Cellular Players in the Herpes Simplex Virus Dependent Apoptosis Balancing Act

Abstract
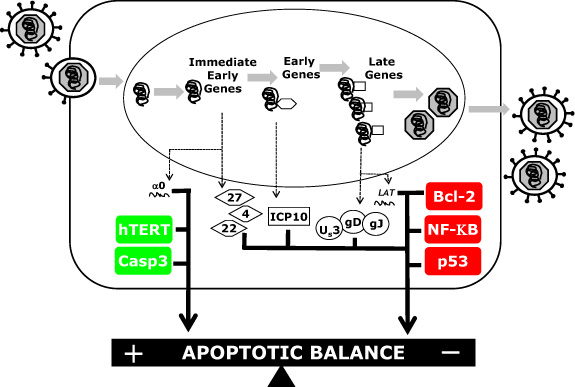
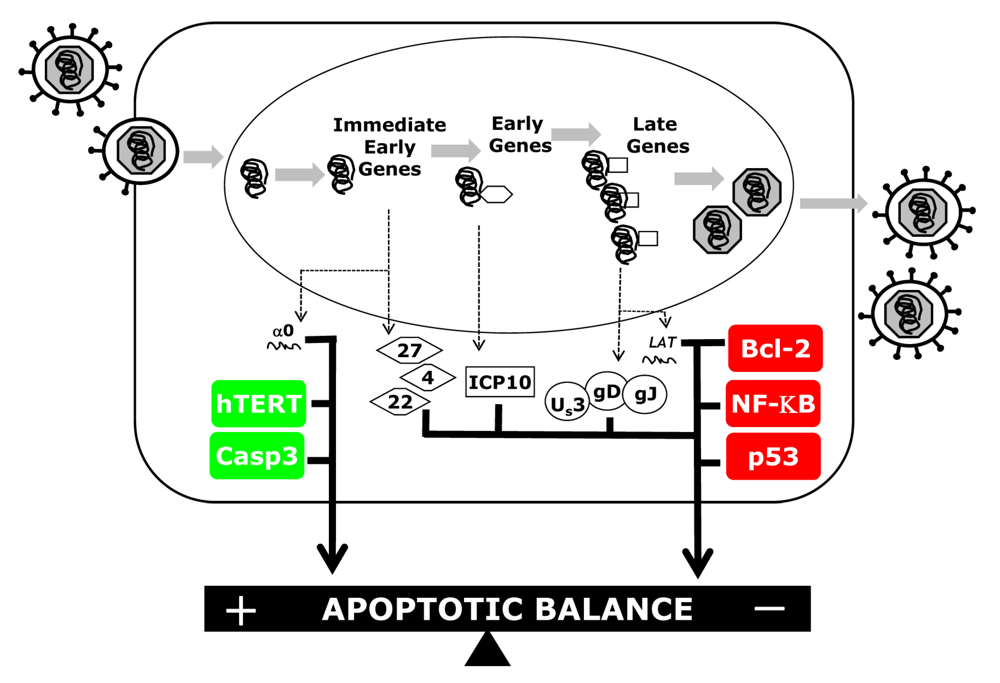
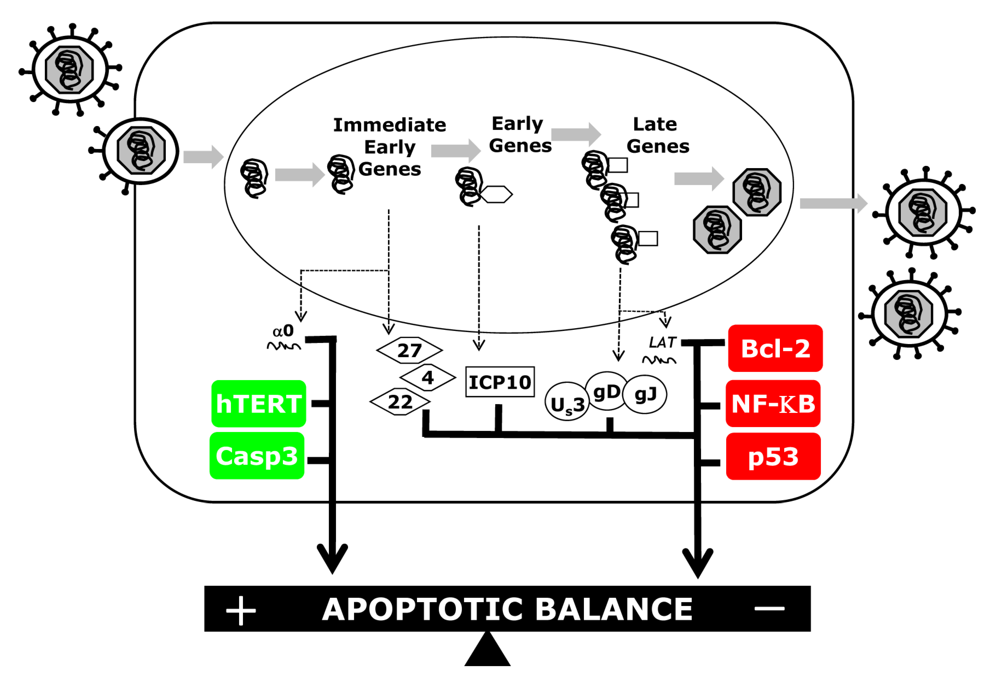
:
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1 Caspases
2.2. Bcl-2 Family Members
2.3. NF-ĸB
2.4. Oncogenic Genes
2.4.1. p53
2.4.2. Telomerase
3. Conclusions
Acknowledgments
References
- Fadok, V.A.; Bratton, D.L.; Frasch, S.C.; Warner, M.L.; Henson, P.M. The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ 1998, 5, 551–562. [Google Scholar] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972, 26, 239–257. [Google Scholar] [PubMed]
- Wyllie, A.H.; Kerr, J.F.; Currie, A.R. Cell death: the significance of apoptosis. Int Rev Cytol 1980, 68, 251–306. [Google Scholar] [CrossRef] [PubMed]
- Nagata, S. Apoptosis by death factor. Cell 1997, 88, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.M.; MacFarlane, M.; Zhuang, J.; Wolf, B.B.; Green, D.R.; Cohen, G.M. Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J Biol Chem 1999, 274, 5053–5060. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazi, A.; Dixit, V.M. Death receptors: signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Evan, G.I. A matter of life and death. Cancer cell 2002, 1, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Green, D.; Kroemer, G. The central executioners of apoptosis: caspases or mitochondria? Trends Cell Biol 1998, 8, 267–271. [Google Scholar] [CrossRef]
- Petit, P.X.; Susin, S.A.; Zamzami, N.; Mignotte, B.; Kroemer, G. Mitochondria and programmed cell death: back to the future. FEBS letters 1996, 396, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Koyama, A.H.; Adachi, A.; Irie, H. Physiological significance of apoptosis during animal virus infection. Int Rev Immunol 2003, 22, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Blaho, J.A. Virus infection and apoptosis (issue II) an introduction: cheating death or death as a fact of life? Int Rev Immunol 2004, 23, 1–6. [Google Scholar] [CrossRef]
- Querido, E.; Marcellus, R.C.; Lai, A.; Charbonneau, R.; Teodoro, J.G.; Ketner, G.; Branton, P.E. Regulation of p53 levels by the E1B 55-kilodalton protein and E4orf6 in adenovirus-infected cells. J Virol 1997, 71, 3788–3798. [Google Scholar] [PubMed]
- Scheffner, M.; Munger, K.; Byrne, J.C.; Howley, P.M. The state of the p53 and retinoblastoma genes in human cervical carcinoma cell lines. Proc Natl Acad Sci U S A 1991, 88, 5523–5527. [Google Scholar] [CrossRef] [PubMed]
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79. [Google Scholar] [PubMed]
- Clem, R.J.; Fechheimer, M.; Miller, L.K. Prevention of apoptosis by a baculovirus gene during infection of insect cells. Science 1991, 254, 1388–1390. [Google Scholar] [PubMed]
