HCV Animal Models: A Journey of More than 30 Years

{kind=link}
Abstract
:1. Introduction
2. Primate models
2.1. The chimpanzee
2.2. Callithricidae
2.3. Tree shrews
3. Rodent Models
3.1. Rats
3.2. Transgenic mice
3.3. Trimera mice
3.4. Chimeric uPA-SCID mice
3.4.1. Characterization
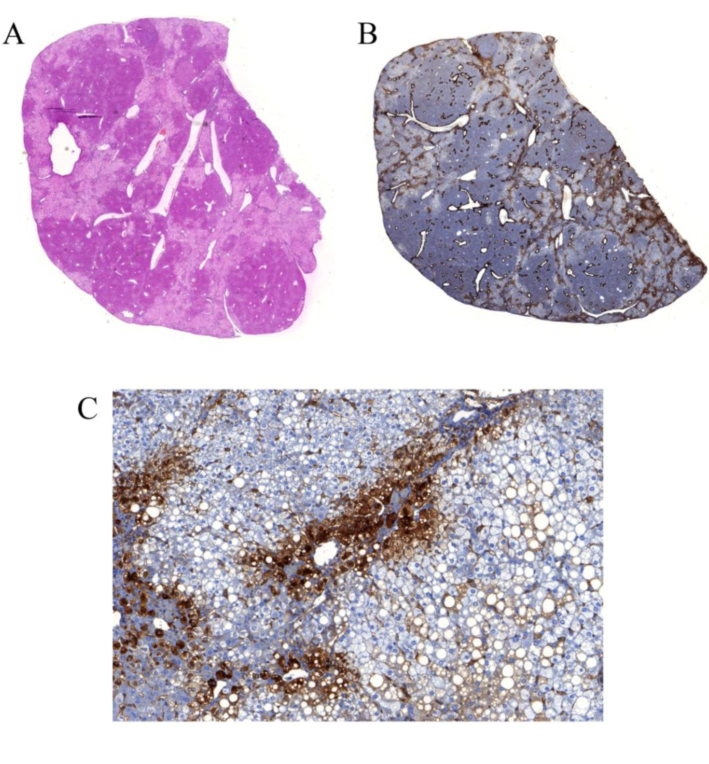
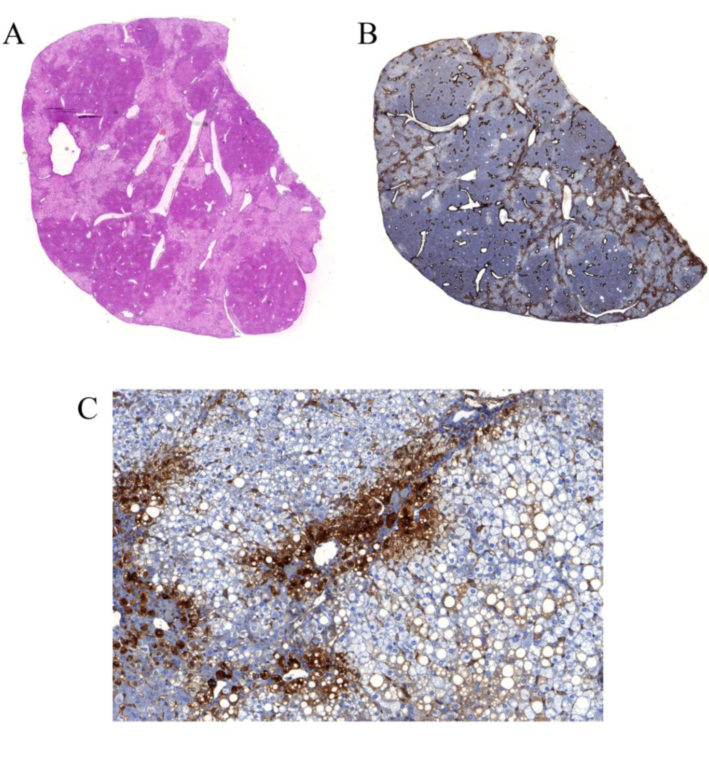
3.4.2. HCV infection
3.4.3. Study of entry inhibitors
4. Conclusions and future perspectives
Acknowledgments
References and Notes
- Prince, A.M.; Brotman, B.; Grady, G.F.; Kuhns, W.J.; Hazzi, C.; Levine, R.W.; Millian, S.J. Long-incubation post-transfusion hepatitis without serological evidence of exposure to hepatitis-B virus. Lancet 1974, 2, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Feinstone, S.M.; Kapikian, A.Z.; Purcell, R.H.; Alter, H.J.; Holland, P.V. Transfusion-associated hepatitis not due to viral hepatitis type A or B. N. Engl. J. Med. 1975, 292, 767–770. [Google Scholar] [PubMed]
- Choo, Q.L.; Kuo, G.; Weiner, A.J.; Overby, L.R.; Bradley, D.W.; Houghton, M. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science 1989, 244, 359–362. [Google Scholar] [PubMed]
- Lohmann, V.; Korner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; Rice, C.M. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. U S A 2005, 102, 9294–9299. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; Bartenschlager, R.; Liang, T.J. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, J.M.; Scheel, T.K.; Jensen, T.B.; Lademann, J.B.; Prentoe, J.C.; Knudsen, M.L.; Hoegh, A.M.; Bukh, J. Development and characterization of hepatitis C virus genotype 1-7 cell culture systems: role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 2009, 49, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Olsavsky, K.M.; Page, J.L.; Johnson, M.C.; Zarbl, H.; Strom, S.C.; Omiecinski, C.J. Gene expression profiling and differentiation assessment in primary human hepatocyte cultures, established hepatoma cell lines, and human liver tissues. Toxicol. Appl. Pharmacol. 2007, 222, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Bassett, S.E.; Brasky, K.M.; Lanford, R.E. Analysis of hepatitis C virus-inoculated chimpanzees reveals unexpected clinical profiles. J. Virol. 1998, 72, 2589–2599. [Google Scholar] [PubMed]
- Abe, K.; Inchauspe, G.; Shikata, T.; Prince, A.M. Three different patterns of hepatitis C virus infection in chimpanzees. Hepatology 1992, 15, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Forns, X.; Bukh, J.; Purcell, R.H. The challenge of developing a vaccine against hepatitis C virus. J. Hepatol. 2002, 37, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Major, M.E.; Dahari, H.; Mihalik, K.; Puig, M.; Rice, C.M.; Neumann, A.U.; Feinstone, S.M. Hepatitis C virus kinetics and host responses associated with disease and outcome of infection in chimpanzees. Hepatology 2004, 39, 1709–1720. [Google Scholar] [CrossRef] [PubMed]
- Hoofnagle, J.H.; di Bisceglie, A.M. The treatment of chronic viral hepatitis. N. Engl. J. Med. 1997, 336, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Muchmore, E.; Popper, H.; Peterson, D.A.; Miller, M.F.; Lieberman, H.M. Non-A, non-B hepatitis-related hepatocellular carcinoma in a chimpanzee. J. Med. Primatol. 1988, 17, 235–246. [Google Scholar] [PubMed]
- Shimizu, Y.K.; Weiner, A.J.; Rosenblatt, J.; Wong, D.C.; Shapiro, M.; Popkin, T.; Houghton, M.; Alter, H.J.; Purcell, R.H. Early events in hepatitis C virus infection of chimpanzees. Proc. Natl. Acad. Sci. U S A 1990, 87, 6441–6444. [Google Scholar] [CrossRef] [PubMed]
