The Aryl Hydrocarbon Receptor Pathway: A Key Component of the microRNA-Mediated AML Signalisome

Abstract
:
1. Introduction
1.1. miRNA Background
1.2. Acute Myeloid Leukemia (AML) Background
1.3. Pathways Modulated in Leukemia
1.4. Previous Studies on miRNAs in AML
1.5. Study Aim
2. Methods
2.1. Identifying the AML-Associated Gene Expression Signature
2.2. Identifying AML-Associated miRNAs and Their Transcriptional Targets
2.3. Network, Pathway, and Functional Enrichment Analysis
3. Results
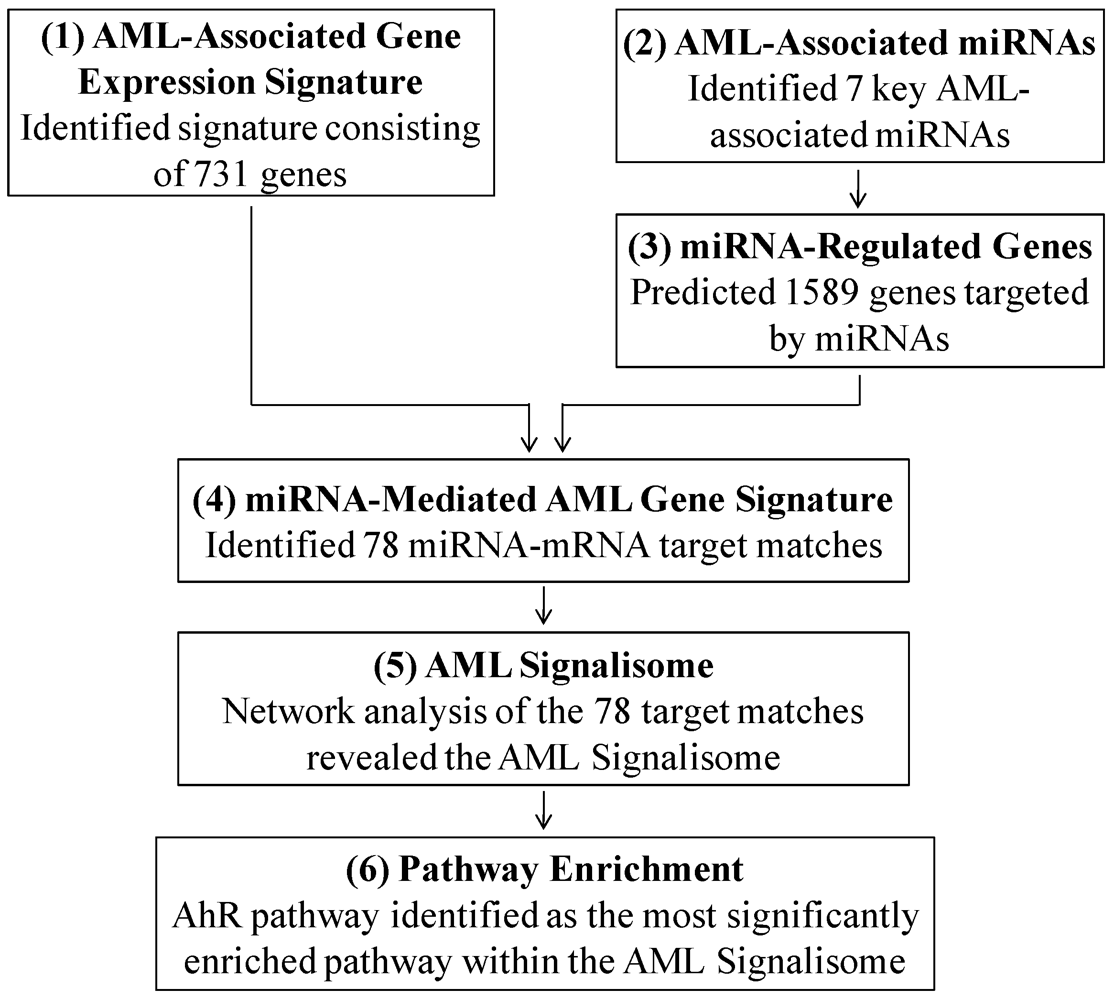
3.1. Identifying the AML Signalisome

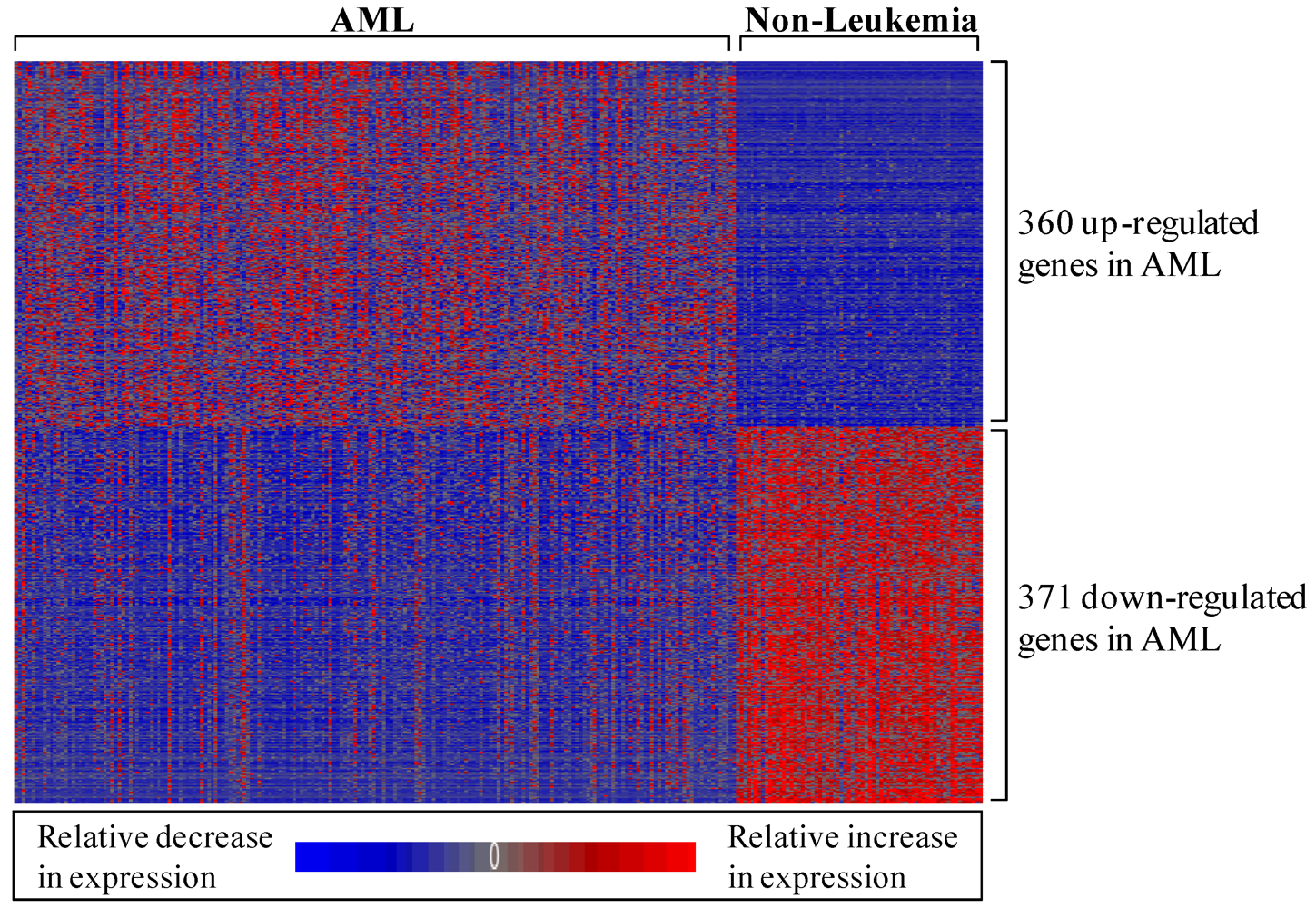
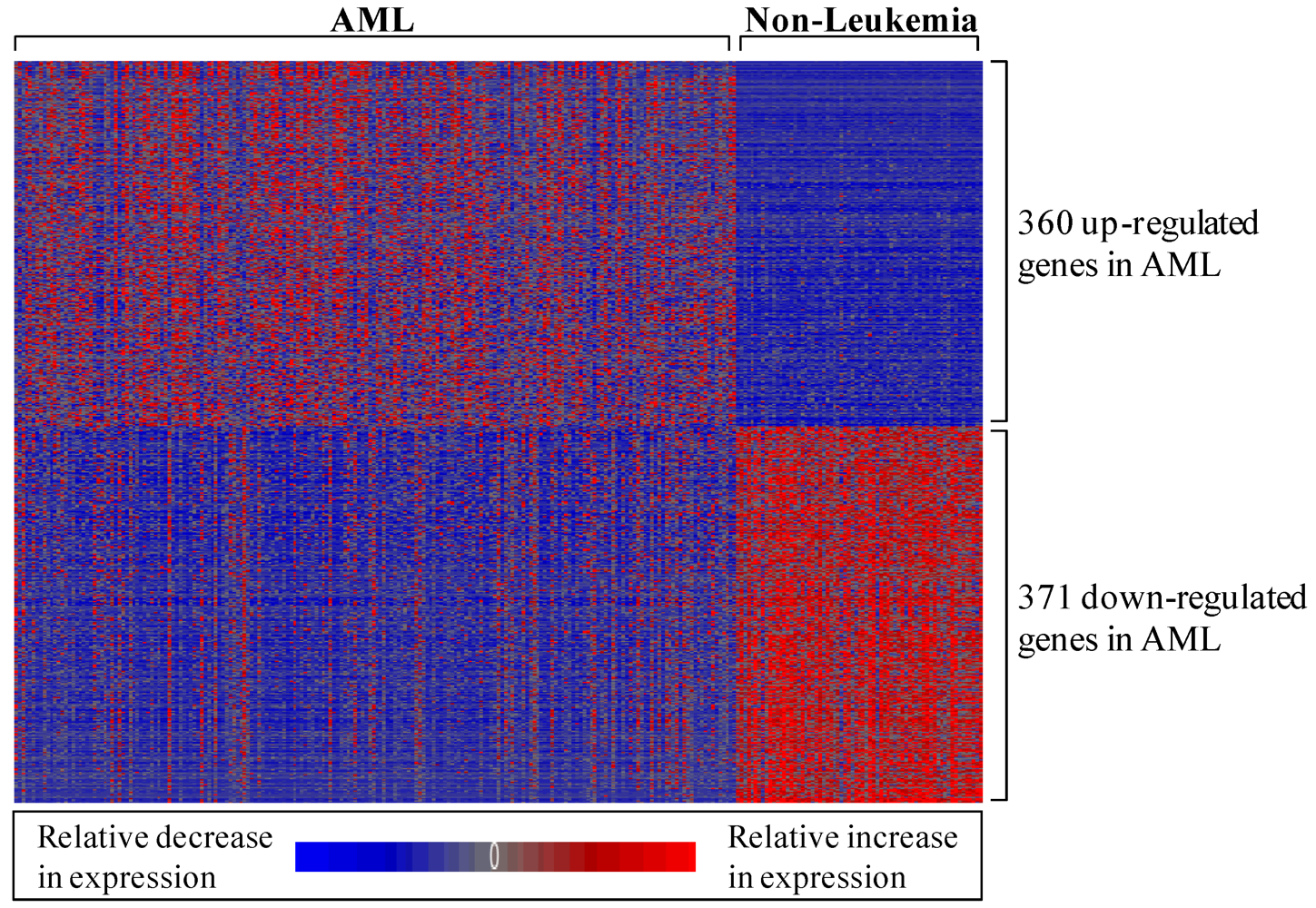
3.1.1. AML-Associated Gene Expression Signature Is Identified
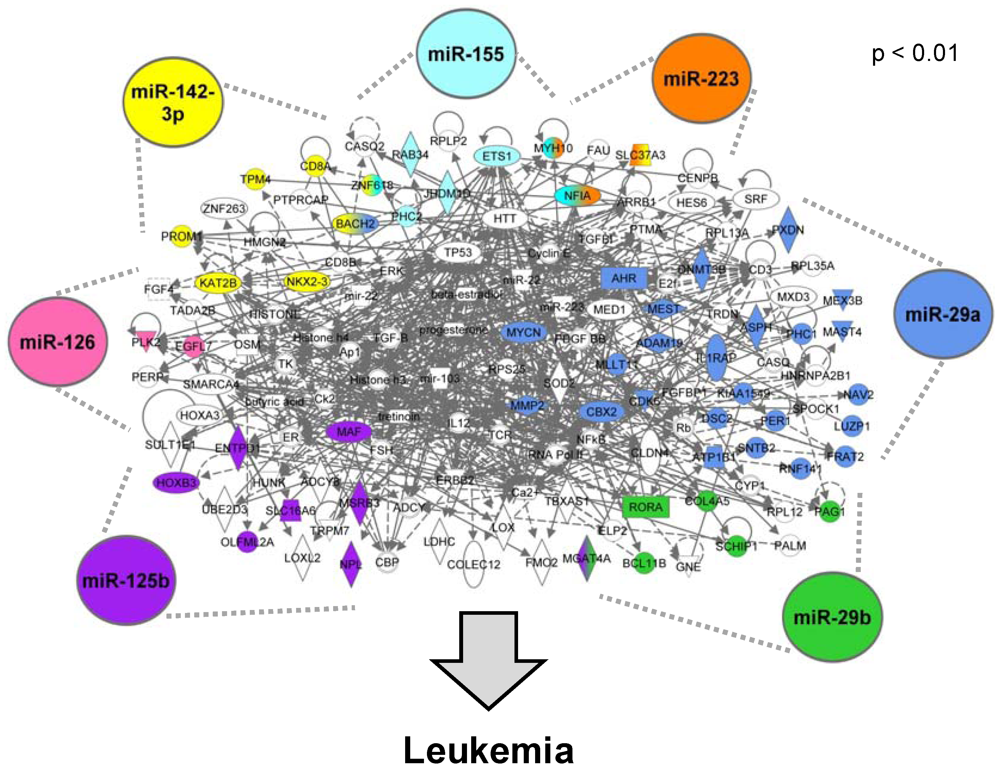
3.1.2. AML-Associated miRNAs Are Identified

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3.1.3. miRNA-Regulated Genes Are Predicted