- Cheng, E.H.; Nicholas, J.; Bellows, D.S.; Hayward, G.S.; Guo, H.G.; Reitz, M.S.; Hardwick, J.M. A Bcl-2 homolog encoded by Kaposi sarcoma-associated virus, human herpesvirus 8, inhibits apoptosis but does not heterodimerize with Bax or Bak. Proc Natl Acad Sci U S A 1997, 94, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Sarid, R.; Sato, T.; Bohenzky, R.A.; Russo, J.J.; Chang, Y. Kaposi's sarcoma-associated herpesvirus encodes a functional bcl-2 homologue. Nat Med 1997, 3, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Cazals-Hatem, D.L.; Kitada, S.; Tanaka, S.; Miyashita, T.; Hovey, L.R.; Huen, D.; Rickinson, A.; Veerapandian, P.; Krajewski, S. Evolutionary conservation of function among mammalian, avian, and viral homologs of the Bcl-2 oncoprotein. DNA Cell Biol 1994, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Aubert, M.; Blaho, J.A. The herpes simplex virus type 1 regulatory protein ICP27 is required for the prevention of apoptosis in infected human cells. J Virol 1999, 73, 2803–2813. [Google Scholar] [PubMed]
- Koyama, A.H.; Adachi, A. Induction of apoptosis by herpes simplex virus type 1. J Gen Virol 1997, 78, 2909–2912. [Google Scholar] [PubMed]
- Sanfilippo, C.M.; Blaho, J.A. ICP0 gene expression is a herpes simplex virus type 1 apoptotic trigger. J Virol 2006, 80, 6810–6821. [Google Scholar] [CrossRef] [PubMed]
- Sanfilippo, C.M.; Chirimuuta, F.N.; Blaho, J.A. Herpes Simplex Virus Type 1 Immediate-Early Gene Expression Is Required for the Induction of Apoptosis in Human Epithelial HEp-2 Cells. J Virol 2004, 78, 224–239. [Google Scholar] [CrossRef] [PubMed]
- Galvan, V.; Roizman, B. Herpes simplex virus 1 induces and blocks apoptosis at multiple steps during infection and protects cells from exogenous inducers in a cell- type-dependent manner. Proc Natl Acad Sci U S A 1998, 95, 3931–3936. [Google Scholar] [CrossRef] [PubMed]
- Koyama, A.H.; Miwa, Y. Suppression of apoptotic DNA fragmentation in herpes simplex virus type 1-infected cells. J Virol 1997, 71, 2567–2571. [Google Scholar] [PubMed]
- Aubert, M.; Blaho, J.A. Modulation of apoptosis during HSV infection in human cells. Microbes Infect 2001, 3, 859–866. [Google Scholar] [CrossRef]
- Leopardi, R.; Roizman, B. The herpes simplex virus major regulatory protein ICP4 blocks apoptosis induced by the virus or by hyperthermia. Proc Natl Acad Sci U S A 1996, 93, 9583–9587. [Google Scholar] [CrossRef] [PubMed]
- Han, J.Y.; Miller, S.A.; Wolfe, T.M.; Pourhassan, H.; Jerome, K.R. Cell type-specific induction and inhibition of apoptosis by Herpes Simplex virus type 2 ICP10. J Virol 2009, 83, 2765–2769. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.; Gyure, K.A.; Pereira, E.F.; Aurelian, L. Herpes simplex virus type 1-induced encephalitis has an apoptotic component associated with activation of c-Jun N-terminal kinase. J Neurovirol 2003, 9, 101–111. [Google Scholar] [PubMed]
- Jerome, K.R.; Fox, R.; Chen, Z.; Sears, A.E.; Lee, H.; Corey, L. Herpes simplex virus inhibits apoptosis through the action of two genes, Us5 and Us3. J Virol 1999, 73, 8950–8957. [Google Scholar] [PubMed]
