Effects of Smoking and Genotype on the PSR Index of Periodontal Disease in Adults Aged 18–49

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Demographic and Descriptive Information about the Sample

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PSR1 | PSR2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Predictor | Level | OR | 95% CI | Wald χ2 a | OR | 95% CI | Wald χ2 a | |
| Age | 10 year increments | 1.35 | 1.01–1.81 | 4.04 * | 1.59 | 1.25–2.01 | 14.57 *** | |
| Smoking | Never | 1.00 | 3.29 | 1.00 | 13.29 ** | |||
| Former | 1.07 | 0.60–1.92 | 1.15 | 0.72–1.84 | ||||
| Current | 1.57 | 0.92–2.70 | 2.13 | 1.36–3.31 | ||||
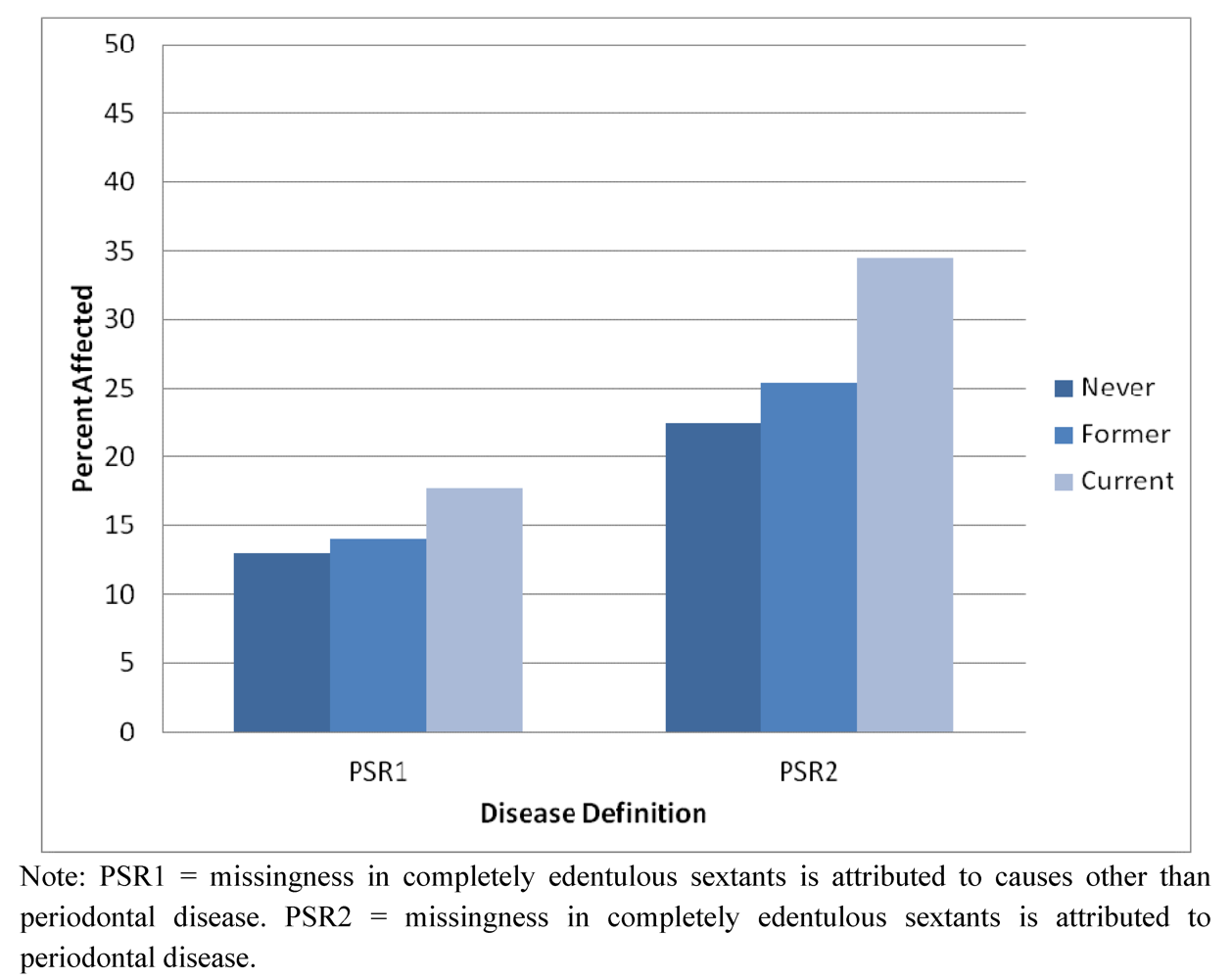
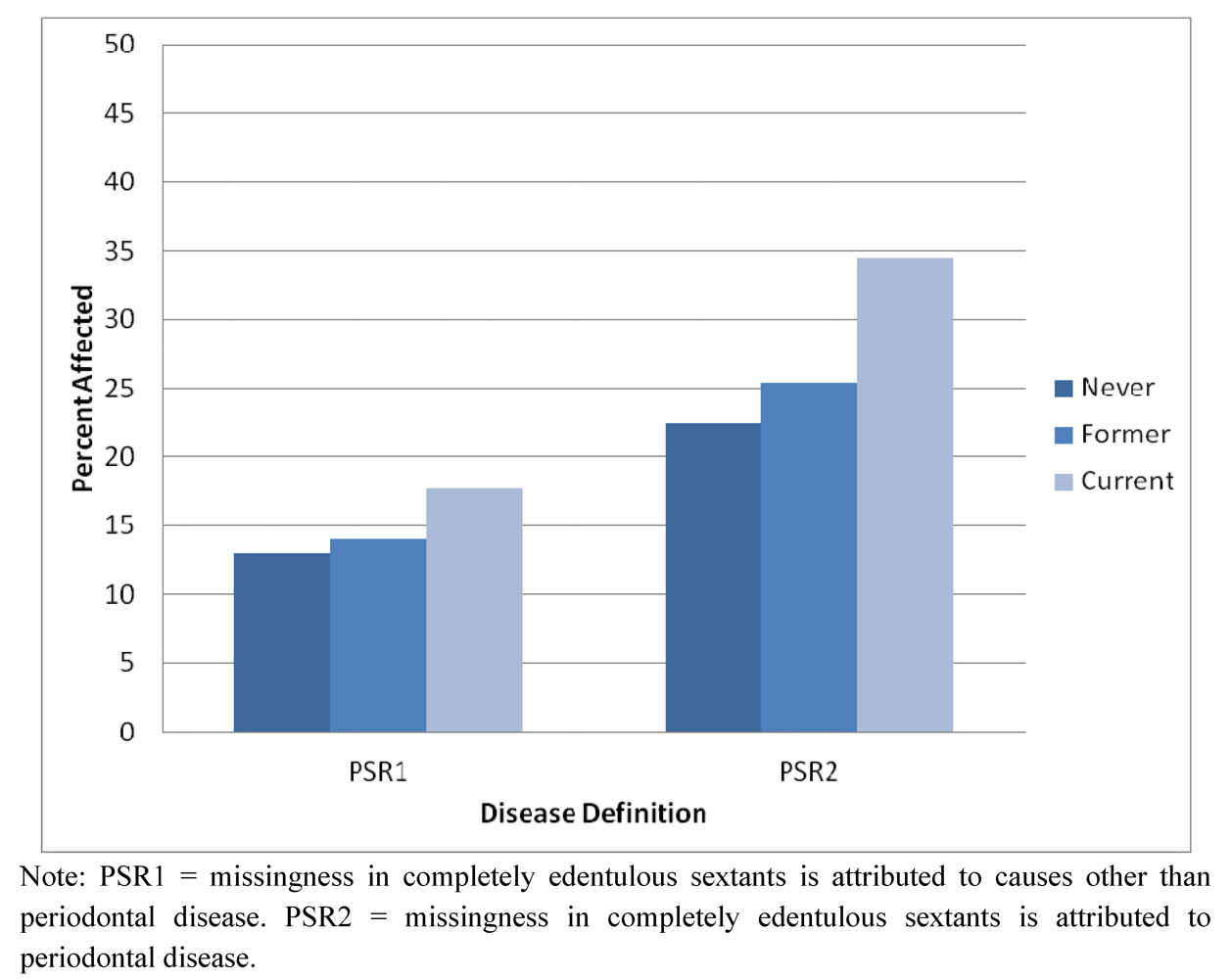
2.2. Results Supporting an Additive Relationship
2.3. Results Supporting a Unique Relationship for Genotype but Not Smoking Status
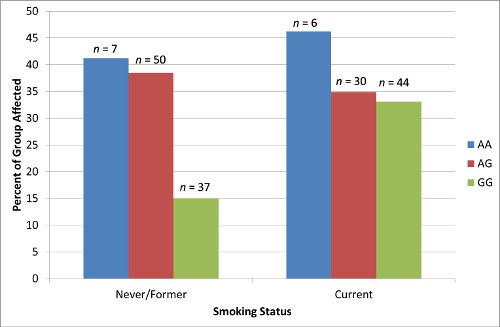
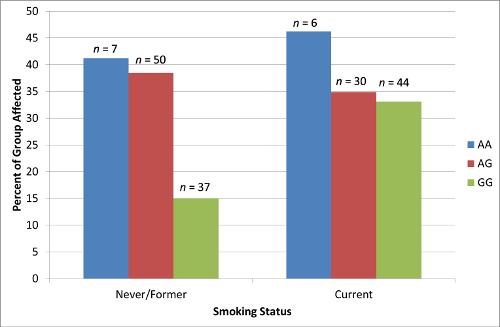
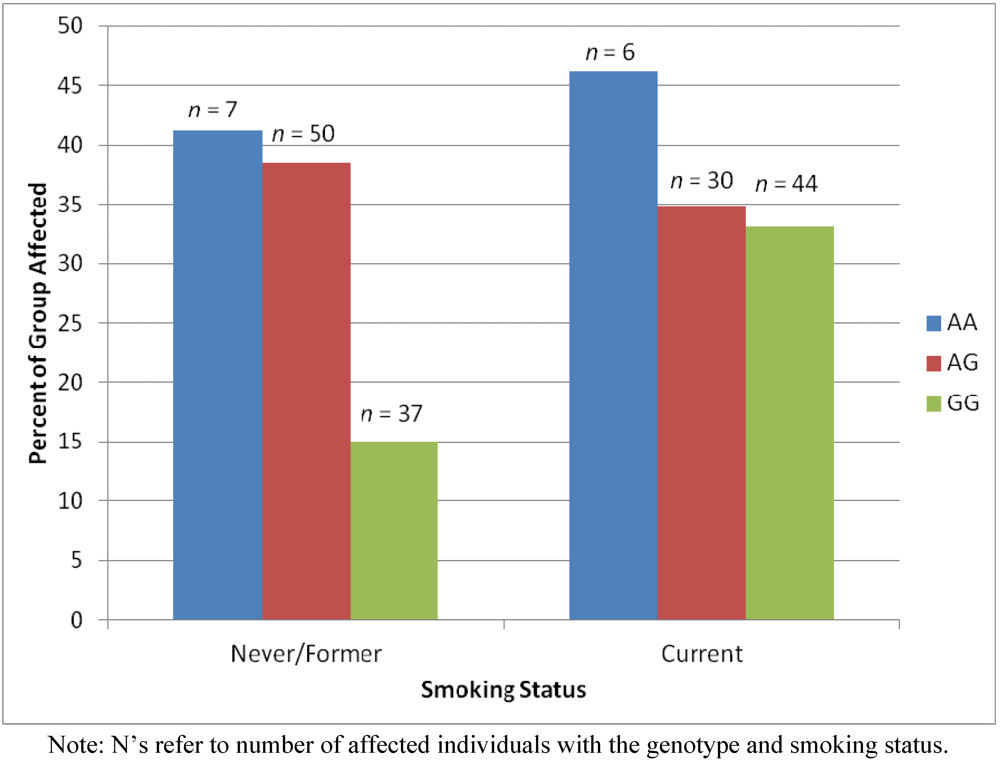