- Farci, P.; London, W.T.; Wong, D.C.; Dawson, G.J.; Vallari, D.S.; Engle, R.; Purcell, R.H. The natural history of infection with hepatitis C virus (HCV) in chimpanzees: comparison of serologic responses measured with first- and second-generation assays and relationship to HCV viremia. J. Infect. Dis. 1992, 165, 1006–1011. [Google Scholar] [PubMed]
- Cooper, S.; Erickson, A.L.; Adams, E.J.; Kansopon, J.; Weiner, A.J.; Chien, D.Y.; Houghton, M.; Parham, P.; Walker, C.M. Analysis of a successful immune response against hepatitis C virus. Immunity 1999, 10, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Bigger, C.B.; Brasky, K.M.; Lanford, R.E. DNA microarray analysis of chimpanzee liver during acute resolving hepatitis C virus infection. J. Virol. 2001, 75, 7059–7066. [Google Scholar] [CrossRef] [PubMed]
- Thimme, R.; Bukh, J.; Spangenberg, H.C.; Wieland, S.; Pemberton, J.; Steiger, C.; Govindarajan, S.; Purcell, R.H.; Chisari, F.V. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc. Natl. Acad. Sci. U S A 2002, 99, 15661–15668. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Pezacki, J.P.; Wodicka, L.; Brideau, A.D.; Supekova, L.; Thimme, R.; Wieland, S.; Bukh, J.; Purcell, R.H.; Schultz, P.G.; Chisari, F.V. Genomic analysis of the host response to hepatitis C virus infection. Proc. Natl. Acad. Sci. U S A 2002, 99, 15669–15674. [Google Scholar] [CrossRef] [PubMed]
- Shoukry, N.H.; Sidney, J.; Sette, A.; Walker, C.M. Conserved hierarchy of helper T cell responses in a chimpanzee during primary and secondary hepatitis C virus infections. J. Immunol. 2004, 172, 483–492. [Google Scholar] [PubMed]
- Bukh, J.; Forns, X.; Emerson, S.U.; Purcell, R.H. Studies of hepatitis C virus in chimpanzees and their importance for vaccine development. Intervirology 2001, 44, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Leroux-Roels, G. Development of prophylactic and therapeutic vaccines against hepatitis C virus. Expert Rev. Vaccines 2005, 4, 351–371. [Google Scholar] [CrossRef]
- Folgori, A.; Capone, S.; Ruggeri, L.; Meola, A.; Sporeno, E.; Ercole, B.B.; Pezzanera, M.; Tafi, R.; Arcuri, M.; Fattori, E.; Lahm, A.; Luzzago, A.; Vitelli, A.; Colloca, S.; Cortese, R.; Nicosia, A. A T-cell HCV vaccine eliciting effective immunity against heterologous virus challenge in chimpanzees. Nat. Med. 2006, 12, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Kurata, T.; Teramoto, Y.; Shiga, J.; Shikata, T. Lack of susceptibility of various primates and woodchucks to hepatitis C virus. J. Med. Primatol. 1993, 22, 433–434. [Google Scholar] [PubMed]
- Feinstone, S.M.; Alter, H.J.; Dienes, H.P.; Shimizu, Y.; Popper, H.; Blackmore, D.; Sly, D.; London, W.T.; Purcell, R.H. Non-A, non-B hepatitis in chimpanzees and marmosets. J. Infect. Dis. 1981, 144, 588–598. [Google Scholar] [PubMed]
- Karayiannis, P.; Scheuer, P.J.; Bamber, M.; Cohn, D.; Hurn, B.A.; Thomas, H.C. Experimental infection of Tamarins with human non-A, non-B hepatitis virus. J. Med. Virol. 1983, 11, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Katagiri, J.; Kojima, H.; Kamimura, T.; Ichida, F.; Ashida, M.; Hamada, C.; Shibayama, T. Studies on transmission of human non-A, non-B hepatitis to marmosets. J. Med. Virol. 1987, 22, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Tabor, E. Nonhuman primate models for non-A, non-B hepatitis. Cancer Detect. Prev. 1989, 14, 221–225. [Google Scholar] [PubMed]
- Garson, J.A.; Whitby, K.; Watkins, P.; Morgan, A.J. Lack of susceptibility of the cottontop tamarin to hepatitis C infection. J. Med. Virol. 1997, 52, 286–288. [Google Scholar] [CrossRef] [PubMed]
