1. Introduction
After the “Bronze Age”, modern society has evolved into the “Plastic Age”. There are many types and properties of plastics, for multiple purposes. Almost every plastic is difficult to degrade, requiring a long time, and causing environmental harm. Thus, raw plastic material from natural resources, without any environment problems, is necessary. In modern times, new plastic research or inventions focus on raw materials which are biodegradable or originated from nature. Therefore, it is necessary to find another natural resource, which is not used as food, have enough amount for industrial applications, and rarely or never used before and lignin may be a good candidate.
Wood is composed of three components; cellulose, hemicellulose, and lignin mixtures, which is called the ‘lignin-carbohydrate-complex’ (LCC) at the micro-scale. The purpose of any kind of pulping process is to breakup this LCC and use only cellulose [
1]. Cellulose, hemicellulose, and every kind of materials which is extracted from wood are used, but not lignin. Kraft lignin is extracted from solution because it cannot be treated the way it is. Specifically, kraft lignin is treated with solutions containing high concentrations of alkali ions and neutralized by a strong acid to create a powder phase mixture which is composed of mostly lignin. Generally, this powder is burnt to supply the energy requirement of the production process [
2]. Different from the kraft lignin which most of the materials are incinerated, lignosulfonate, organosolv lignin, and other lignins are used as feed binders, adhesives, emulsifiers, etc. However, all of these lignins are hard to use as wide-use materials, like synthetic polymers, due to its unique molecular structure and properties [
3].
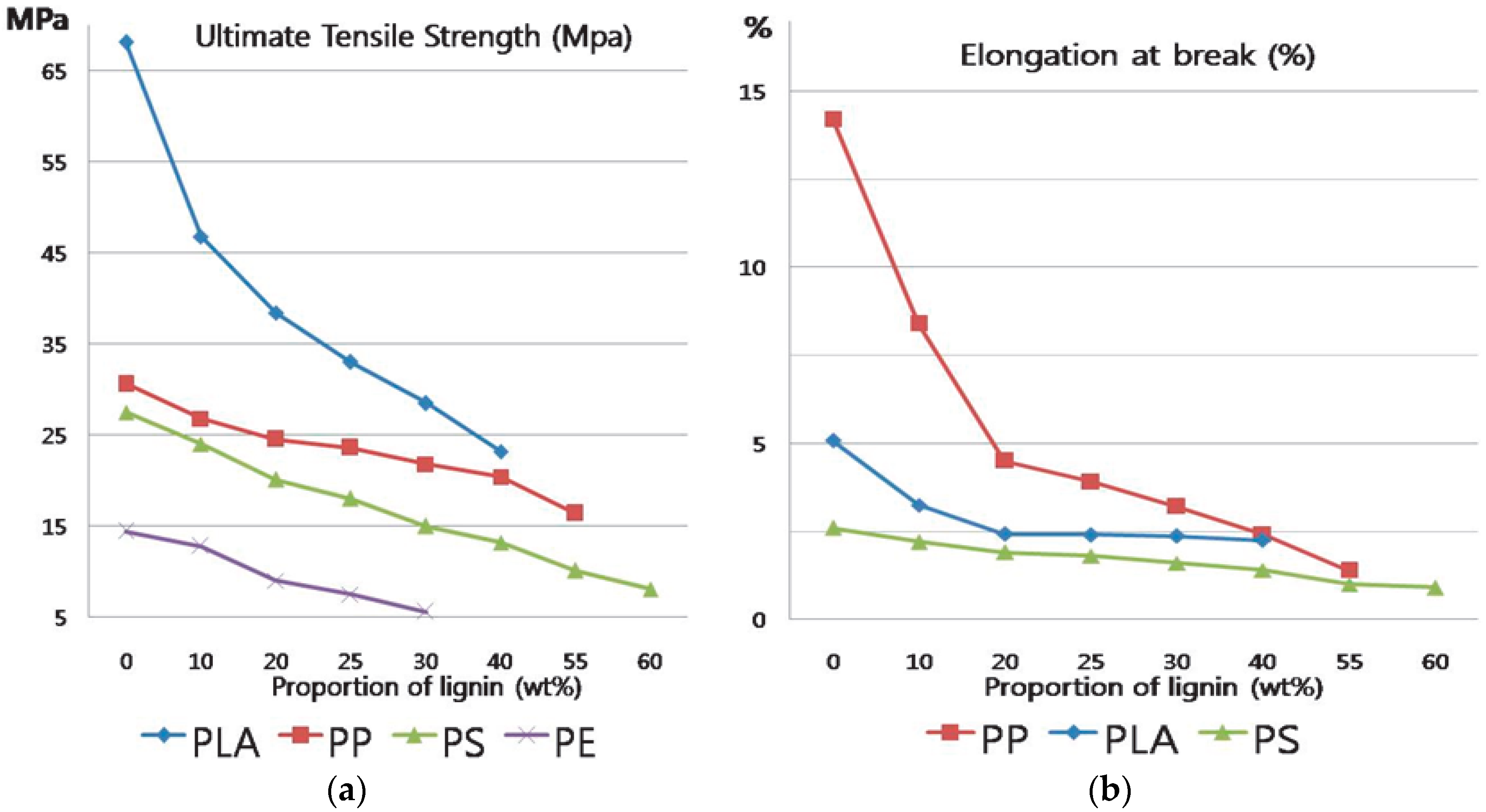
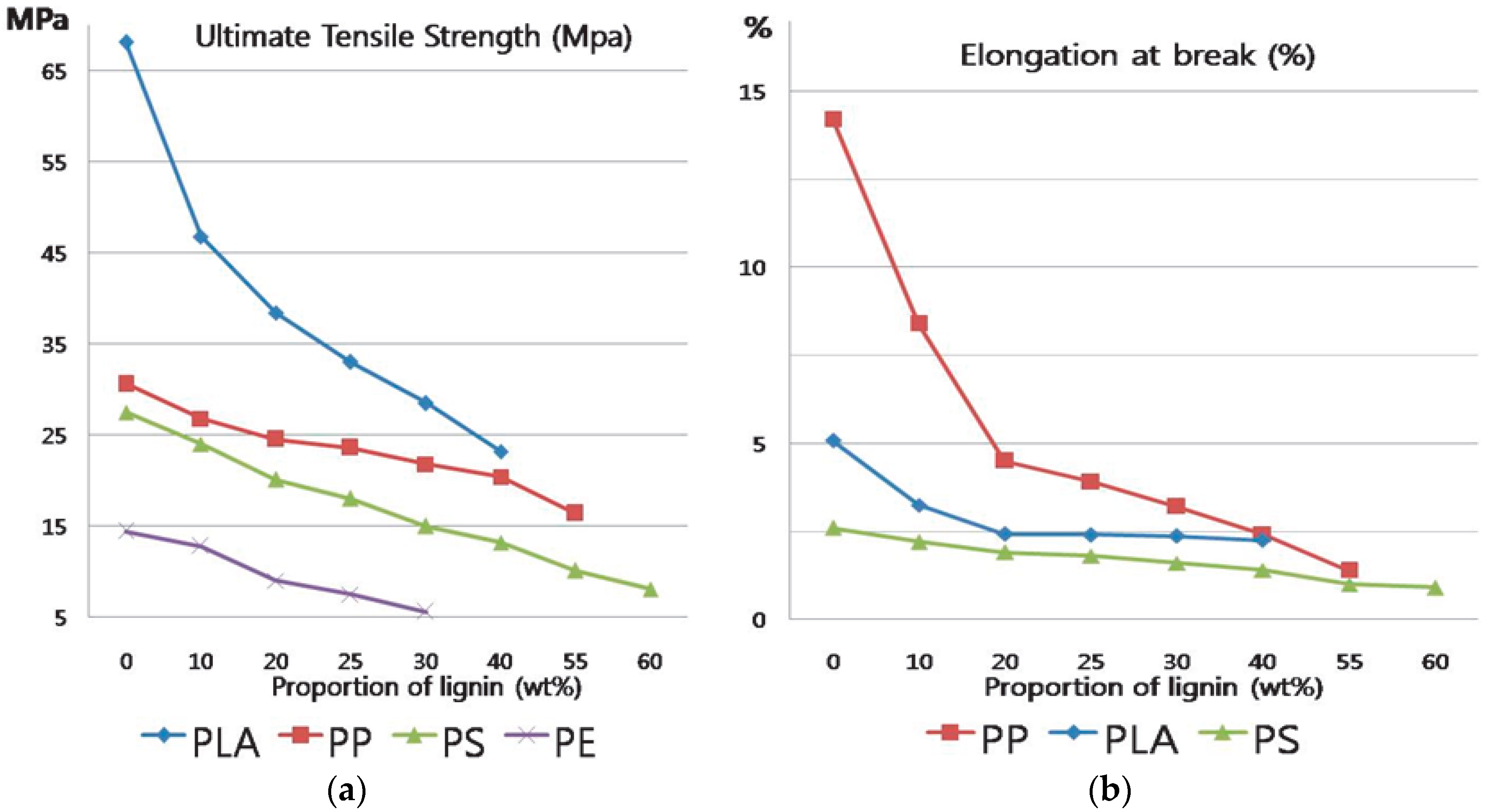
Many researchers have reported that it is possible to produce thermoplastic materials which contain a large amount of lignin [
4]. However, the elongation of such materials tends to decrease dramatically with increasing lignin content in fact, the physical properties of thermoplastic blends are significantly degraded when more than 10%~25% of lignin is incorporated into synthetic polymers, as shown in