3.1.4. The miRNA-Mediated AML Gene Signature Is Identified
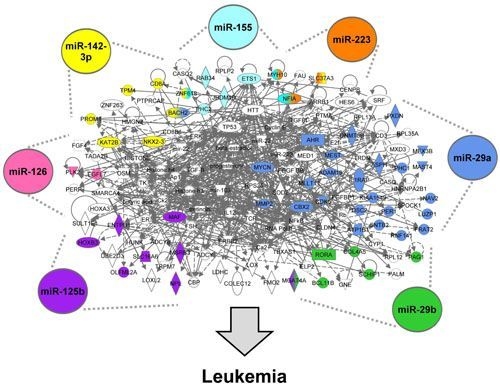
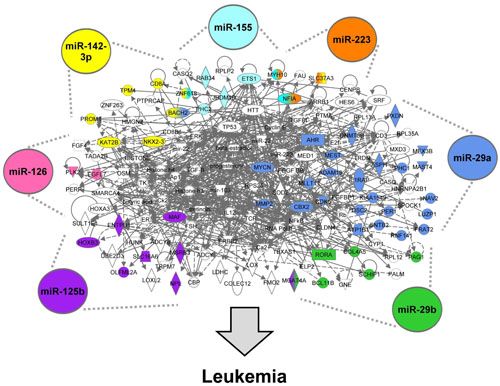
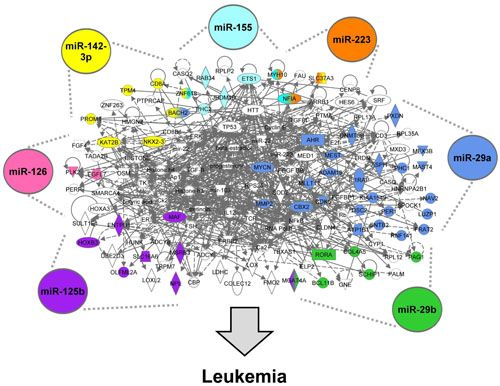
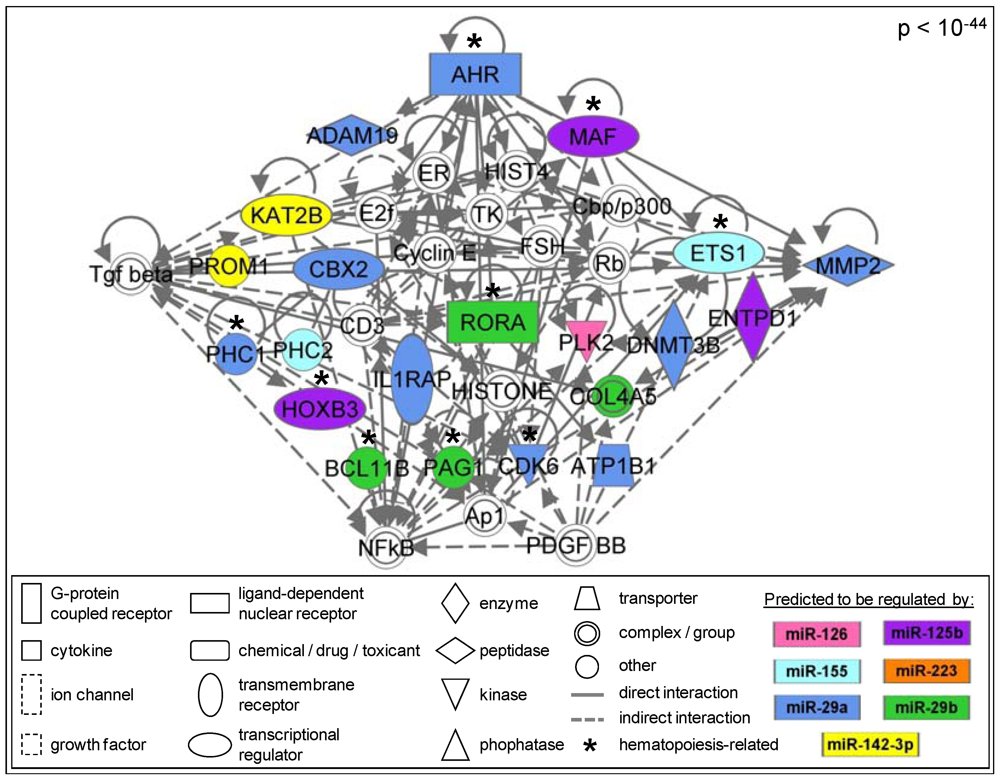
3.1.5. The AML Signalisome Is Identified

3.1.6. Members of the AhR Pathway Are Enriched within the AML Signalisome
| Pathway | p value |
|---|---|
| Aryl Hydrocarbon Receptor Signaling | 0.017 |
| Hepatic Fibrosis/Hepatic Stellate Cell Activation | 0.019 |
| Airway Pathology in Chronic Obstructive Pulmonary Disease | 0.032 |
| Calcium Signaling | 0.032 |
| Aminosugars Metabolism | 0.032 |
| HER-2 Signaling in Breast Cancer | 0.038 |
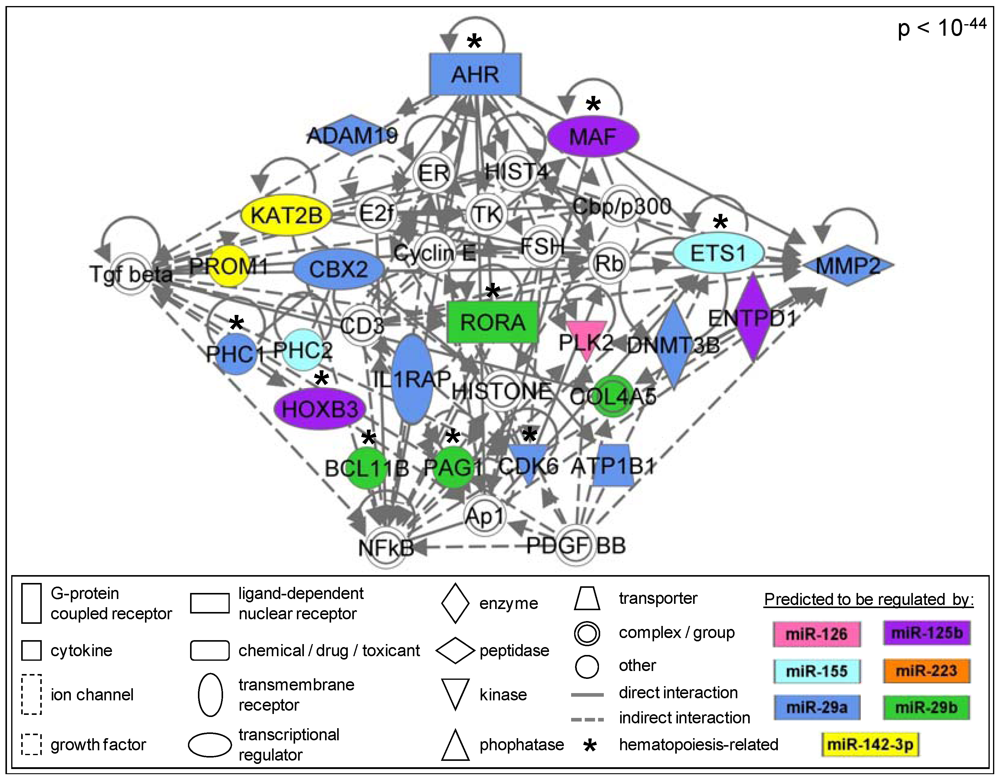
4. Discussion
5. Conclusions
Acknowledgements
Conflict of Interest
References
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar]