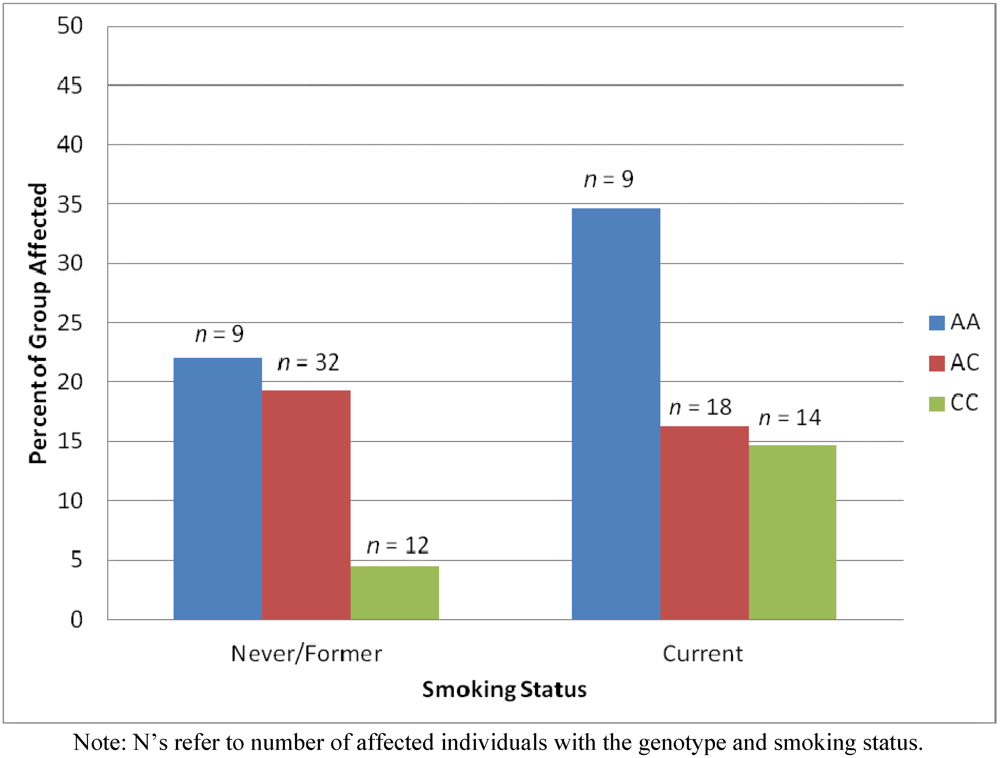
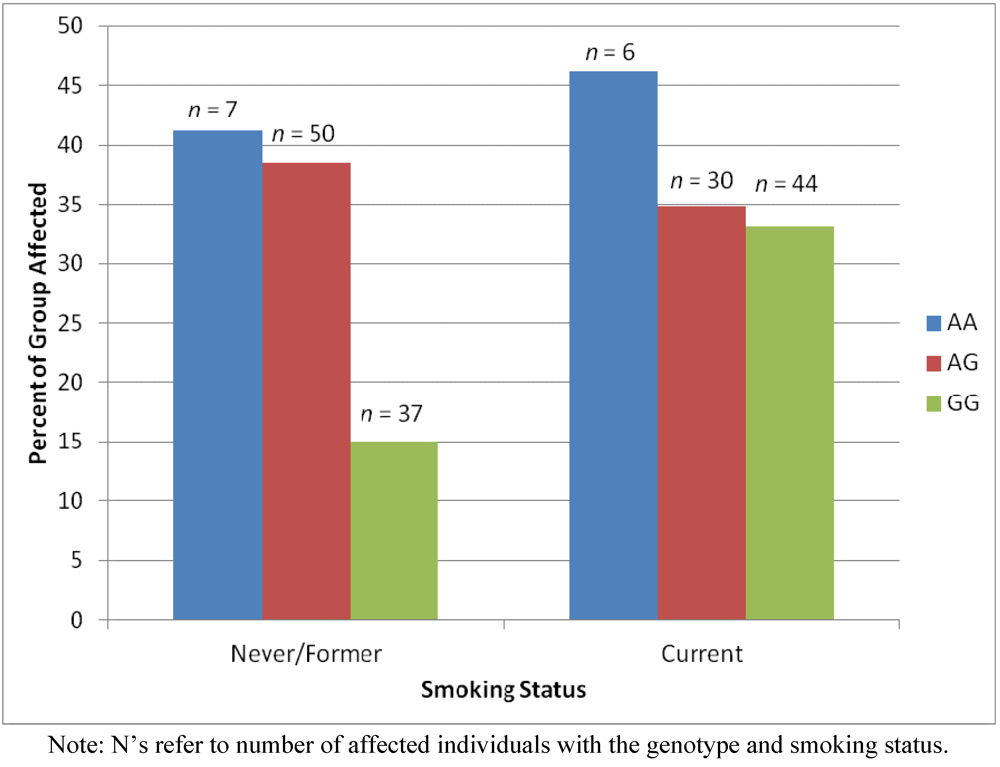
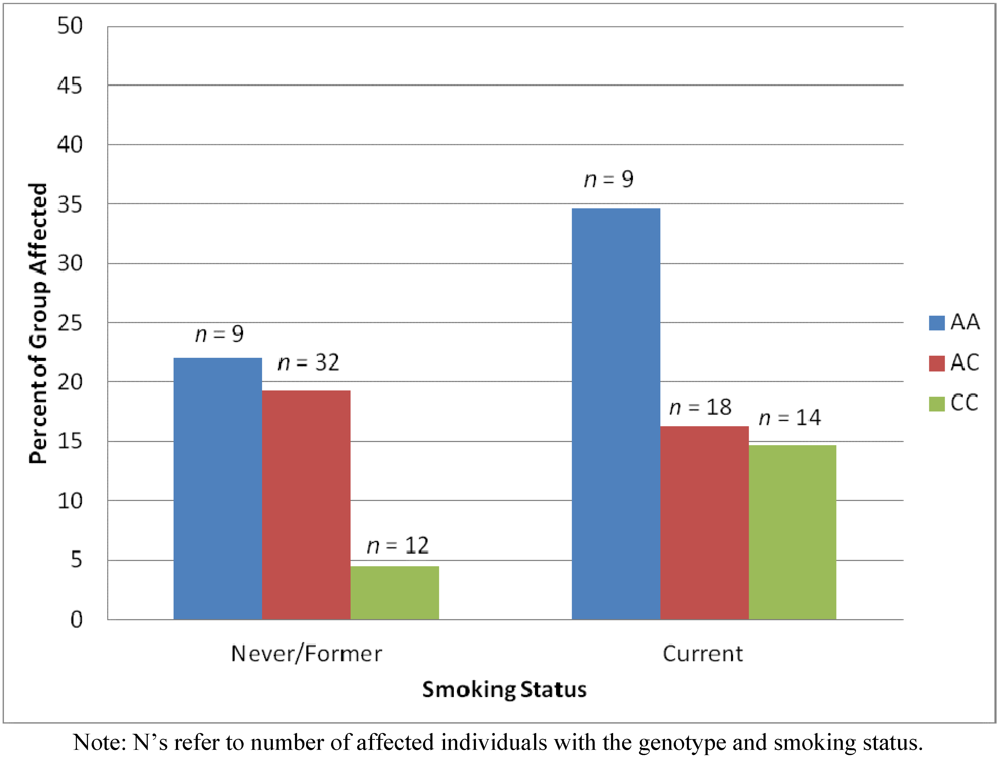
2.4. Results Supporting an Interaction of Genotype by Smoking Status
3. Experimental Section
3.1. Participant Recruitment
3.2. Phenotype and Covariate Assessment
3.3. Data Analysis
| SNP | Outcome Associated With | Strength of Association | Chromosome, Coordinate | Nearby Genes |
|---|---|---|---|---|
| rs10457525 | PSR1 | 5.72 × 10−07 | 6, 129872966 | LAMA2, ARHGAP18 (SENEX) |
| rs733048 | PSR1 | 1.07 × 10−06 | 4, 13242797 | HSP90AB2P, RAB28, BOD1L, NKX3-2 |
| rs10457526 | PSR1 | 1.17 × 10−06 | 6, 129896501 | LAMA2, ARHGAP18 (SENEX) |
| rs733048 | PSR2 | 6.15 × 10−06 | 4, 13242797 | HSP90AB2P, RAB28, BOD1L, NKX3-2 |
| rs12630931 | PSR2 | 7.32 × 10−06 | 3, 31981767 | OSBPL10, ZNF860, GPD1L, CMTM8, STT3B |
| rs3870371 | PSR1 | 8.79 × 10−06 | 8, 122697132 | HAS2, HAS2AS |
3.4. Missing Data
4. Conclusions
Strengths and Limitations
Acknowledgments
Conflict of Interest
References