- Leopardi, R.; Van Sant, C.; Roizman, B. The herpes simplex virus 1 protein kinase US3 is required for protection from apoptosis induced by the virus. Proc Natl Acad Sci U S A 1997, 94, 7891–7896. [Google Scholar] [CrossRef] [PubMed]
- Perng, G.C.; Jones, C.; Ciacci-Zanella, J.; Stone, M.; Henderson, G.; Yukht, A.; Slanina, S.M.; Hofman, F.M.; Ghiasi, H.; Nesburn, A.B.; Wechsler, S.L. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 2000, 287, 1500–1503. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Galvan, V.; Campadelli-Fiume, G.; Roizman, B. Glycoprotein D or J Delivered in trans Blocks Apoptosis in SK-N-SH Cells Induced by a Herpes Simplex Virus 1 Mutant Lacking Intact Genes Expressing Both Glycoproteins. J Virol 2000, 74, 11782–11791. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Roizman, B. The domains of glycoprotein D required to block apoptosis depend on whether glycoprotein D is present in the virions carrying herpes simplex virus 1 genome lacking the gene encoding the glycoprotein. J Virol 2001, 75, 6166–6172. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.; Athmanathan, S.; Thakur, A.; Willcox, M. A novel apoptotic interaction between HSV-1 and human corneal epithelial cells. Curr Eye Res 2003, 26, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Atherton, S. Apoptosis and increased expression of Fas ligand after uniocular anterior chamber (AC) inoculation of HSV-1. Curr Eye Res 2003, 26, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.E.; Pedroza, L.; Beuerman, R.; Hill, J.M. Herpes simplex virus type-1 infection of corneal epithelial cells induces apoptosis of the underlying keratocytes. Exp Eye Res 1997, 64, 775–779. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.H.; Willcox, M.D.; Athmanathan, S. Ocular and neuronal cell apoptosis during HSV-1 infection: a review. Curr Eye Res 2004, 29, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Silverman, R.H.; Zhou, A.; Goto, T.; Kwon, B.S.; Kaufman, H.E.; Hill, J.M. Increased severity of HSV-1 keratitis and mortality in mice lacking the 2-5A-dependent RNase L gene. Invest Ophthalmol Vis Sci 2001, 42, 120–126. [Google Scholar] [PubMed]
- DeBiasi, R.L.; Kleinschmidt-DeMasters, B.K.; Weinberg, A.; Tyler, K.L. Use of PCR for the diagnosis of herpesvirus infections of the central nervous system. J Clin Virol 2002, 25, S5–11. [Google Scholar] [CrossRef] [PubMed]
- Sabri, F.; Granath, F.; Hjalmarsson, A.; Aurelius, E.; Skoldenberg, B. Modulation of sFas indicates apoptosis in human herpes simplex encephalitis. J Neuroimmunol 2006, 171, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Geiger, K.D.; Nash, T.C.; Sawyer, S.; Krahl, T.; Patstone, G.; Reed, J.C.; Krajewski, S.; Dalton, D.; Buchmeier, M.J.; Sarvetnick, N. Interferon-gamma protects against herpes simplex virus type 1-mediated neuronal death. Virology 1997, 238, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Aubert, M.; Blaho, J.A. Viral oncoapoptosis of human tumor cells. Gene Ther 2003, 10, 1437–1445. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.L.; Kraft, R.M.; Blaho, J.A. Susceptibility of cancer cells to herpes simplex virus-dependent apoptosis. J Gen Virol 2007, 88, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; Lock, M.; Miller, C.G.; Fraser, N.W. Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. J Virol 2002, 76, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Asano, S.; Honda, T.; Goshima, F.; Nishiyama, Y.; Sugiura, Y. US3 protein kinase of herpes simplex virus protects primary afferent neurons from virus-induced apoptosis in ICR mice. Neurosci Lett 2000, 294, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Branco, F.J.; Fraser, N.W. Herpes simplex virus type 1 latency-associated transcript expression protects trigeminal ganglion neurons from apoptosis. J Virol 2005, 79, 9019–9025. [Google Scholar] [CrossRef] [PubMed]
- Aubert, M.; Pomeranz, L.E.; Blaho, J.A. Herpes simplex virus blocks apoptosis by precluding mitochondrial cytochrome c release independent of caspase activation in infected human epithelial cells. Apoptosis 2007, 12, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Galvan, V.; Brandimarti, R.; Munger, J.; Roizman, B. Bcl-2 blocks a caspase-dependent pathway of apoptosis activated by herpes simplex virus 1 infection in HEp-2 cells. J Virol 2000, 74, 1931–1938. [Google Scholar] [CrossRef] [PubMed]
- Kraft, R.M.; Nguyen, M.L.; Yang, X.H.; Thor, A.D.; Blaho, J.A. Caspase 3 activation during herpes simplex virus 1 infection. Virus Res 2006, 120, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Zachos, G.; Koffa, M.; Preston, C.M.; Clements, J.B.; Conner, J. Herpes simplex virus type 1 blocks the apoptotic host cell defense mechanisms that target Bcl-2 and manipulates activation of p38 mitogen-activated protein kinase to improve viral replication. J Virol 2001, 75, 2710–2728. [Google Scholar] [CrossRef] [PubMed]
- Janicke, R.U.; Sprengart, M.L.; Wati, M.R.; Porter, A.G. Caspase-3 is required for DNA fragmentation and morphological changes associated with apoptosis. J Biol Chem 1998, 273, 9357–9360. [Google Scholar] [CrossRef] [PubMed]
- Wurzer, W.J.; Planz, O.; Ehrhardt, C.; Giner, M.; Silberzahn, T.; Pleschka, S.; Ludwig, S. Caspase 3 activation is essential for efficient influenza virus propagation. Embo J 2003, 22, 2717–2728. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.; Hagglund, R.; Roizman, B. Infected cell protein No. 22 is subject to proteolytic cleavage by caspases activated by a mutant that induces apoptosis. Virology 2003, 305, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Mechanisms of Bcl-2 family protein function and dysfunction in health and disease. Behring Institute Mitteilungen 1996, 72–100. [Google Scholar] [PubMed]