- Chen, C.Z.; Lodish, H.F. MicroRNAs as regulators of mammalian hematopoiesis. Semin. Immunol. 2005, 17, 155–165. [Google Scholar] [CrossRef]
- Marcucci, G.; Mrózek, K.; Radmacher, M.D.; Garzon, R.; Bloomfield, C.D. The prognostic and functional role of microRNAs in acute myeloid leukemia. Blood 2011, 117, 1121–1129. [Google Scholar]
- Murray, M.Y.; Rushworth, S.A.; MacEwan, D.J. Micro RNAs as a new therapeutic target towards leukaemia signalling. Cell Signal 2012, 24, 363–368. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar]
- Lagos-Quintana, M.; Rauhut, R.; Lendeckel, W.; Tuschl, T. Identification of novel genes coding for small expressed RNAs. Science 2001, 294, 853–858. [Google Scholar]
- Lau, N.C.; Lim, L.P.; Weinstein, E.G.; Bartel, D.P. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science 2001, 294, 858–862. [Google Scholar]
- Iorio, M.V.; Piovan, C.; Croce, C.M. Interplay between microRNAs and the epigenetic machinery: An intricate network. Biochim. Biophys. Acta 1799, 694–701. [Google Scholar]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Neyman, N.; Aminou, R.; Waldron, W.; Altekruse, S.F.; Kosary, C.L.; Ruhl, J.; Tatalovich, Z.; et al. SEER Cancer Statistics Review, 1975-2008; National Cancer Institute: Bethesda, MD, USA, 2011. [Google Scholar]
- Estey, E.; Döhner, H. Acute myeloid leukaemia. Lancet 2006, 368, 1894–1907. [Google Scholar] [CrossRef]
- Guzman, M.L.; Neering, S.J.; Upchurch, D.; Grimes, B.; Howard, D.S.; Rizzieri, D.A.; Luger, S.M.; Jordan, C.T. Nuclear factor-kappaB is constitutively activated in primitive human acute myelogenous leukemia cells. Blood 2001, 98, 2301–2307. [Google Scholar]
- Towatari, M.; Iida, H.; Tanimoto, M.; Iwata, H.; Hamaguchi, M.; Saito, H. Constitutive activation of mitogen-activated protein kinase pathway in acute leukemia cells. Leukemia 1997, 11, 479–484. [Google Scholar]
