An Anthracene-Based Tripodal Chemosensor for Anion Sensing

Abstract
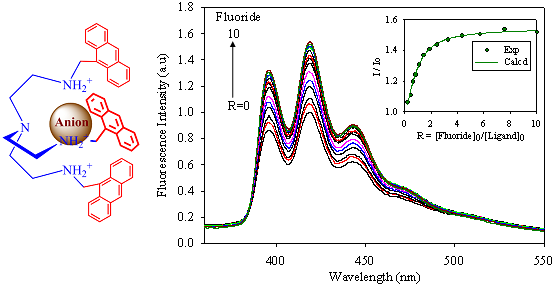
:
1. Introduction
2. Results and Discussion
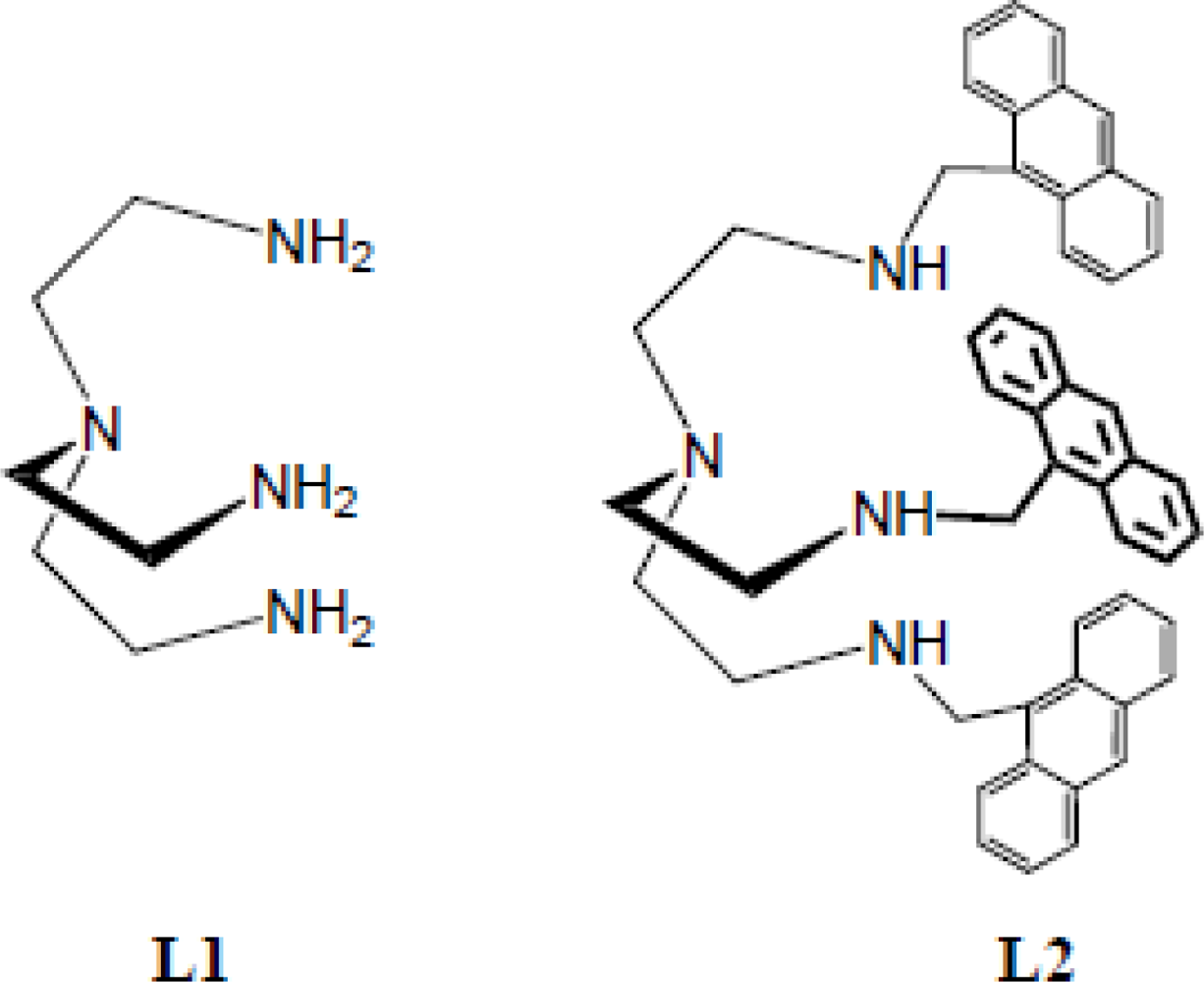
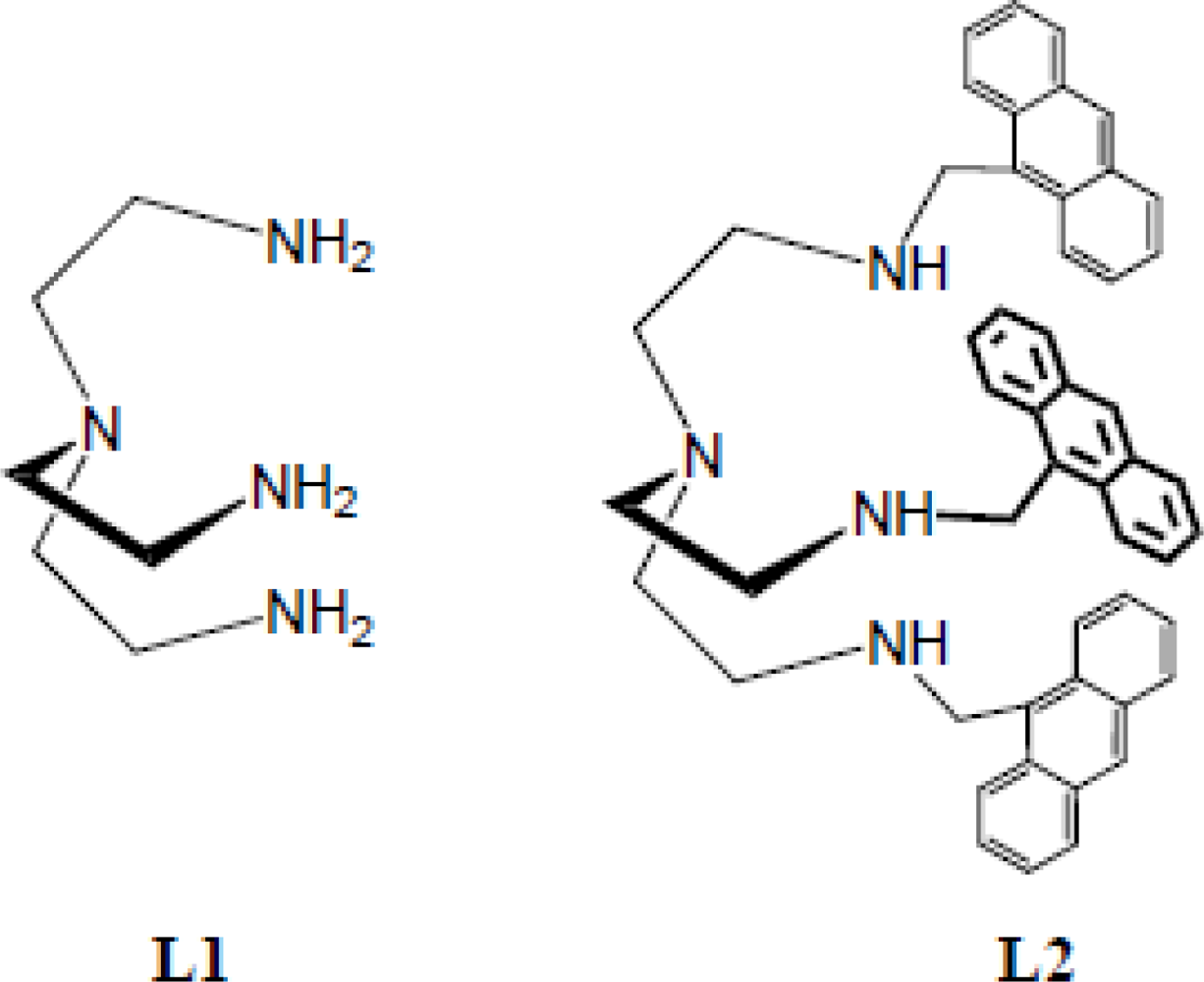