Figure 1 [
5,
6,
7,
8]. Therefore, maximizing the amount of lignin in thermoplastic materials while minimizing the corresponding degradation of mechanical properties is a very important research topic. The development of materials with such properties could reduce the price of production and environmental pollution.
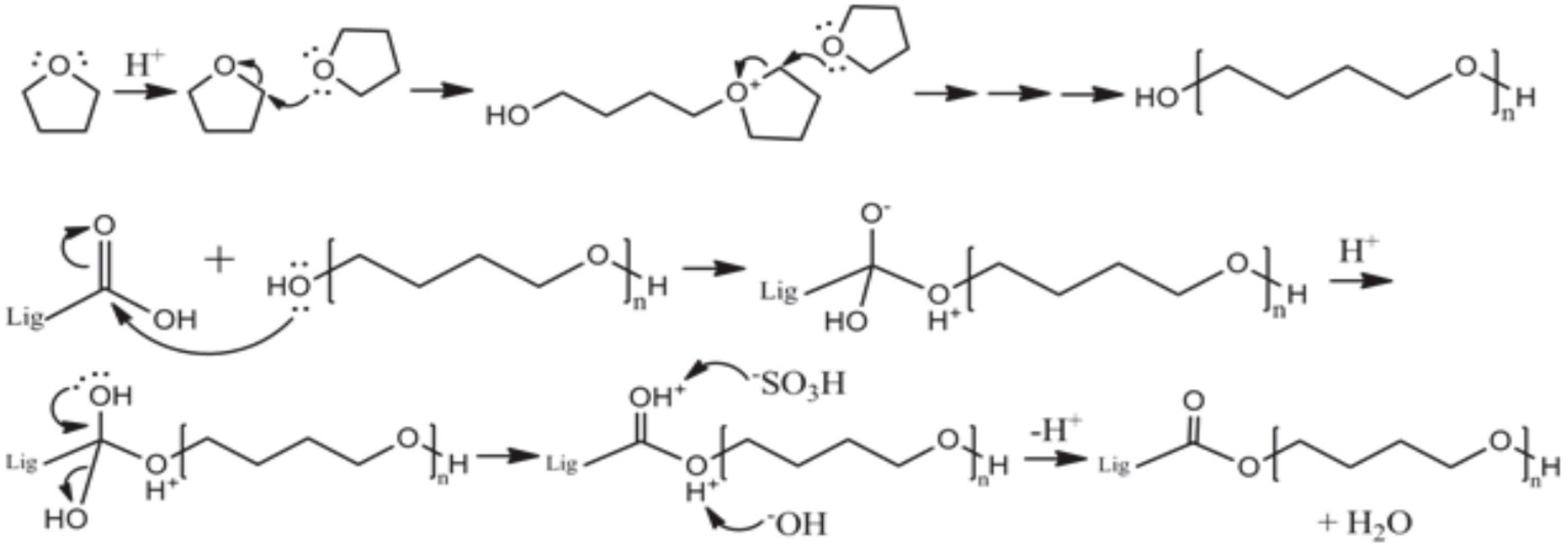
Chemical modification of lignin is a major issue in the area of lignin research. Chemical modification of lignin is performed to replace the carboxyl group and hydroxyl group of the lignin by alkylation, acylation, and hydroxyalkylation [
9]. A new method is proposed in this study where the lignin functional group is replaced by the proper molecular weight of an aliphatic chain. Replacing a lignin functional group is similar to another lignin modification method like alkylation and/or acylation, but the limitation of these methods is that those with molecular weight which are lower than one hundred Dalton can be substituted. The modification method used in this study faced difficulty in dramatically increasing compatibility between matrixes.
2. Experimental
2.1. Materials
Tetrahydrofuran and γ-butyrolactone were used in this study. These reagents and sulfuric acid which were used as catalysts were purchased from Junsei Co., Tokyo, Japan, GR grade. The Indulin AT® (pine kraft lignin) used in this study was purchased from MeadWestVaco Co., North Charleston, SC, USA. Ingeo™. Polyethylene terephthalate (PET) (Mw = 44,650, Mn = 23,410, PDI = 1.91) with a low melting temperature—in order to blend with γ-butyrolactone modified lignin (BLL), polypropylene (PP) (Mw = 210,000, Mn = 49,400, PDI = 4.25)—in order to blend with tetrahydrofuran-modified lignin (THFL), were industrial grades and used as they were received. Sabouraud dextrose agar (model No: 210950) purchased from Becton and Dickinson Difco Co., Ltd. (Franklin Lakes, NJ, USA) (BD Difco) was used as a medium and Aspergillus awamori fungus (ATCC 6970) from the Korean Collection for Type Cultures (KCTC) (Daejeon, Korea) and were used for the degradation test.