- Munger, J.; Roizman, B. The US3 protein kinase of herpes simplex virus 1 mediates the posttranslational modification of BAD and prevents BAD-induced programmed cell death in the absence of other viral proteins. Proc Natl Acad Sci U S A 2001, 98, 10410–10415. [Google Scholar] [CrossRef] [PubMed]
- Ogg, P.D.; McDonell, P.J.; Ryckman, B.J.; Knudson, C.M.; Roller, R.J. The HSV-1 Us3 protein kinase is sufficient to block apoptosis induced by overexpression of a variety of Bcl-2 family members. Virology 2004, 319, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Cartier, A.; Komai, T.; Masucci, M.G. The Us3 protein kinase of herpes simplex virus 1 blocks apoptosis and induces phosporylation of the Bcl-2 family member Bad. Exp Cell Res 2003, 291, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Sciortino, M.T.; Perri, D.; Medici, M.A.; Grelli, S.; Serafino, A.; Borner, C.; Mastino, A. Role of Bcl-2 expression for productive herpes simplex virus 2 replication. Virology 2006, 356, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Liou, H.C. Regulation of the immune system by NF-kappaB and IkappaB. J Biochem Mol Bio 2002, 35, 537–546. [Google Scholar] [CrossRef]
- Chen, Z.J.; Parent, L.; Maniatis, T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell 1996, 84, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Baltimore, D. Activation in vitro of NF-kappa B by phosphorylation of its inhibitor I kappa B. Nature 1990, 344, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Pahl, H.L. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 1999, 18, 6853–6866. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Mayo, M.W.; Korneluk, R.G.; Goeddel, D.V.; Baldwin Jr., A.S. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science 1998, 281, 1680–1683. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Hanson, J.; McLean, T.I.; Olgiate, J.; Hilton, M.; Miller, W.E.; Bachenheimer, S.L. Herpes simplex type 1 induction of persistent NF-kappa B nuclear translocation increases the efficiency of virus replication. Virology 1998, 247, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Goodkin, M.L.; Ting, A.T.; Blaho, J.A. NF-kappaB is required for apoptosis prevention during herpes simplex virus type 1 infection. J Virol 2003, 77, 7261–7280. [Google Scholar] [CrossRef] [PubMed]
- Gregory, D.; Hargett, D.; Holmes, D.; Money, E.; Bachenheimer, S.L. Efficient replication by herpes simplex virus type 1 involves activation of the IkappaB kinase-IkappaB-p65 pathway. J Virol 2004, 78, 13582–13590. [Google Scholar] [CrossRef] [PubMed]
- Medici, M.A.; Sciortino, M.T.; Perri, D.; Amici, C.; Avitabile, E.; Ciotti, M.; Balestrieri, E.; De Smaele, E.; Franzoso, G.; Mastino, A. Protection by herpes simplex virus glycoprotein D against Fas-mediated apoptosis: role of nuclear factor kappaB. J Biol Chem 2003, 278, 36059–36067. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, B.; Luo, T.R.; Zhang, W.; Roizman, B. Activation of NF-kappaB in cells productively infected with HSV-1 depends on activated protein kinase R and plays no apparent role in blocking apoptosis. Proc Natl Acad Sci U S A 2003, 100, 12408–12413. [Google Scholar] [CrossRef] [PubMed]