- Albandar, J.M.; Brunelle, J.A.; Kingman, A. Destructive periodontal disease in adults 30 years of age and older in the United States, 1988–1994. J. Periodontol. 1999, 70, 13–29. [Google Scholar]
- Stabholz, A.; Soskolne, A.; Shapira, L. Genetic and environmental risk factors for chronic periodontitis and aggressive periodontitis. Periodontology 2000 2010, 53, 138–153. [Google Scholar]
- Ryder, M.I. The influence of smoking on host responses in periodontal infections. Periodontology 2000 2007, 43, 267–277. [Google Scholar] [CrossRef]
- Hujoel, P.P.; Bergstrom, J.; del Aguilla, M.A.; DeRouen, T.A. A hidden periodontitis epidemic during the 20th century? Community Dent. Oral Epidemiol. 2003, 31, 1–6. [Google Scholar]
- Hyman, J.J.; Reid, B.C. Epidemiologic risk factors for periodontal attachment loss among adults in the United States. J. Clin. Periodontol. 2003, 30, 230–237. [Google Scholar] [CrossRef]
- Kendler, K.S.; Eaves, L.J. Models for the joint effect of genotype and environment on liability to psychiatric illness. Am. J. Psychiatry 1986, 143, 279–289. [Google Scholar]
- Meisel, P.; Siegemund, A.; Dombrowa, S.; Sawaf, H.; Fanghaenel, J.; Kocher, T. Smoking and polymorphisms of the interleukin-1 gene cluster (IL-1alpha, IL-1beta, and IL-1RN) in patients with periodontal diseas. J. Periodontol. 2002, 73, 27–32. [Google Scholar]
- Meisel, P.; Schwahn, C.; Gesch, D.; Bernhardt, O.; John, U.; Kocher, T. Dose-effect relation of smoking and the interleukin-1 gene polymorphism in periodontal disease. J. Periodontol. 2004, 75, 236–242. [Google Scholar]
- Wang, X.J.; Polk, D.E.; Feingold, E.; Shaffer, J.R.; Weyant, R.J.; Crout, R.J.; McNeil, D.W.; Marazita, M.L. GWAS of PSR Index of periodontal disease in adults ages 18-49. J. Dent. Res. under review.
- Landry, R.G.; Jean, M. Periodontal Screening and Recording (PSR) Index: Precursors, utility, and limitations in a clinical setting. Int. Dent. J. 2002, 52, 35–40. [Google Scholar] [CrossRef]