- Bukh, J.; Apgar, C.L.; Yanagi, M. Toward a surrogate model for hepatitis C virus: An infectious molecular clone of the GB virus-B hepatitis agent. Virology 1999, 262, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.; Bodola, F.; Sangar, D.V.; Goettge, K.; Popov, V.; Rijnbrand, R.; Lanford, R.E.; Lemon, S.M. Chronic hepatitis associated with GB virus B persistence in a tamarin after intrahepatic inoculation of synthetic viral RNA. Proc. Natl. Acad. Sci. U S A 2003, 100, 9962–9967. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.H.; Faulk, K.; Engle, R.E.; Govindarajan, S.; St Claire, M.; Bukh, J. In vivo analysis of the 3' untranslated region of GB virus B after in vitro mutagenesis of an infectious cDNA clone: persistent infection in a transfected tamarin. J. Virol. 2004, 78, 9389–9399. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Chavez, D.; Notvall, L.; Brasky, K.M. Comparison of tamarins and marmosets as hosts for GBV-B infections and the effect of immunosuppression on duration of viremia. Virology 2003, 311, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Bright, H.; Carroll, A.R.; Watts, P.A.; Fenton, R.J. Development of a GB virus B marmoset model and its validation with a novel series of hepatitis C virus NS3 protease inhibitors. J. Virol. 2004, 78, 2062–2071. [Google Scholar] [CrossRef] [PubMed]
- Weatherford, T.; Chavez, D.; Brasky, K.M.; Lanford, R.E. The marmoset model of GB virus B infections: adaptation to host phenotypic variation. J. Virol. 2009, 83, 5806–5814. [Google Scholar] [CrossRef] [PubMed]
- Bukh, J.; Engle, R.E.; Govindarajan, S.; Purcell, R.H. Immunity against the GBV-B hepatitis virus in tamarins can prevent productive infection following rechallenge and is long-lived. J. Med. Virol. 2008, 80, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Rijnbrand, R.; Yang, Y.; Beales, L.; Bodola, F.; Goettge, K.; Cohen, L.; Lanford, R.E.; Lemon, S.M.; Martin, A. A chimeric GB virus B with 5' nontranslated RNA sequence from hepatitis C virus causes hepatitis in tamarins. Hepatology 2005, 41, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Haqshenas, G.; Dong, X.; Netter, H.; Torresi, J.; Gowans, E.J. A chimeric GB virus B encoding the hepatitis C virus hypervariable region 1 is infectious in vivo. J. Gen. Virol. 2007, 88, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.; Trowbridge, R.; Thommes, P.; Parry, N.; Rowlands, D.; Harris, M.; Bright, H. Chimeric GB virus B genomes containing hepatitis C virus p7 are infectious in vivo. J. Hepatol. 2008, 49, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Weatherford, T.; Chavez, D.; Brasky, K.M.; Lemon, S.M.; Martin, A.; Lanford, R.E. Lack of adaptation of chimeric GB virus B/hepatitis C virus in the marmoset model: possible effects of bottleneck. J. Virol. 2009, 83, 8062–8075. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.D. Are tree shrews primates? In Primate Origins and Evolution: A Phylogenetic Reconstruction, Martin, R.D., Ed. 1990; Chapman & Hall: London, UK. [Google Scholar]
- Xie, Z.C.; Riezu-Boj, J.I.; Lasarte, J.J.; Guillen, J.; Su, J.H.; Civeira, M.P.; Prieto, J. Transmission of hepatitis C virus infection to tree shrews. Virology 1998, 244, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, H.; Cao, X.; Ben, K. Efficient infection of tree shrew (Tupaia belangeri) with hepatitis C virus grown in cell culture or from patient plasma. J. Gen. Virol. 2007, 88, 2504–2512. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.X.; Tang, E.H.; Xie, G.Z.; Zhang, X.S.; Xui, W.M.; Lao, Q.S.; Liu, M.Y.; Wen, Y.L.; Zhu, B.C.; Lu, Y.J. Investigation on herpes virus of tree shrews. Chin. J. Micro. Immunol 1983, 3, 33–36. [Google Scholar]