- Simon, M.; Grandage, V.L.; Linch, D.C.; Khwaja, A. Constitutive activation of the Wnt/beta-catenin signalling pathway in acute myeloid leukaemia. Oncogene 2005, 24, 2410–2420. [Google Scholar] [CrossRef]
- Casado, F.L.; Singh, K.P.; Gasiewicz, T.A. The aryl hydrocarbon receptor: Regulation of hematopoiesis and involvement in the progression of blood diseases. Blood Cells Mol. Dis. 2010, 44, 199–206. [Google Scholar] [CrossRef]
- Hayashibara, T.; Yamada, Y.; Mori, N.; Harasawa, H.; Sugahara, K.; Miyanishi, T.; Kamihira, S.; Tomonaga, M. Possible involvement of aryl hydrocarbon receptor (AhR) in adult T-cell leukemia (ATL) leukemogenesis: Constitutive activation of AhR in ATL. Biochem. Biophys. Res. Commun. 2003, 300, 128–134. [Google Scholar] [CrossRef]
- McHale, C.M.; Zhang, L.; Smith, M.T. Current understanding of the mechanism of benzene-induced leukemia in humans: Implications for risk assessment. Carcinogenesis 2012, 33, 240–252. [Google Scholar]
- Schwind, S.; Maharry, K.; Radmacher, M.D.; Mrózek, K.; Holland, K.B.; Margeson, D.; Whitman, S.P.; Hickey, C.; Becker, H.; Metzeler, K.H.; et al. Prognostic significance of expression of a single microRNA, miR-181a, in cytogenetically normal acute myeloid leukemia: A cancer and leukemia group B study. J. Clin. Oncol. 2010, 28, 5257–5264. [Google Scholar]
- Havelange, V.; Stauffer, N.; Heaphy, C.C.; Volinia, S.; Andreeff, M.; Marcucci, G.; Croce, C.M.; Garzon, R. Functional implications of microRNAs in acute myeloid leukemia by integrating microRNA and messenger RNA expression profiling. Cancer 2011, 117, 4696–4706. [Google Scholar]
- National Center for Biotechnology Information. Gene Expression Omnibus GSE15061. 2011. Available online: http://www.ncbi.nlm.nih.gov/geo/ (accessed on 19 December 2011).