- Taddeo, B.; Zhang, W.; Lakeman, F.; Roizman, B. Cells lacking NF-kappaB or in which NF-kappaB is not activated vary with respect to ability to sustain herpes simplex virus 1 replication and are not susceptible to apoptosis induced by a replication-incompetent mutant virus. J Virol 2004, 78, 11615–11621. [Google Scholar] [CrossRef] [PubMed]
- Yedowitz, J.C.; Blaho, J.A. Herpes simplex virus 2 modulates apoptosis and stimulates NF-kappaB nuclear translocation during infection in human epithelial HEp-2 cells. Virology 2005, 342, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Goodkin, M.L.; Epstein, S.; Asbell, P.A.; Blaho, J.A. Nuclear translocation of NF-kappaB precedes apoptotic poly(ADP-ribose) polymerase cleavage during productive HSV-1 replication in corneal epithelial cells. Invest Ophthalmol vis Sci 2007, 48, 4980–4988. [Google Scholar] [CrossRef] [PubMed]
- Morton, E.R.; Blaho, J.A. Herpes simplex virus blocks Fas-mediated apoptosis independent of viral activation of NF-kappaB in human epithelial HEp-2 cells. J Interferon Cytokine Res 2007, 27, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.L.; Kraft, R.M.; Blaho, J.A. African green monkey kidney Vero cells require de novo protein synthesis for efficient herpes simplex virus 1-dependent apoptosis. Virology 2005, 336, 274–290. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.C.; DiMaio, D. Repression of human papillomavirus oncogenes in HeLa cervical carcinoma cells causes the orderly reactivation of dormant tumor suppressor pathways. Proc Natl Acad Sci U S A 2000, 97, 12513–12518. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.; Ono, T.; Ishimoto, A.; Dowhanick, J.J.; Frizzell, M.A.; Howley, P.M.; Sakai, H. Mechanisms of human papillomavirus E2-mediated repression of viral oncogene expression and cervical cancer cell growth inhibition. J Virol 2000, 74, 3752–3760. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.J.; Suh, E.J.; Kang, H.T.; Im, J.S.; Um, S.J.; Park, J.S.; Hwang, E.S. Induction of senescence-like state and suppression of telomerase activity through inhibition of HPV E6/E7 gene expression in cells immortalized by HPV16 DNA. Exp Cell Res 2002, 277, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Psyrri, A.; DeFilippis, R.A.; Edwards, A.P.; Yates, K.E.; Manuelidis, L.; DiMaio, D. Role of the retinoblastoma pathway in senescence triggered by repression of the human papillomavirus E7 protein in cervical carcinoma cells. Cancer Res 2004, 64, 3079–3086. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; DeFilippis, R.A.; Manuelidis, L.; DiMaio, D. Repression of the human papillomavirus E6 gene initiates p53-dependent, telomerase-independent senescence and apoptosis in HeLa cervical carcinoma cells. J Virol 2004, 78, 4063–4073. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, E.C.; DiMaio, D. Induced senescence in HeLa cervical carcinoma cells containing elevated telomerase activity and extended telomeres. Cell Growth Differ 2001, 12, 525–534. [Google Scholar] [PubMed]