- Wu, G.Y.; Konishi, M.; Walton, C.M.; Olive, D.; Hayashi, K.; Wu, C.H. A novel immunocompetent rat model of HCV infection and hepatitis. Gastroenterology 2005, 128, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Koike, K.; Moriya, K.; Ishibashi, K.; Matsuura, Y.; Suzuki, T.; Saito, I.; Iino, S.; Kurokawa, K.; Miyamura, T. Expression of hepatitis C virus envelope proteins in transgenic mice. J. Gen. Virol. 1995, 76, 3031–3038. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Furusaka, A.; Koziel, M.J.; Chung, R.T.; Wang, T.C.; Schmidt, E.V.; Liang, T.J. Transgenic expression of hepatitis C virus structural proteins in the mouse. Hepatology 1997, 25, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Pasquinelli, C.; Shoenberger, J.M.; Chung, J.; Chang, K.M.; Guidotti, L.G.; Selby, M.; Berger, K.; Lesniewski, R.; Houghton, M.; Chisari, F.V. Hepatitis C virus core and E2 protein expression in transgenic mice. Hepatology 1997, 25, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Moriya, K.; Yotsuyanagi, H.; Shintani, Y.; Fujie, H.; Ishibashi, K.; Matsuura, Y.; Miyamura, T.; Koike, K. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol. 1997, 78, 1527–1531. [Google Scholar] [PubMed]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef]
- Lerat, H.; Honda, M.; Beard, M.R.; Loesch, K.; Sun, J.; Yang, Y.; Okuda, M.; Gosert, R.; Xiao, S.Y.; Weinman, S.A.; Lemon, S.M. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology 2002, 122, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Alonzi, T.; Agrati, C.; Costabile, B.; Cicchini, C.; Amicone, L.; Cavallari, C.; Rocca, C.D.; Folgori, A.; Fipaldini, C.; Poccia, F.; Monica, N.L.; Tripodi, M. Steatosis and intrahepatic lymphocyte recruitment in hepatitis C virus transgenic mice. J. Gen. Virol. 2004, 85, 1509–1520. [Google Scholar] [CrossRef] [PubMed]
- Kamegaya, Y.; Hiasa, Y.; Zukerberg, L.; Fowler, N.; Blackard, J.T.; Lin, W.; Choe, W.H.; Schmidt, E.V.; Chung, R.T. Hepatitis C virus acts as a tumor accelerator by blocking apoptosis in a mouse model of hepatocarcinogenesis. Hepatology 2005, 41, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Moriishi, K.; Mochizuki, R.; Moriya, K.; Miyamoto, H.; Mori, Y.; Abe, T.; Murata, S.; Tanaka, K.; Miyamura, T.; Suzuki, T.; Koike, K.; Matsuura, Y. Critical role of PA28gamma in hepatitis C virus-associated steatogenesis and hepatocarcinogenesis. Proc. Natl. Acad. Sci. U S A 2007, 104, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Moriya, K.; Kiyosawa, K.; Koike, K.; Aoyama, T. Hepatitis C virus core protein induces spontaneous and persistent activation of peroxisome proliferator-activated receptor alpha in transgenic mice: implications for HCV-associated hepatocarcinogenesis. Int. J. Cancer 2008, 122, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Moriya, K.; Kiyosawa, K.; Koike, K.; Gonzalez, F.J.; Aoyama, T. PPARalpha activation is essential for HCV core protein-induced hepatic steatosis and hepatocellular carcinoma in mice. J. Clin. Invest. 2008, 118, 683–694. [Google Scholar] [PubMed]
- Klopstock, N.; Katzenellenbogen, M.; Pappo, O.; Sklair-Levy, M.; Olam, D.; Mizrahi, L.; Potikha, T.; Galun, E.; Goldenberg, D. HCV tumor promoting effect is dependent on host genetic background. PLoS ONE 2009, 4, e5025. [Google Scholar] [CrossRef] [PubMed]
- Majumder, M.; Ghosh, A.K.; Steele, R.; Zhou, X.Y.; Phillips, N.J.; Ray, R.; Ray, R.B. Hepatitis C virus NS5A protein impairs TNF-mediated hepatic apoptosis, but not by an anti-FAS antibody, in transgenic mice. Virology 2002, 294, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Frelin, L.; Brenndorfer, E.D.; Ahlen, G.; Weiland, M.; Hultgren, C.; Alheim, M.; Glaumann, H.; Rozell, B.; Milich, D.R.; Bode, J.G.; Sallberg, M. The hepatitis C virus and immune evasion: non-structural 3/4A transgenic mice are resistant to lethal tumour necrosis factor alpha mediated liver disease. Gut 2006, 55, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Ernst, E.; Schonig, K.; Bugert, J.J.; Blaker, H.; Pfaff, E.; Stremmel, W.; Encke, J. Generation of inducible hepatitis C virus transgenic mouse lines. J. Med. Virol. 2007, 79, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Lubin, I.; Faktorowich, Y.; Lapidot, T.; Gan, Y.; Eshhar, Z.; Gazit, E.; Levite, M.; Reisner, Y. Engraftment and development of human T and B cells in mice after bone marrow transplantation. Science 1991, 252, 427–431. [Google Scholar] [PubMed]