- Mills, K.I.; Kohlmann, A.; Williams, P.M.; Wieczorek, L.; Liu, W.M.; Li, R.; Wei, W.; Bowen, D.T.; Loeffler, H.; Hernandez, J.M.; et al. Microarray-based classifiers and prognosis models identify subgroups with distinct clinical outcomes and high risk of AML transformation of myelodysplastic syndrome. Blood 2009, 114, 1063–1072. [Google Scholar]
- Haferlach, T.; Kohlmann, A.; Wieczorek, L.; Basso, G.; Kronnie, G.T.; Béné, M.C.; De Vos, J.; Hernández, J.M.; Hofmann, W.K.; Mills, K.I.; et al. Clinical utility of microarray-based gene expression profiling in the diagnosis and subclassification of leukemia: Report from the International Microarray Innovations in Leukemia Study Group. J. Clin. Oncol. 2010, 28, 2529–2537. [Google Scholar]
- Liu, W.M.; Li, R.; Sun, J.Z.; Wang, J.; Tsai, J.; Wen, W.; Kohlmann, A.; Williams, P.M. PQN and DQN: Algorithms for expression microarrays. J. Theor. Biol. 2006, 243, 273–278. [Google Scholar] [CrossRef]
- Partek Incorporated. Partek® Genomics SuiteTM Software. 2011. Available online: http://www.partek.com/partekgs (accessed on 19 December 2011).
- Storey, J.D. The positive false discovery rate: A Bayesian interpretation and the q-value. Ann. Stat. 2003, 31, 2013–2035. [Google Scholar] [CrossRef]
- Rager, J.E.; Lichtveld, K.; Ebersviller, S.; Smeester, L.; Jaspers, I.; Sexton, K.G.; Fry, R.C. A toxicogenomic comparison of primary and photochemically altered air pollutant mixtures. Environ. Health Perspect. 2011, 119, 1583–1589. [Google Scholar]
- Smeester, L.; Rager, J.E.; Bailey, K.A.; Guan, X.; Smith, N.; García-Vargas, G.; del Razo, L.M.; Drobná, Z.; Kelkar, H.; Stýblo, M.; et al. Epigenetic changes in individuals with arsenicosis. Chem. Res. Toxicol. 2011, 24, 165–167. [Google Scholar] [CrossRef]
- Bousquet, M.; Quelen, C.; Rosati, R.; Mansat-De Mas, V.; La Starza, R.; Bastard, C.; Lippert, E.; Talmant, P.; Lafage-Pochitaloff, M.; Leroux, D.; et al. Myeloid cell differentiation arrest by miR-125b-1 in myelodysplastic syndrome and acute myeloid leukemia with the t(2;11)(p21;q23) translocation. J. Exp. Med. 2008, 205, 2499–2506. [Google Scholar]
- Cammarata, G.; Augugliaro, L.; Salemi, D.; Agueli, C.; La Rosa, M.; Dagnino, L.; Civiletto, G.; Messana, F.; Marfia, A.; Bica, M.G.; et al. Differential expression of specific microRNA and their targets in acute myeloid leukemia. Am. J. Hematol. 2010, 85, 331–339. [Google Scholar]
- Garzon, R.; Volinia, S.; Liu, C.G.; Fernandez-Cymering, C.; Palumbo, T.; Pichiorri, F.; Fabbri, M.; Coombes, K.; Alder, H.; Nakamura, T.; et al. MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood 2008, 111, 3183–3189. [Google Scholar] [CrossRef]
- Han, Y.C.; Park, C.Y.; Bhagat, G.; Zhang, J.; Wang, Y.; Fan, J.B.; Liu, M.; Zou, Y.; Weissman, I.L.; Gu, H. microRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. J. Exp. Med. 2010, 207, 475–489. [Google Scholar]
- O’Connell, R.M.; Rao, D.S.; Chaudhuri, A.A.; Boldin, M.P.; Taganov, K.D.; Nicoll, J.; Paquette, R.L.; Baltimore, D. Sustained expression of microRNA-155 in hematopoietic stem cells causes a myeloproliferative disorder. J. Exp. Med. 2008, 205, 585–594. [Google Scholar]
- Wang, F.; Wang, X.S.; Yang, G.H.; Zhai, P.F.; Xiao, Z.; Xia, L.Y.; Chen, L.R.; Wang, Y.; Wang, X.Z.; Bi, L.X.; et al. miR-29a and miR-142-3p downregulation and diagnostic implication in human acute myeloid leukemia. Mol Biol Rep 2012, 39, 2713–2722. [Google Scholar]
- Whitehead Institute for Biomedical Research. TargetScanHuman Release 5.2. 2011. Available online: http://www.targetscan.org (accessed on 15 December 2011).
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Ingenuity Systems®. Ingenuity Pathway Analysis. 2012. Available online: http://www.ingenuity.com (accessed on 4 January 2012).