- Nguyen, M.L.; Kraft, R.M.; Aubert, M.; Goodwin, E.; DiMaio, D.; Blaho, J.A. p53 and hTERT determine sensitivity to viral apoptosis. J Virol 2007, 81, 12985–12995. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of human papillomavirus-induced oncogenesis. J Virol 2004, 78, 11451–11460. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. Embo J 1991, 10, 4129–4135. [Google Scholar] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins. Mol Cell Biol 1993, 13, 4918–4927. [Google Scholar] [PubMed]
- Hachiya, M.; Chumakov, A.; Miller, C.W.; Akashi, M.; Said, J.; Koeffler, H.P. Mutant p53 proteins behave in a dominant, negative fashion in vivo. Anticancer Res 1994, 14, 1853–1859. [Google Scholar] [PubMed]
- Shaulian, E.; Zauberman, A.; Ginsberg, D.; Oren, M. Identification of a minimal transforming domain of p53: negative dominance through abrogation of sequence-specific DNA binding. Mol Cell Biol 1992, 12, 5581–5592. [Google Scholar] [PubMed]
- Boutell, C.; Everett, R.D. The herpes simplex virus type 1 (HSV-1) regulatory protein ICP0 interacts with and Ubiquitinates p53. J Biol Chem 2003, 278, 36596–36602. [Google Scholar] [CrossRef] [PubMed]
- Boutell, C.; Everett, R.D. Herpes simplex virus type 1 infection induces the stabilization of p53 in a USP7- and ATM-independent manner. J Virol 2004, 78, 8068–8077. [Google Scholar] [CrossRef] [PubMed]
- Hobbs 2nd, W.E.; DeLuca, N.A. Perturbation of cell cycle progression and cellular gene expression as a function of herpes simplex virus ICP0. J Virol 1999, 73, 8245–8255. [Google Scholar] [PubMed]
- Harrington, L.; Zhou, W.; McPhail, T.; Oulton, R.; Yeung, D.S.; Mar, V.; Bass, M.B.; Robinson, M.O. Human telomerase contains evolutionarily conserved catalytic and structural subunits. Genes & development 1997, 11, 3109–3115. [Google Scholar] [CrossRef]
- Kilian, A.; Bowtell, D.D.; Abud, H.E.; Hime, G.R.; Venter, D.J.; Keese, P.K.; Duncan, E.L.; Reddel, R.R.; Jefferson, R.A. Isolation of a candidate human telomerase catalytic subunit gene, which reveals complex splicing patterns in different cell types. Hum Mol Genet 1997, 6, 2011–2019. [Google Scholar] [CrossRef] [PubMed]
- Meyerson, M.; Counter, C.M.; Eaton, E.N.; Ellisen, L.W.; Steiner, P.; Caddle, S.D.; Ziaugra, L.; Beijersbergen, R.L.; Davidoff, M.J.; Liu, Q.; Bacchetti, S.; Haber, D.A.; Weinberg, R.A. hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 1997, 90, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.M.; Morin, G.B.; Chapman, K.B.; Weinrich, S.L.; Andrews, W.H.; Lingner, J.; Harley, C.B.; Cech, T.R. Telomerase catalytic subunit homologs from fission yeast and human. Science 1997, 277, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef] [PubMed]