- Lubin, I.; Segall, H.; Marcus, H.; David, M.; Kulova, L.; Steinitz, M.; Erlich, P.; Gan, J.; Reisner, Y. Engraftment of human peripheral blood lymphocytes in normal strains of mice. Blood 1994, 83, 2368–2381. [Google Scholar] [PubMed]
- Ilan, E.; Arazi, J.; Nussbaum, O.; Zauberman, A.; Eren, R.; Lubin, I.; Neville, L.; Ben-Moshe, O.; Kischitzky, A.; Litchi, A.; Margalit, I.; Gopher, J.; Mounir, S.; Cai, W.; Daudi, N.; Eid, A.; Jurim, O.; Czerniak, A.; Galun, E.; Dagan, S. The hepatitis C virus (HCV)-Trimera mouse: a model for evaluation of agents against HCV. J. Infect. Dis. 2002, 185, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Eren, R.; Landstein, D.; Terkieltaub, D.; Nussbaum, O.; Zauberman, A.; Ben-Porath, J.; Gopher, J.; Buchnick, R.; Kovjazin, R.; Rosenthal-Galili, Z.; Aviel, S.; Ilan, E.; Shoshany, Y.; Neville, L.; Waisman, T.; Ben-Moshe, O.; Kischitsky, A.; Foung, S.K.; Keck, Z.Y.; Pappo, O.; Eid, A.; Jurim, O.; Zamir, G.; Galun, E.; Dagan, S. Preclinical evaluation of two neutralizing human monoclonal antibodies against hepatitis C virus (HCV): a potential treatment to prevent HCV reinfection in liver transplant patients. J. Virol. 2006, 80, 2654–2664. [Google Scholar] [CrossRef] [PubMed]
- Galun, E.; Burakova, T.; Ketzinel, M.; Lubin, I.; Shezen, E.; Kahana, Y.; Eid, A.; Ilan, Y.; Rivkind, A.; Pizov, G.; Shouval, D.; Reisner, Y. Hepatitis C virus viremia in SCID-->BNX mouse chimera. J. J. Infect. Dis. 1995, 172, 25–30. [Google Scholar] [PubMed]
- Ilan, E.; Burakova, T.; Dagan, S.; Nussbaum, O.; Lubin, I.; Eren, R.; Ben-Moshe, O.; Arazi, J.; Berr, S.; Neville, L.; Yuen, L.; Mansour, T.S.; Gillard, J.; Eid, A.; Jurim, O.; Shouval, D.; Reisner, Y.; Galun, E. The hepatitis B virus-trimera mouse: a model for human HBV infection and evaluation of anti-HBV therapeutic agents. Hepatology 1999, 29, 553–562. [Google Scholar] [CrossRef] [PubMed]
- Heckel, J.L.; Sandgren, E.P.; Degen, J.L.; Palmiter, R.D.; Brinster, R.L. Neonatal bleeding in transgenic mice expressing urokinase-type plasminogen activator. Cell 1990, 62, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Sandgren, E.P.; Palmiter, R.D.; Heckel, J.L.; Daugherty, C.C.; Brinster, R.L.; Degen, J.L. Complete hepatic regeneration after somatic deletion of an albumin-plasminogen activator transgene. Cell 1991, 66, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Rhim, J.A.; Sandgren, E.P.; Degen, J.L.; Palmiter, R.D.; Brinster, R.L. Replacement of diseased mouse liver by hepatic cell transplantation. Science 1994, 263, 1149–1152. [Google Scholar] [PubMed]
- Rhim, J.A.; Sandgren, E.P.; Palmiter, R.D.; Brinster, R.L. Complete reconstitution of mouse liver with xenogeneic hepatocytes. Proc. Natl. Acad. Sci. U S A 1995, 92, 4942–4946. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Dandri, M.; Gupta, S.; Rogler, C.E. Liver repopulation with xenogenic hepatocytes in B and T cell-deficient mice leads to chronic hepadnavirus infection and clonal growth of hepatocellular carcinoma. Proc. Natl. Acad. Sci. U S A 1998, 95, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Dandri, M.; Burda, M.R.; Gocht, A.; Torok, E.; Pollok, J.M.; Rogler, C.E.; Will, H.; Petersen, J. Woodchuck hepatocytes remain permissive for hepadnavirus infection and mouse liver repopulation after cryopreservation. Hepatology 2001, 34, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Dandri, M.; Burda, M.R.; Torok, E.; Pollok, J.M.; Iwanska, A.; Sommer, G.; Rogiers, X.; Rogler, C.E.; Gupta, S.; Will, H.; Greten, H.; Petersen, J. Repopulation of mouse liver with human hepatocytes and in vivo infection with hepatitis B virus. Hepatology 2001, 33, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Mercer, D.F.; Schiller, D.E.; Elliott, J.F.; Douglas, D.N.; Hao, C.; Rinfret, A.; Addison, W.R.; Fischer, K.P.; Churchill, T.A.; Lakey, J.R.; Tyrrell, D.L.; Kneteman, N.M. Hepatitis C virus replication in mice with chimeric human livers. Nat. Med. 2001, 7, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Vanlandschoot, P.; Leroux-Roels, G. A simple and rapid method to determine the zygosity of uPA-transgenic SCID mice. Biochem. Biophys. Res. Commun. 2003, 308, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Tateno, C.; Yoshizane, Y.; Saito, N.; Kataoka, M.; Utoh, R.; Yamasaki, C.; Tachibana, A.; Soeno, Y.; Asahina, K.; Hino, H.; Asahara, T.; Yokoi, T.; Furukawa, T.; Yoshizato, K. Near completely humanized liver in mice shows human-type metabolic responses to drugs. Am. J. Pathol. 2004, 165, 901–912. [Google Scholar] [PubMed]
- Meuleman, P.; Libbrecht, L.; De Vos, R.; de Hemptinne, B.; Gevaert, K.; Vandekerckhove, J.; Roskams, T.; Leroux-Roels, G. Morphological and biochemical characterization of a human liver in a uPA-SCID mouse chimera. Hepatology 2005, 41, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, R.; Baldysiak-Figiel, A.; Krugel, V.; Ueberham, E.; Gaunitz, F. Hepatocellular expression of glutamine synthetase: an indicator of morphogen actions as master regulators of zonation in adult liver. Prog. Histochem. Cytochem. 2007, 41, 201–266. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Steyaert, S.; Libbrecht, L.; Couvent, S.; Van Houtte, F.; Clinckspoor, F.; de Hemptinne, B.; Roskams, T.; Vanlandschoot, P.; Leroux-Roels, G. Human hepatocytes secrete soluble CD14, a process not directly influenced by HBV and HCV infection. Clin. Chim. Acta 2006, 366, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Pozo, O.J.; Van Eenoo, P.; Deventer, K.; Lootens, L.; Grimalt, S.; Sancho, J.V.; Hernandez, F.; Meuleman, P.; Leroux-Roels, G.; Delbeke, F.T. Detection and structural investigation of metabolites of stanozolol in human urine by liquid chromatography tandem mass spectrometry. Steroids 2009, 74, 837–852. [Google Scholar] [CrossRef] [PubMed]
- Lootens, L.; Meuleman, P.; Pozo, O.J.; Van Eenoo, P.; Leroux-Roels, G.; Delbeke, F.T. uPA+/+-SCID Mouse with Humanized Liver as a Model for in Vivo Metabolism of Exogenous Steroids: Methandienone as a Case Study. Clin. Chem. 2009, in press. [Google Scholar]
- Lindenbach, B.D.; Meuleman, P.; Ploss, A.; Vanwolleghem, T.; Syder, A.J.; McKeating, J.A.; Lanford, R.E.; Feinstone, S.M.; Major, M.E.; Leroux-Roels, G.; Rice, C.M. Cell culture-grown hepatitis C virus is infectious in vivo and can be recultured in vitro. Proc. Natl. Acad. Sci. U S A 2006, 103, 3805–3809. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Verney, G.; Dreux, M.; Donot, P.; Morice, Y.; Penin, F.; Pawlotsky, J.M.; Lavillette, D.; Cosset, F.L. An interplay between hypervariable region 1 of the hepatitis C virus E2 glycoprotein, the scavenger receptor BI, and high-density lipoprotein promotes both enhancement of infection and protection against neutralizing antibodies. J. Virol. 2005, 79, 8217–8229. [Google Scholar] [CrossRef] [PubMed]