- Metzeler, K.H.; Hummel, M.; Bloomfield, C.D.; Spiekermann, K.; Braess, J.; Sauerland, M.C.; Heinecke, A.; Radmacher, M.; Marcucci, G.; Whitman, S.P.; et al. An 86-probe-set gene-expression signature predicts survival in cytogenetically normal acute myeloid leukemia. Blood 2008, 112, 4193–4201. [Google Scholar]
- Bousquet, M.; Harris, M.H.; Zhou, B.; Lodish, H.F. MicroRNA miR-125b causes leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 21558–21563. [Google Scholar]
- Shen, W.F.; Hu, Y.L.; Uttarwar, L.; Passegue, E.; Largman, C. MicroRNA-126 regulates HOXA9 by binding to the homeobox. Mol. Cell. Biol. 2008, 28, 4609–4619. [Google Scholar]
- Lv, M.; Zhang, X.; Jia, H.; Li, D.; Zhang, B.; Zhang, H.; Hong, M.; Jiang, T.; Jiang, Q.; Lu, J.; et al. An oncogenic role of miR-142-3p in human T-cell acute lymphoblastic leukemia (T-ALL) by targeting glucocorticoid receptor-α and cAMP/PKA pathways. Leukemia 2012, 26, 769–777. [Google Scholar] [CrossRef]
- Kuchenbauer, F.; Mah, S.M.; Heuser, M.; McPherson, A.; Rüschmann, J.; Rouhi, A.; Berg, T.; Bullinger, L.; Argiropoulos, B.; Morin, R.D.; et al. Comprehensive analysis of mammalian miRNA* species and their role in myeloid cells. Blood 2011, 118, 3350–3358. [Google Scholar]
- Garzon, R.; Heaphy, C.E.; Havelange, V.; Fabbri, M.; Volinia, S.; Tsao, T.; Zanesi, N.; Kornblau, S.M.; Marcucci, G.; Calin, G.A.; et al. MicroRNA 29b functions in acute myeloid leukemia. Blood 2009, 114, 5331–5341. [Google Scholar]
- Pulikkan, J.A.; Dengler, V.; Peramangalam, P.S.; Peer Zada, A.A.; Müller-Tidow, C.; Bohlander, S.K.; Tenen, D.G.; Behre, G. Cell-cycle regulator E2F1 and microRNA-223 comprise an autoregulatory negative feedback loop in acute myeloid leukemia. Blood 2010, 115, 1768–1778. [Google Scholar]
- Liu, S.; Wu, L.C.; Pang, J.; Santhanam, R.; Schwind, S.; Wu, Y.Z.; Hickey, C.J.; Yu, J.; Becker, H.; Maharry, K.; et al. Sp1/NFkappaB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell 2010, 17, 333–347. [Google Scholar] [CrossRef]
- Marcucci, G.; Radmacher, M.D.; Maharry, K.; Mrózek, K.; Ruppert, A.S.; Paschka, P.; Vukosavljevic, T.; Whitman, S.P.; Baldus, C.D.; Langer, C.; et al. MicroRNA expression in cytogenetically normal acute myeloid leukemia. N. Engl. J. Med. 2008, 358, 1919–1928. [Google Scholar]
- Barouki, R.; Coumoul, X.; Fernandez-Salguero, P.M. The aryl hydrocarbon receptor, more than a xenobiotic-interacting protein. FEBS Lett. 2007, 581, 3608–3615. [Google Scholar] [CrossRef]
- Mulero-Navarro, S.; Carvajal-Gonzalez, J.M.; Herranz, M.; Ballestar, E.; Fraga, M.F.; Ropero, S.; Esteller, M.; Fernandez-Salguero, P.M. The dioxin receptor is silenced by promoter hypermethylation in human acute lymphoblastic leukemia through inhibition of Sp1 binding. Carcinogenesis 2006, 27, 1099–1104. [Google Scholar]
Supplementary Files
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rager, J.E.; Fry, R.C. The Aryl Hydrocarbon Receptor Pathway: A Key Component of the microRNA-Mediated AML Signalisome. Int. J. Environ. Res. Public Health 2012, 9, 1939-1953. https://doi.org/10.3390/ijerph9051939
Rager JE, Fry RC. The Aryl Hydrocarbon Receptor Pathway: A Key Component of the microRNA-Mediated AML Signalisome. International Journal of Environmental Research and Public Health. 2012; 9(5):1939-1953. https://doi.org/10.3390/ijerph9051939
Chicago/Turabian StyleRager, Julia E., and Rebecca C. Fry. 2012. "The Aryl Hydrocarbon Receptor Pathway: A Key Component of the microRNA-Mediated AML Signalisome" International Journal of Environmental Research and Public Health 9, no. 5: 1939-1953. https://doi.org/10.3390/ijerph9051939