- Chao, H.; Spicer, A.P. Natural antisense mRNAs to hyaluronan synthase 2 inhibit hyaluronan biosynthesis and cell proliferation. J. Biol. Chem. 2005, 280, 27513–27522. [Google Scholar]
- Slevin, M.; Kumar, S.; Gaffney, J. Angiogenic oligosaccharides of hyaluronan induce multiple signaling pathways affecting vascular endothelial cell mitogenic and wound healing responses. J. Biol. Chem. 2002, 277, 41046–41059. [Google Scholar]
- Polk, D.E.; Weyant, R.J.; Crout, R.J.; McNeil, D.W.; Tarter, R.E.; Thomas, J.G.; Marazita, M.L. Study protocol of the Center for Oral Health Research in Appalachia (COHRA) etiology study. BMC Oral Health 2008, 8. [Google Scholar] [CrossRef]
- Nasi, J.H. Background to, and implementation of, the Periodontal Screening and Recording (PSR) procedure in the USA. Int. Dent. J. 1994, 44, 585–588. [Google Scholar]
- Tarter, R. Evaluation and treatment of adolescent substance abuse: A decision tree method. Am. J. Drug Alcohol Abuse 1990, 16, 1–46. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Polk, D.E.; Wang, X.; Feingold, E.; Shaffer, J.R.; Weeks, D.E.; Weyant, R.J.; Crout, R.J.; McNeil, D.W.; Marazita, M.L. Effects of Smoking and Genotype on the PSR Index of Periodontal Disease in Adults Aged 18–49. Int. J. Environ. Res. Public Health 2012, 9, 2839-2850. https://doi.org/10.3390/ijerph9082839
Polk DE, Wang X, Feingold E, Shaffer JR, Weeks DE, Weyant RJ, Crout RJ, McNeil DW, Marazita ML. Effects of Smoking and Genotype on the PSR Index of Periodontal Disease in Adults Aged 18–49. International Journal of Environmental Research and Public Health. 2012; 9(8):2839-2850. https://doi.org/10.3390/ijerph9082839
Chicago/Turabian StylePolk, Deborah E., Xiaojing Wang, Eleanor Feingold, John R. Shaffer, Daniel E. Weeks, Robert J. Weyant, Richard J. Crout, Daniel W. McNeil, and Mary L. Marazita. 2012. "Effects of Smoking and Genotype on the PSR Index of Periodontal Disease in Adults Aged 18–49" International Journal of Environmental Research and Public Health 9, no. 8: 2839-2850. https://doi.org/10.3390/ijerph9082839