proteins that package mRNA in herpes simplex virions, one (VP22) transports the mRNA to uninfected cells for expression prior to viral infection. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 8318–8323. [Google Scholar] [CrossRef]
- Zheng, C.F.; Brownlie, R.; Huang, D.Y.; Babiuk, L.A.; van Drunen Littel-van den Hurk, S. Intercellular trafficking of the major tegument protein VP22 of bovine herpesvirus-1 and its application to improve a DNA vaccine. Arch. Virol. 2006, 151, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Lemken, M.L.; Wolf, C.; Wybranietz, W.A.; Schmidt, U.; Smirnow, I.; Buhring, H.J.; Mack, A.F.; Lauer, U.M.; Bitzer, M. Evidence for intercellular trafficking of VP22 in living cells. Mol. Ther. 2007, 15, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Kretz, A.; Wybranietz, W.A.; Hermening, S.; Lauer, U.M.; Isenmann, S. HSV-1 VP22 augments adenoviral gene transfer to CNS neurons in the retina and striatum in vivo. Mol. Ther. 2003, 7, 659–669. [Google Scholar] [CrossRef] [PubMed]
- del Rio, T.; Werner, H.C.; Enquist, L.W. The pseudorabies virus VP22 homologue (UL49) is dispensable for virus growth in vitro and has no effect on virulence and neuronal spread in rodents. J. Virol. 2002, 76, 774–782. [Google Scholar] [CrossRef]
- Duffy, C.; Lavail, J.H.; Tauscher, A.N.; Wills, E.G.; Blaho, J.A.; Baines, J.D. Characterization of a UL49-null mutant: VP22 of herpes simplex virus type 1 facilitates viral spread in cultured cells and the mouse cornea. J. Virol. 2006, 80, 8664–8675. [Google Scholar] [CrossRef]
- Liu, M.; Schmidt, E.E.; Halford, W.P. ICP0 dismantles microtubule networks in herpes simplex virus-infected cells. PLoS ONE 2010, 5, e10975. [Google Scholar] [CrossRef]
- Guo, L.; Zhang, Y.; Che, Y.; Wu, W.; Li, W.; Wang, L.; Liao, Y.; Liu, L.; Li, Q. Interactions of the HSV-1 UL25 capsid protein with cellular microtubule-associated protein. Virologica Sinica 2008, 23, 211–217. [Google Scholar] [CrossRef]
- Takakuwa, H.; Goshima, F.; Koshizuka, T.; Murata, T.; Daikoku, T.; Nishiyama, Y. Herpes simplex virus encodes a virion-associated protein which promotes long cellular processes in over-expressing cells. Genes Cells 2001, 6, 955–966. [Google Scholar] [CrossRef]
- Pei, Y.; Xiang, Y.F.; Chen, J.N.; Lu, C.H.; Hao, J.; Du, Q.; Lai, C.C.; Qu, C.; Li, S.; Ju, H.Q.; et al. Pentagalloylglucose downregulates cofilin1 and inhibits HSV-1 infection. Antivir. Res. 2011, 89, 98–108. [Google Scholar] [CrossRef]


© 2011 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zaichick, S.V.; Bohannon, K.P.; Smith, G.A. Alphaherpesviruses and the Cytoskeleton in Neuronal Infections. Viruses 2011, 3, 941-981. https://doi.org/10.3390/v3070941
Zaichick SV, Bohannon KP, Smith GA. Alphaherpesviruses and the Cytoskeleton in Neuronal Infections. Viruses. 2011; 3(7):941-981. https://doi.org/10.3390/v3070941
Chicago/Turabian StyleZaichick, Sofia V., Kevin P. Bohannon, and Gregory A. Smith. 2011. "Alphaherpesviruses and the Cytoskeleton in Neuronal Infections" Viruses 3, no. 7: 941-981. https://doi.org/10.3390/v3070941
APA StyleZaichick, S. V., Bohannon, K. P., & Smith, G. A. (2011). Alphaherpesviruses and the Cytoskeleton in Neuronal Infections. Viruses, 3(7), 941-981. https://doi.org/10.3390/v3070941