2.2. Preparation of Modified Lignin
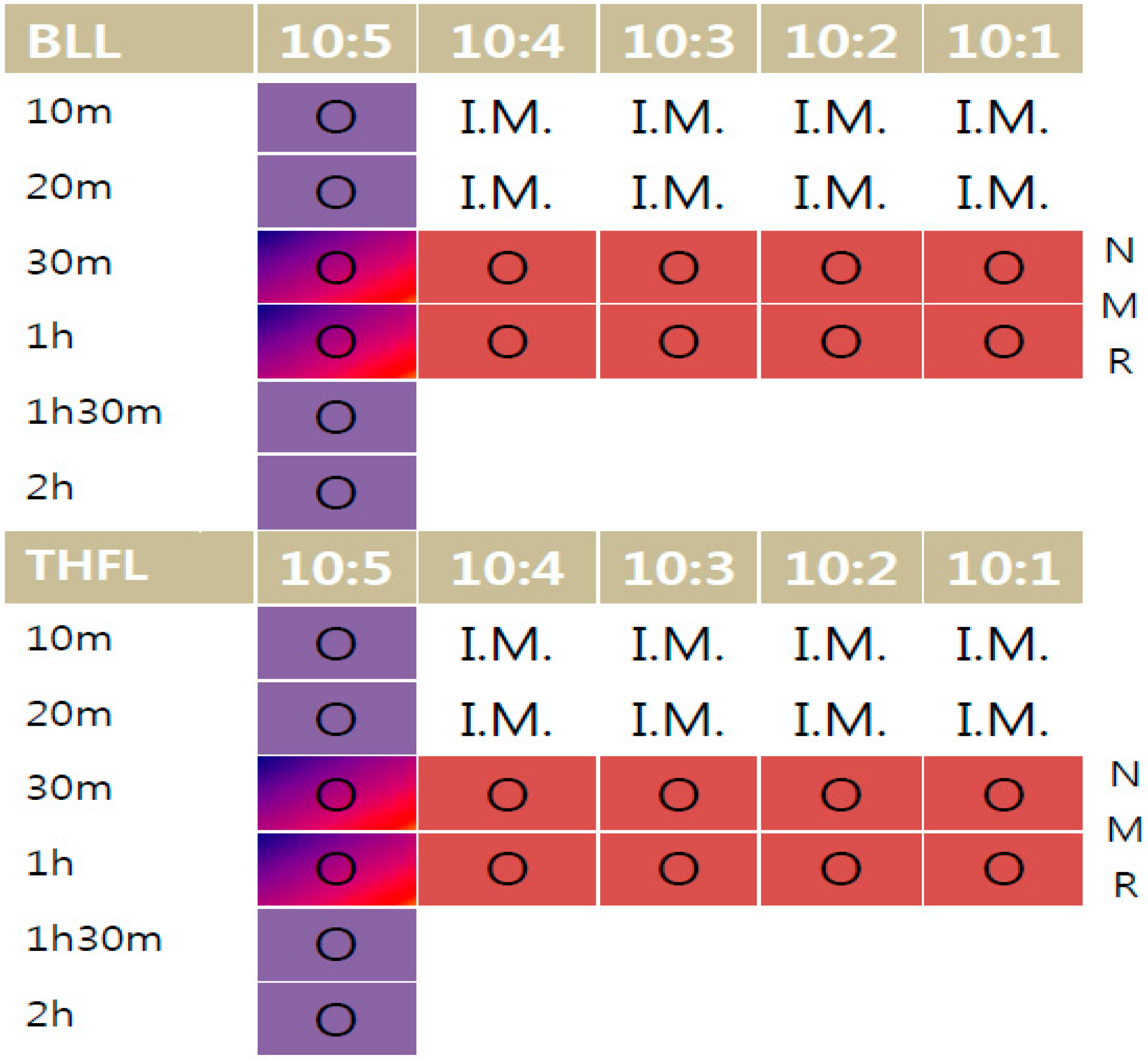
A modification procedure was started using fixed 5 mL reagent and 0.5 g~2.5 g of lignin, according to the 0.5 g variation, with charge ratio from 10:1 to 10:5. The reaction started with sulfuric acid as a catalyst and was put into a 1 wt % lignin/reagent mixture. BLL was reacted at 200 °C in the first half reaction time. After that, the temperature was increased to 250 °C and reacted for another half reaction time. The THFL was reacted first at 100 °C, and then reacted at 150 °C, like BLL. Agitation speed was 900 rpm for the entire process. The time variations were 10, 20, 30 min, 1 h, 1 h and 30 min, and 2 h, respectively, under evaporation in order to remove the byproduct and volatilized reagent. During the two hours of reaction, the viscosity of the mixture was increased rapidly in the last stage.
After following the determined time procedure, the reacted sample was washed with excess distilled water. The modified lignin sinking occurred due to its insolubility and higher density than the distilled water. A small amount of unreacted reagent, unreacted lignin, and catalyst were present as a suspended solid in the supernatant. After sinking, the supernatant was removed and unreacted material suspended to separate from modified lignin. The washed sample needed to be dried under vacuum conditions for use. In order to prevent a side reaction or post reaction due to the minimum quantity of remaining catalyst or reagent when the drying temperature is more than 100 °C, 12 h of vacuum drying must be proceeded at 80 °C with 30 in. hg, or 762 torr of pressure. To dry the sample completely, it was mixed in 3 h intervals.
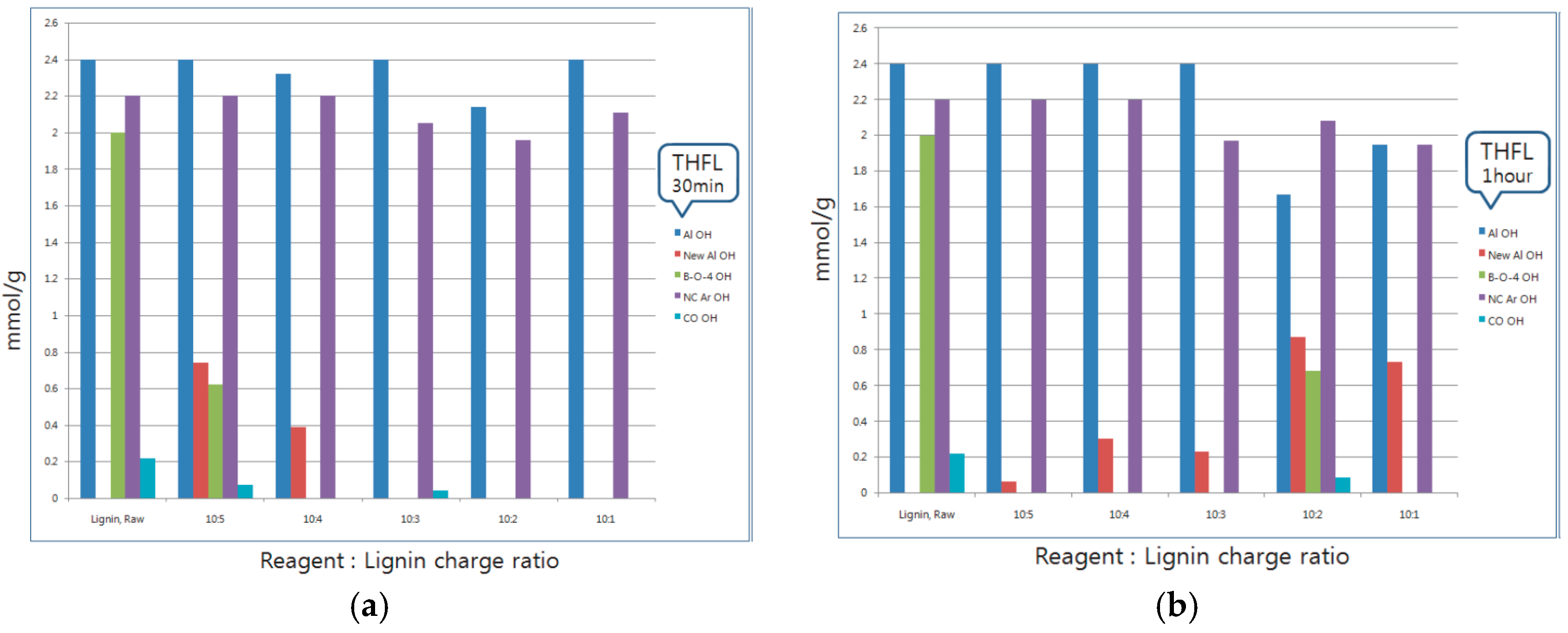
Figure 2 shows a modification time and reaction ratio variation. 10:X is the fixed reagent ratio versus lignin ratio, time is the whole reaction time of the sample. I.M. means ‘impossible mission’ due to too many unreacted materials. 31P-NMR analysis was performed on 30-min and one-hour reacted samples (red marked) and FT-IR analysis and solubility tests were performed on all samples.