- von Hahn, T.; Lindenbach, B.D.; Boullier, A.; Quehenberger, O.; Paulson, M.; Rice, C.M.; McKeating, J.A. Oxidized low-density lipoprotein inhibits hepatitis C virus cell entry in human hepatoma cells. Hepatology 2006, 43, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Wunschmann, S.; Muller, H.M.; Stipp, C.S.; Hemler, M.E.; Stapleton, J.T. In vitro interaction between hepatitis C virus (HCV) envelope glycoprotein E2 and serum lipoproteins (LPs) results in enhanced cellular binding of both HCV E2 and LPs. J. Infect. Dis. 2006, 194, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Walters, K.A.; Joyce, M.A.; Thompson, J.C.; Smith, M.W.; Yeh, M.M.; Proll, S.; Zhu, L.F.; Gao, T.J.; Kneteman, N.M.; Tyrrell, D.L.; Katze, M.G. Host-specific response to HCV infection in the chimeric SCID-beige/Alb-uPA mouse model: role of the innate antiviral immune response. PLoS Pathog. 2006, 2, e59. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.A.; Walters, K.A.; Lamb, S.E.; Yeh, M.M.; Zhu, L.F.; Kneteman, N.; Doyle, J.S.; Katze, M.G.; Tyrrell, D.L. HCV induces oxidative and ER stress, and sensitizes infected cells to apoptosis in SCID/Alb-uPA mice. PLoS Pathog. 2009, 5, e1000291. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Libbrecht, L.; Wieland, S.; De Vos, R.; Habib, N.; Kramvis, A.; Roskams, T.; Leroux-Roels, G. Immune suppression uncovers endogenous cytopathic effects of the hepatitis B virus. J. Virol. 2006, 80, 2797–2807. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.; Tanaka, Y.; Kurbanov, F.; Maruyama, I.; Shimada, T.; Takahashi, S.; Shirai, T.; Hino, K.; Sakaida, I.; Mizokami, M. Direct cytopathic effects of particular hepatitis B virus genotypes in severe combined immunodeficiency transgenic with urokinase-type plasminogen activator mouse with human hepatocytes. Gastroenterology 2009, 136, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Logvinoff, C.; Major, M.E.; Oldach, D.; Heyward, S.; Talal, A.; Balfe, P.; Feinstone, S.M.; Alter, H.; Rice, C.M.; McKeating, J.A. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc. Natl. Acad. Sci. U S A 2004, 101, 10149–10154. [Google Scholar] [CrossRef] [PubMed]
- Pestka, J.M.; Zeisel, M.B.; Blaser, E.; Schurmann, P.; Bartosch, B.; Cosset, F.L.; Patel, A.H.; Meisel, H.; Baumert, J.; Viazov, S.; Rispeter, K.; Blum, H.E.; Roggendorf, M.; Baumert, T.F. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc. Natl. Acad. Sci. U S A 2007, 104, 6025–6030. [Google Scholar] [CrossRef] [PubMed]
- Vanwolleghem, T.; Bukh, J.; Meuleman, P.; Desombere, I.; Meunier, J.C.; Alter, H.; Purcell, R.H.; Leroux-Roels, G. Polyclonal immunoglobulins from a chronic hepatitis C virus patient protect human liver-chimeric mice from infection with a homologous hepatitis C virus strain. Hepatology 2008, 47, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Law, M.; Maruyama, T.; Lewis, J.; Giang, E.; Tarr, A.W.; Stamataki, Z.; Gastaminza, P.; Chisari, F.V.; Jones, I.M.; Fox, R.I.; Ball, J.K.; McKeating, J.A.; Kneteman, N.M.; Burton, D.R. Broadly neutralizing antibodies protect against hepatitis C virus quasispecies challenge. Nat. Med. 2008, 14, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Meuleman, P.; Hesselgesser, J.; Paulson, M.; Vanwolleghem, T.; Desombere, I.; Reiser, H.; Leroux-Roels, G. Anti-CD81 antibodies can prevent a hepatitis C virus infection in vivo. Hepatology 2008, 48, 1761–1768. [Google Scholar] [CrossRef] [PubMed]
- Timpe, J.M.; Stamataki, Z.; Jennings, A.; Hu, K.; Farquhar, M.J.; Harris, H.J.; Schwarz, A.; Desombere, I.; Roels, G.L.; Balfe, P.; McKeating, J.A. Hepatitis C virus cell-cell transmission in hepatoma cells in the presence of neutralizing antibodies. Hepatology 2008, 47, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Witteveldt, J.; Evans, M.J.; Bitzegeio, J.; Koutsoudakis, G.; Owsianka, A.M.; Angus, A.G.; Keck, Z.Y.; Foung, S.K.; Pietschmann, T.; Rice, C.M.; Patel, A.H. CD81 is dispensable for hepatitis C virus cell-to-cell transmission in hepatoma cells. J. Gen. Virol. 2009, 90, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Kaul, A.; Woerz, I.; Meuleman, P.; Leroux-Roels, G.; Bartenschlager, R. Cell culture adaptation of hepatitis C virus and in vivo viability of an adapted variant. J. Virol. 2007, 81, 13168–13179. [Google Scholar] [CrossRef] [PubMed]