- de Lange, T.; Shiue, L.; Myers, R.M.; Cox, D.R.; Naylor, S.L.; Killery, A.M.; Varmus, H.E. Structure and variability of human chromosome ends. Mol Cell Biol 1990, 10, 518–527. [Google Scholar] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Hastie, N.D.; Dempster, M.; Dunlop, M.G.; Thompson, A.M.; Green, D.K.; Allshire, R.C. Telomere reduction in human colorectal carcinoma and with ageing. Nature 1990, 346, 866–868. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Kim, N.W.; Prowse, K.R.; Weinrich, S.L.; Hirsch, K.S.; West, M.D.; Bacchetti, S.; Hirte, H.W.; Counter, C.M.; Greider, C.W.; et al. Telomerase, cell immortality, and cancer. Cold Spring Harbor Symp Quant Biol 1994, 59, 307–31. [Google Scholar] [PubMed]
- Klingelhutz, A.J.; Foster, S.A.; McDougall, J.K. Telomerase activation by the E6 gene product of human papillomavirus type 16. Nature 1996, 380, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, M.; Yamada, O.; Kanda, N.; Akita, S.; Kawano, T.; Ohno, T.; Mizoguchi, H.; Eto, Y.; Anderson, K.C.; Yamada, H. Telomerase overexpression in K562 leukemia cells protects against apoptosis by serum deprivation and double-stranded DNA break inducing agents, but not against DNA synthesis inhibitors. Cancer letters 2002, 178, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Luiten, R.M.; Pene, J.; Yssel, H.; Spits, H. Ectopic hTERT expression extends the life span of human CD4+ helper and regulatory T-cell clones and confers resistance to oxidative stress-induced apoptosis. Blood 2003, 101, 4512–4519. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.G.; Xia, H.L.; Tian, Y.M.; Just, T.; Cai, G.P.; Dai, Y.R. Expression of telomerase inhibits hydroxyl radical-induced apoptosis in normal telomerase negative human lung fibroblasts. FEBS letters 2001, 488, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Mei, H.D. Inhibition of telomerase activity with hTERT antisense increases the effect of CDDP-induced apoptosis in myeloid leukemia. Hematol J 2002, 3, 201–205. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Nguyen, M.L.; Blaho, J.A. Cellular Players in the Herpes Simplex Virus Dependent Apoptosis Balancing Act. Viruses 2009, 1, 965-978. https://doi.org/10.3390/v1030965
Nguyen ML, Blaho JA. Cellular Players in the Herpes Simplex Virus Dependent Apoptosis Balancing Act. Viruses. 2009; 1(3):965-978. https://doi.org/10.3390/v1030965
Chicago/Turabian StyleNguyen, Marie L., and John A. Blaho. 2009. "Cellular Players in the Herpes Simplex Virus Dependent Apoptosis Balancing Act" Viruses 1, no. 3: 965-978. https://doi.org/10.3390/v1030965
APA StyleNguyen, M. L., & Blaho, J. A. (2009). Cellular Players in the Herpes Simplex Virus Dependent Apoptosis Balancing Act. Viruses, 1(3), 965-978. https://doi.org/10.3390/v1030965