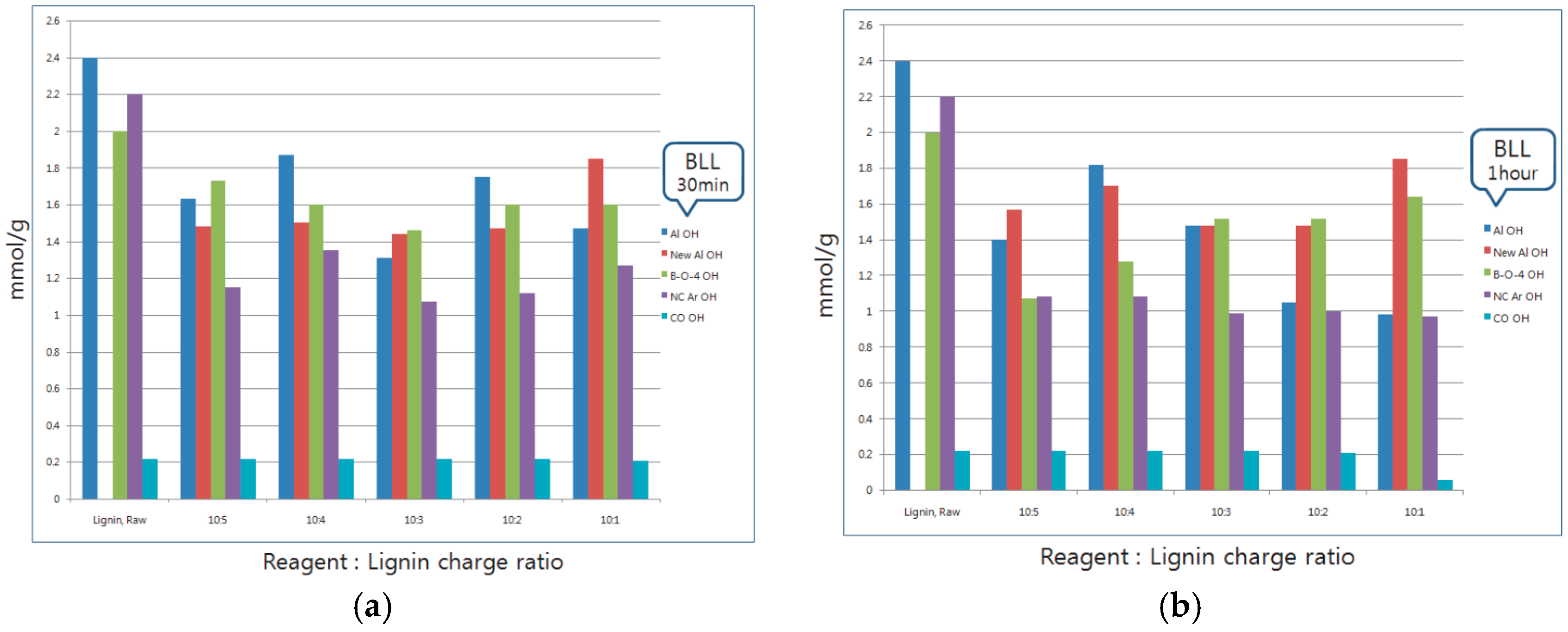
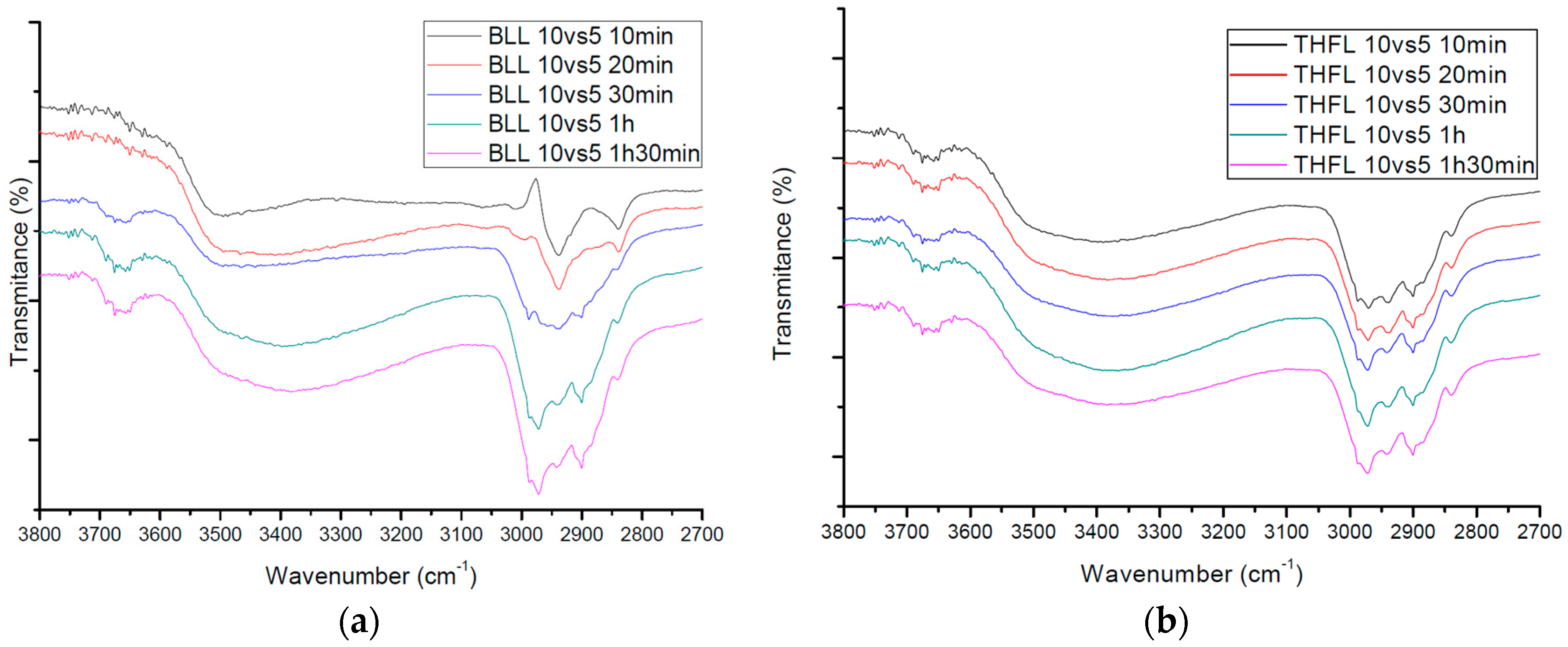
2.3. Analyses of Modified Lignin Functional Group Using 31P-NMR and FT-IR
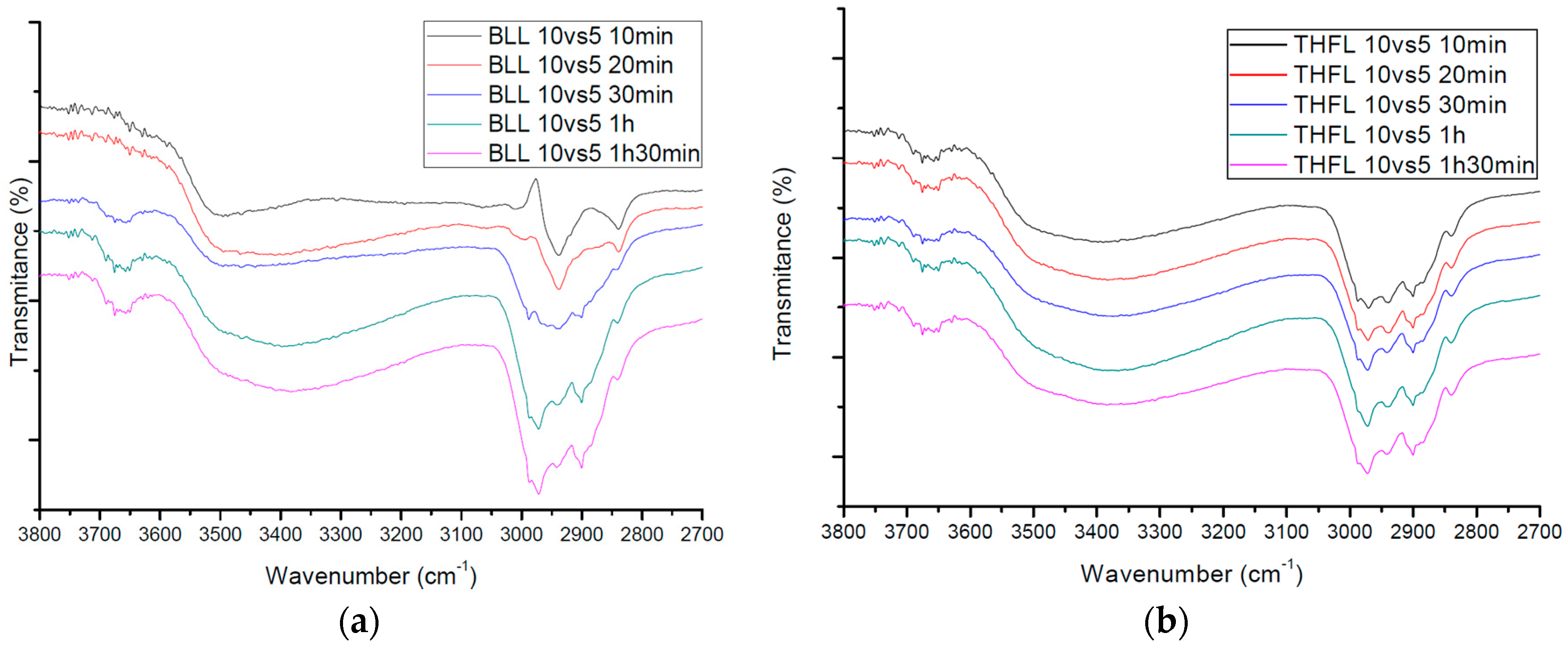
Thirty-minute modified lignin and one-hour modified lignin were monitored and analyzed by quantitative 31P-NMR using published procedures. NMR spectra were acquired using an AVANCE 600 (Bruker, Karlsruhe, Germany), 600 MHz spectrometer equipped with a quad probe dedicated to 1H, 15N, 19F, and 31P-NMR acquisition. To investigate the functional group change of modified lignin that have different reaction charge ratios and times, solid-state FT-IR spectra for the samples that have the fixed charge ratio, and time variations which were 10 min, 20 min, 30 min, one hour, and one hour and 30 min, respectively, were obtained with an FT-IR spectrophotometer (Nicolet Is 5, Thermo Scientific Inc., Waltham, MA, USA).