- Bukh, J.; Pietschmann, T.; Lohmann, V.; Krieger, N.; Faulk, K.; Engle, R.E.; Govindarajan, S.; Shapiro, M.; St Claire, M.; Bartenschlager, R. Mutations that permit efficient replication of hepatitis C virus RNA in Huh-7 cells prevent productive replication in chimpanzees. Proc. Natl. Acad. Sci. U S A 2002, 99, 14416–14421. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Zayas, M.; Meuleman, P.; Long, G.; Appel, N.; Koutsoudakis, G.; Kallis, S.; Leroux-Roels, G.; Lohmann, V.; Bartenschlager, R. Production of Infectious Genotype 1b Virus Particles in Cell Culture and Impairment by Replication Enhancing Mutations. PLoS Pathog. 2009, In Press. [Google Scholar]
- Meuleman, P.; Leroux-Roels, G. The human liver-uPA-SCID mouse: A model for the evaluation of antiviral compounds against HBV and HCV. Antiviral Res. 2008, 80, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Kneteman, N.M.; Weiner, A.J.; O'Connell, J.; Collett, M.; Gao, T.; Aukerman, L.; Kovelsky, R.; Ni, Z. J.; Zhu, Q.; Hashash, A.; Kline, J.; Hsi, B.; Schiller, D.; Douglas, D.; Tyrrell, D.L.; Mercer, D.F. Anti-HCV therapies in chimeric scid-Alb/uPA mice parallel outcomes in human clinical application. Hepatology 2006, 43, 1346–1353. [Google Scholar] [CrossRef] [PubMed]
- Vanwolleghem, T.; Meuleman, P.; Libbrecht, L.; Roskams, T.; De Vos, R.; Leroux-Roels, G. Ultra-rapid cardiotoxicity of the hepatitis C virus protease inhibitor BILN 2061 in the urokinase-type plasminogen activator mouse. Gastroenterology 2007, 133, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Kneteman, N.M.; Howe, A.Y.; Gao, T.; Lewis, J.; Pevear, D.; Lund, G.; Douglas, D.; Mercer, D.F.; Tyrrell, D.L.; Immermann, F.; Chaudhary, I.; Speth, J.; Villano, S.A.; O'Connell, J.; Collett, M. HCV796: A selective nonstructural protein 5B polymerase inhibitor with potent anti-hepatitis C virus activity in vitro, in mice with chimeric human livers, and in humans infected with hepatitis C virus. Hepatology 2009, 49, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Umehara, T.; Ruegg, U.T.; Yasui, F.; Watanabe, T.; Yasuda, H.; Dumont, J.M.; Scalfaro, P.; Yoshiba, M.; Kohara, M. Evaluation of a cyclophilin inhibitor in hepatitis C virus-infected chimeric mice in vivo. Hepatology 2007, 45, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Flisiak, R.; Horban, A.; Gallay, P.; Bobardt, M.; Selvarajah, S.; Wiercinska-Drapalo, A.; Siwak, E.; Cielniak, I.; Higersberger, J.; Kierkus, J.; Aeschlimann, C.; Grosgurin, P.; Nicolas-Metral, V.; Dumont, J.M.; Porchet, H.; Crabbe, R.; Scalfaro, P. The cyclophilin inhibitor Debio-025 shows potent anti-hepatitis C effect in patients coinfected with hepatitis C and human immunodeficiency virus. Hepatology 2008, 47, 817–826. [Google Scholar] [CrossRef] [PubMed]
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Meuleman, P.; Leroux-Roels, G. HCV Animal Models: A Journey of More than 30 Years. Viruses 2009, 1, 222-240. https://doi.org/10.3390/v1020222
Meuleman P, Leroux-Roels G. HCV Animal Models: A Journey of More than 30 Years. Viruses. 2009; 1(2):222-240. https://doi.org/10.3390/v1020222
Chicago/Turabian StyleMeuleman, Philip, and Geert Leroux-Roels. 2009. "HCV Animal Models: A Journey of More than 30 Years" Viruses 1, no. 2: 222-240. https://doi.org/10.3390/v1020222
APA StyleMeuleman, P., & Leroux-Roels, G. (2009). HCV Animal Models: A Journey of More than 30 Years. Viruses, 1(2), 222-240. https://doi.org/10.3390/v1020222