2.4. Analyses of Modified Lignin in Order to Investigate the Changes of Lignin Contents and Solubility
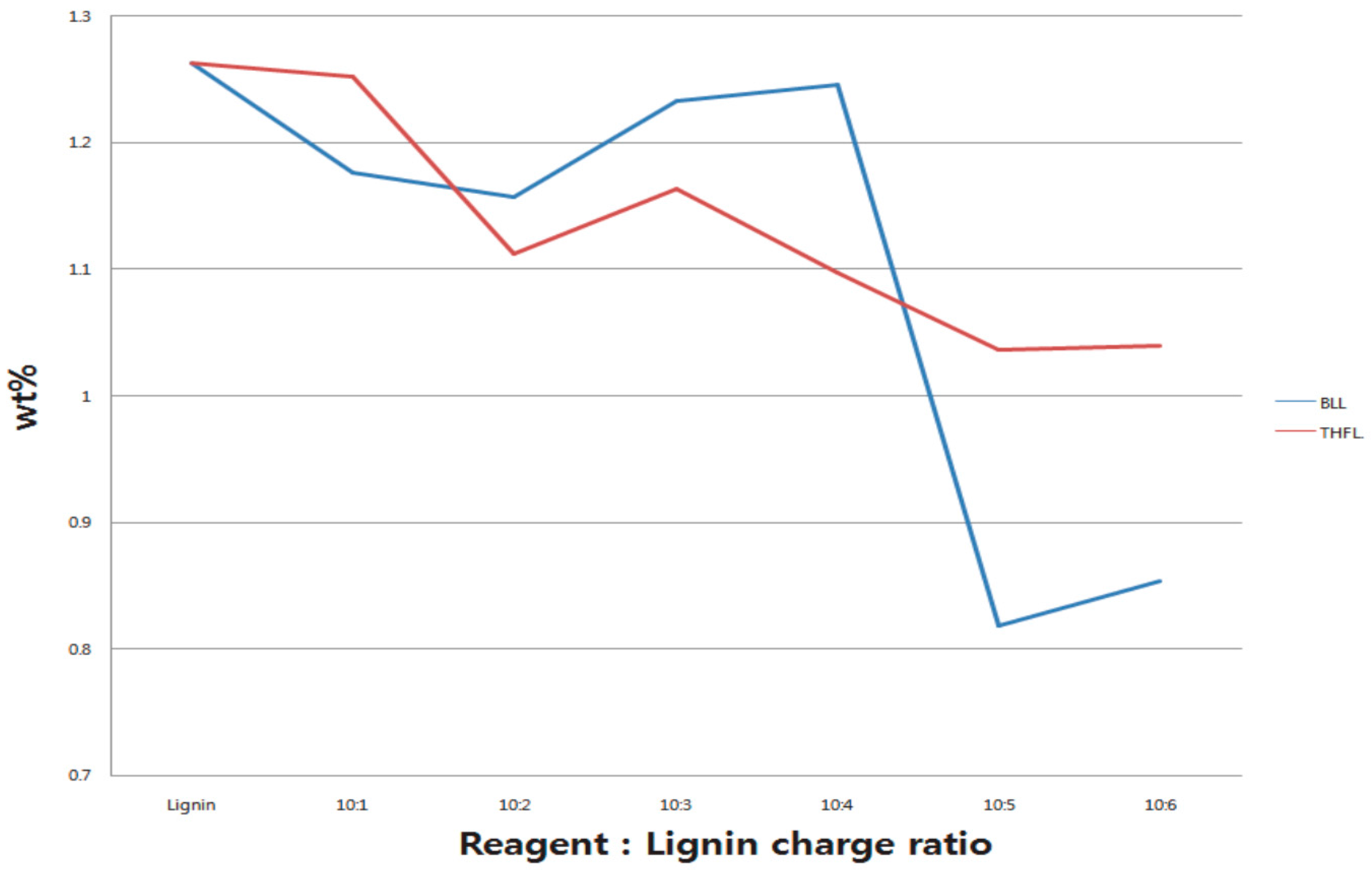
In order to investigate the solubility change of modified lignins that have different reaction charge ratios and times, lignin and modified lignin were dissolved in THF, which did not dissolve lignin. Each sample was dissolved in THF with a total concentration of 2%, (w/v, 100 mg/5 mL) for 5 min and, after that, they were centrifuged for 5 min at 1000 rpm. After removing the supernatant, surplus THF were volatilized in a vacuum chamber. Residues were weighed to quantify the insoluble fraction of modified lignin. The elemental contents of lignin and 2 h modified lignin were analyzed using a Thermo Electron Co., Waltham, MA, USA, Flash EA 1112-Elemental (C,N,S) Analyzer. Synthetic polymers and minerals, like sand or mineral salts, can be measured without error, although only with a few mg samples. However, natural materials like lignin or animal protein are difficult to measure without any error because they do not have regular empirical formulae. Therefore, lignin and modified lignin were weighed to more than 10 mg before measuring in order to reduce the error. Since the reacted reagent is composed of only carbon, oxygen, and hydrogen, and nitrogen only exists in lignin, the elemental analysis was conducted in order to compare the nitrogen content.
2.5. Blending of Modified Lignin and Synthetic Polymers
Modified lignin was mixed with synthetic polymers in proportions of 0, 25.0, and 50.0 wt % of lignin content. The mixtures were melt blended using a table-type kneader (PBV-0.3, Irie Shokai Co., Tokyo, Japan) at 170 ± 2 °C for 20 min. Subsequently, the modified lignin/synthetic polymer composites were compression molded using a two-post manual hydraulic press (#2699, Carver Inc., Wabash, IN, USA) at 170 ± 1 °C and 6.89 MPa for 5 min. The molds were quenched after molding, and then polymer composite sheets were obtained.
2.6. Tensile Test
A universal testing machine (LRX Plus, LLOYD Instruments Inc., Bognor Regis, UK) was used to determine the tensile properties of the modified lignin/synthetic polymer composites and its degraded sample. Tensile tests were performed according to the guidelines of ASTM D638. The range of sample thickness was 1.2 to 1.8 mm, and the strain rate was 10 mm/min. The data are expressed with the averages of a minimum of 10 specimens.
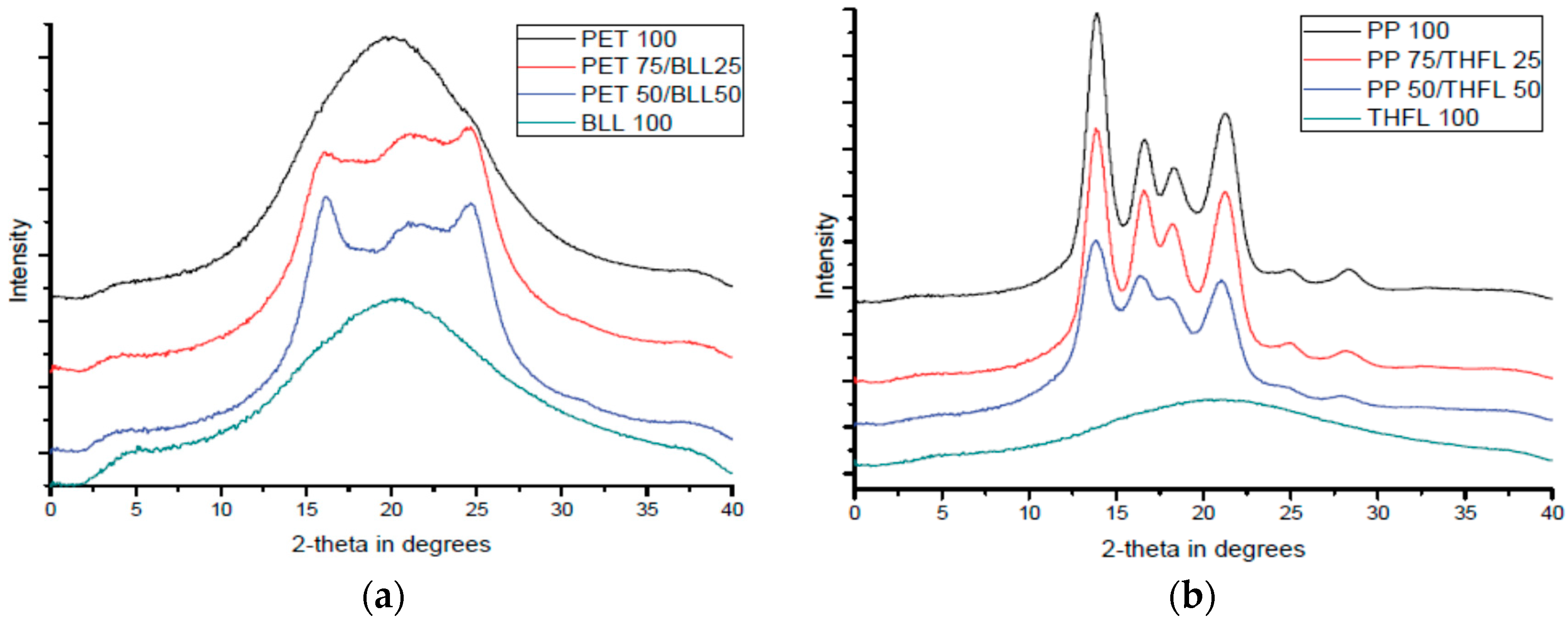
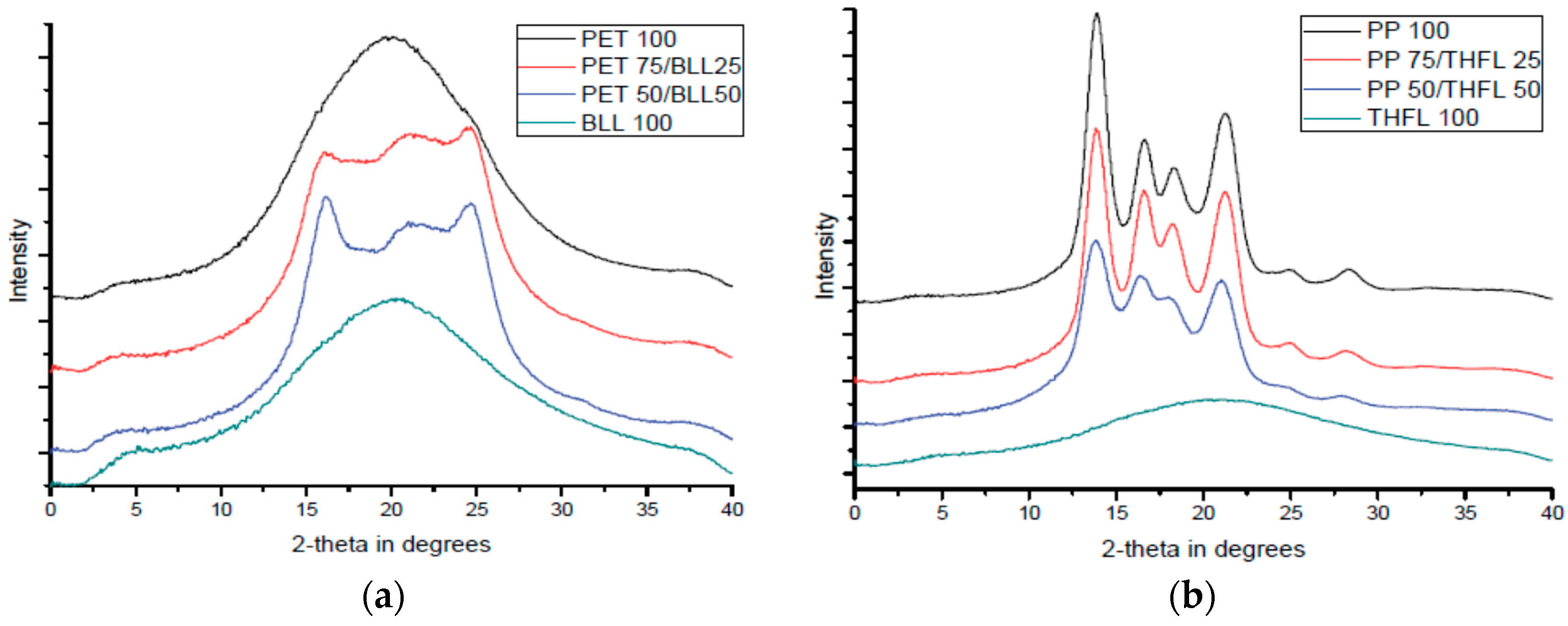
2.7. X-ray Diffraction Analysis (XRD)
X-ray diffraction analysis was used to investigate the change of crystalline area by the modified lignin/matrix ratio (D8-discover with GADDS, Bruker, Germany). Basically, the determination is based on the relative intensity of specific peaks. The XRD method has been previously used to study the polymer structures and directly determine the crystallinity of polymers [
10]. The determination of the degree of crystallinity thus involves the separation of the peaks scattered by the amorphous phase and the peak reflected by the crystalline phase. The degree of crystallinity can be computed using the equation:
where IC and IA are the scattered intensity for the crystalline and the amorphous phase, and XC is the degree of crystallinity of the blend. The degree of crystallinity was calculated by computerized integration values, using baseline integration software (Proteum 2, Bruker, Karlsruhe, Germany).
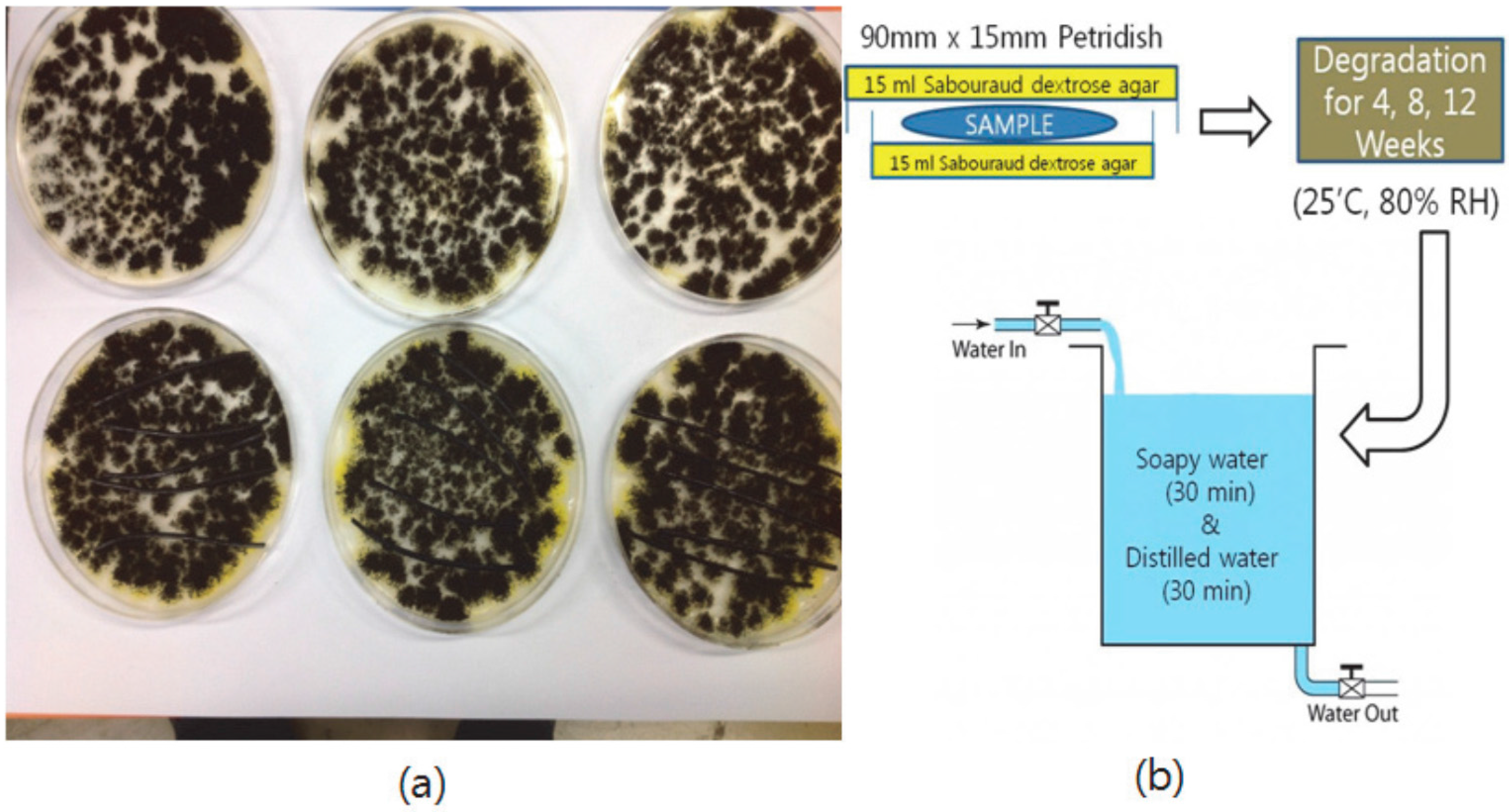
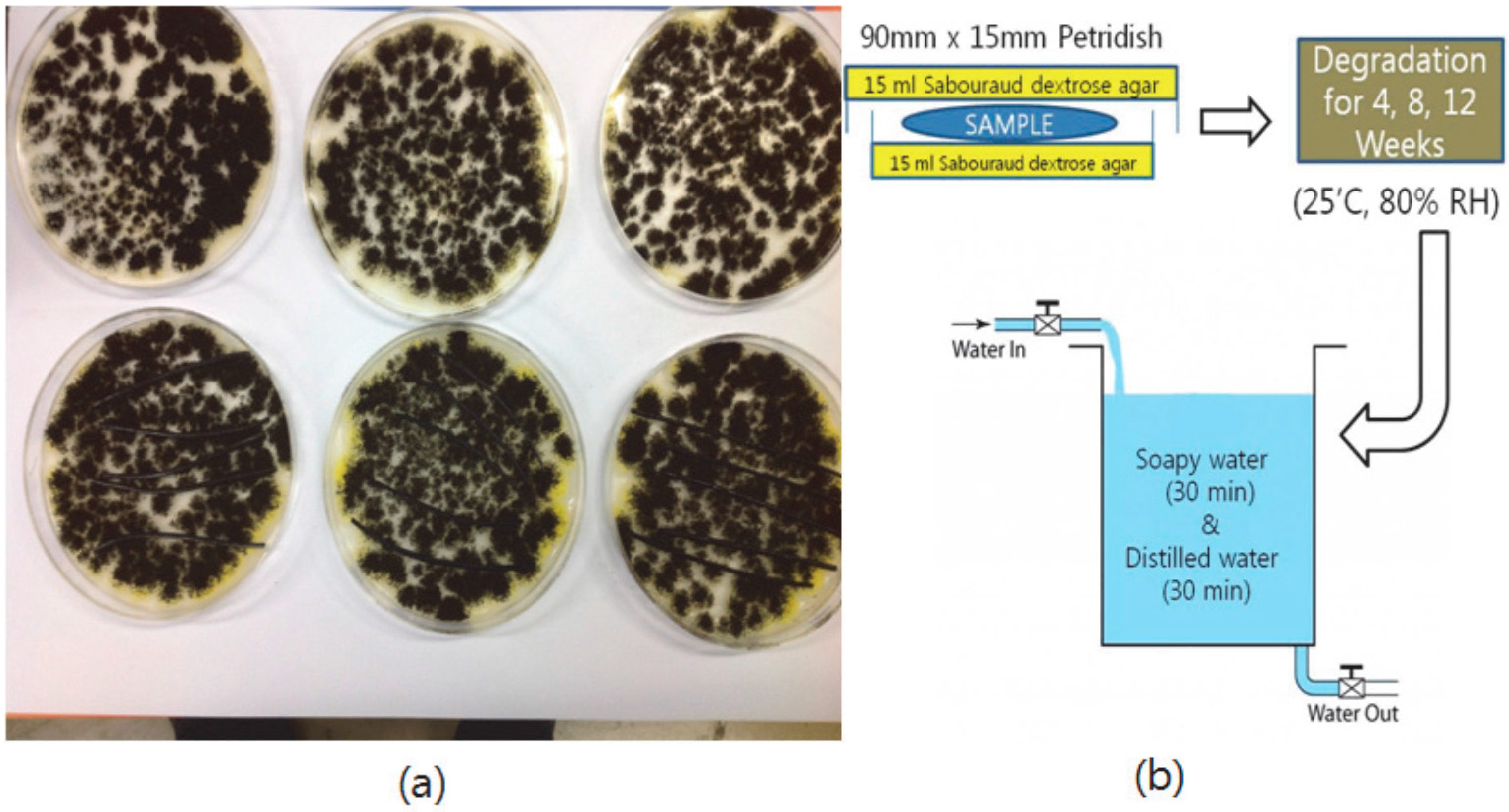
2.8. Degradation Test of Blends
The medium was prepared on the basis of the BD Difco’s manual [
11] and was sterilized in an autoclave. Subsequently, both sides of a Petri dish (90 mm × 15 mm) were coated with 15 mL of medium, and the fungi were seeded. At a temperature of 25 °C and 80% RH (relative humidity), the fungi were grown for 96 h. Afterwards, the blend of 75% polymer and 25% modified lignin was placed on the middle of the medium coated surfaces (number of samples = 5). Fungi were allowed to degrade the sample for four weeks, eight weeks, and 12 weeks (
Figure 3), and the samples were washed in soapy water for 30 min, then washed in distilled water for 30 min. The washed sample was stored in a desiccator for 24 h to remove the residual water. Finally, the change of the sample weight and mechanical properties were measured.
2.9. Scanning Electron Microscope (SEM) Image Analysis of Degraded Blends
The surface of the bio-degraded, modified lignin/synthetic blend was scanned by a SUPRA 55VP, Field-Emission Scanning Electron Microscope, Carl Zeiss, Germany. The degree of biodegradation was analyzed using 1000× images to show how modified lignin has an indirect effect on well-known, non-biodegradable polymers, like PET and PP.
4. Summary and Conclusions
The purpose of this study was to propose an easy and inexpensive method of the modification of lignin. Various analyses were performed using different types of reagents to modify lignin. The reagents were used to mimic the LCC structure. The reaction site of modification was different with respect to the reagent, according to the results of 31P-NMR analysis. BLL was mainly reacted with Al OH and NC Ar OH. A small amount of β-O-4 OH was reacted. THFL did not react with Al OH and NC Ar OH, but reacted with COOH and β-O-4 OH rapidly at the initial modification stage. This is because of the low polarity of THF. β-O-4 linkage of lignin was degraded rapidly at the initial stage of modification, PTMEG substitution occurred at the same time. Polycondensation occurred in the last stage of modification. The yield of modification was higher when the charge ratio of lignin and the reaction time were higher.
XRD results showed the degree of crystallinity of the blend and interactions were different due to the types of the modified lignins and polymers. Tensile results indicate that the material used to modify lignin retains its material properties and affects the mechanical properties of the final blends, probably due to the decreased number of OH bonds in the lignin. In order to show the degradation behavior by addition of BLL and THFL, degradation test was performed for 12 weeks with four-week intervals. It was found that blends using BLL and THFL were degraded. BLL shows more active degradation. From this study, a new method of lignin modification is proposed, and it is found that modified lignin will retain the property of substituted aliphatic chains. This method could be a proper lignin modification method.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}