2.1. Synthesis
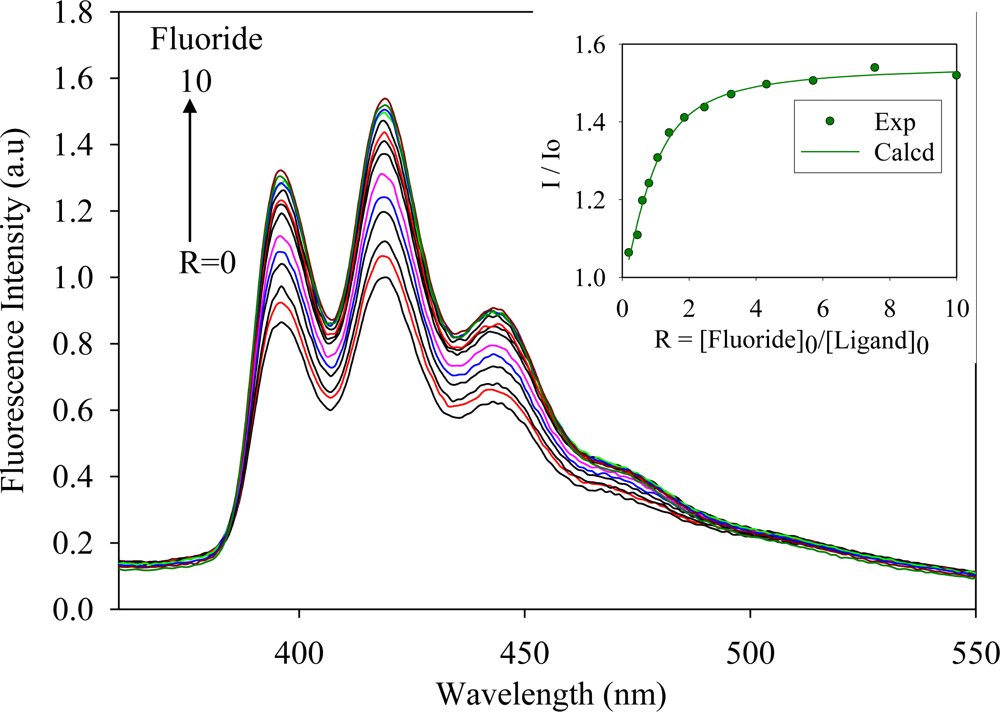
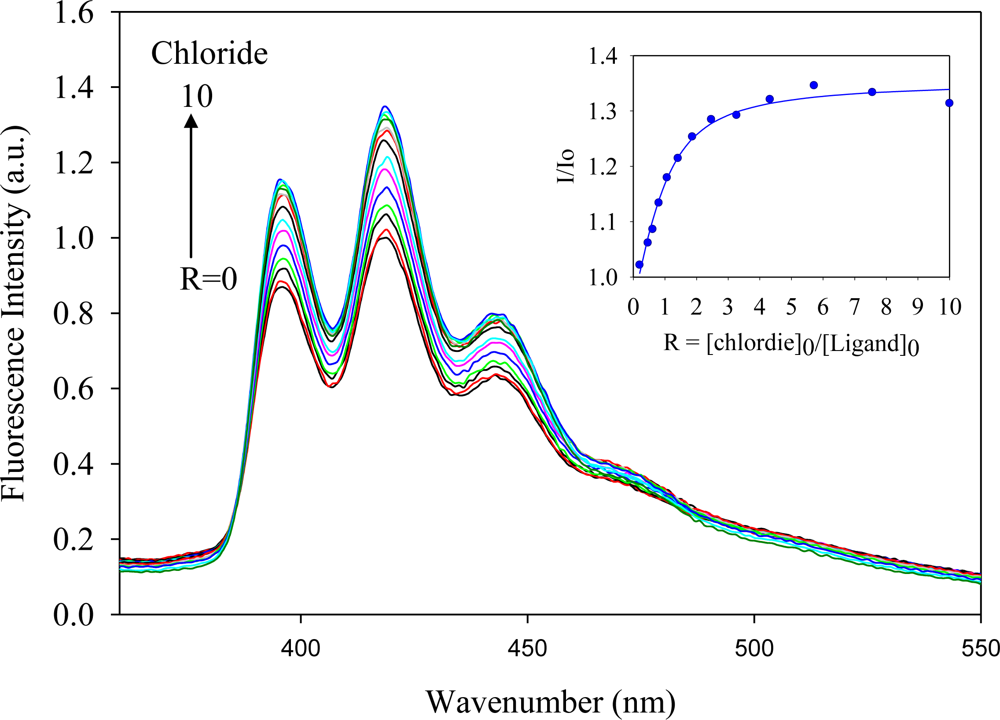
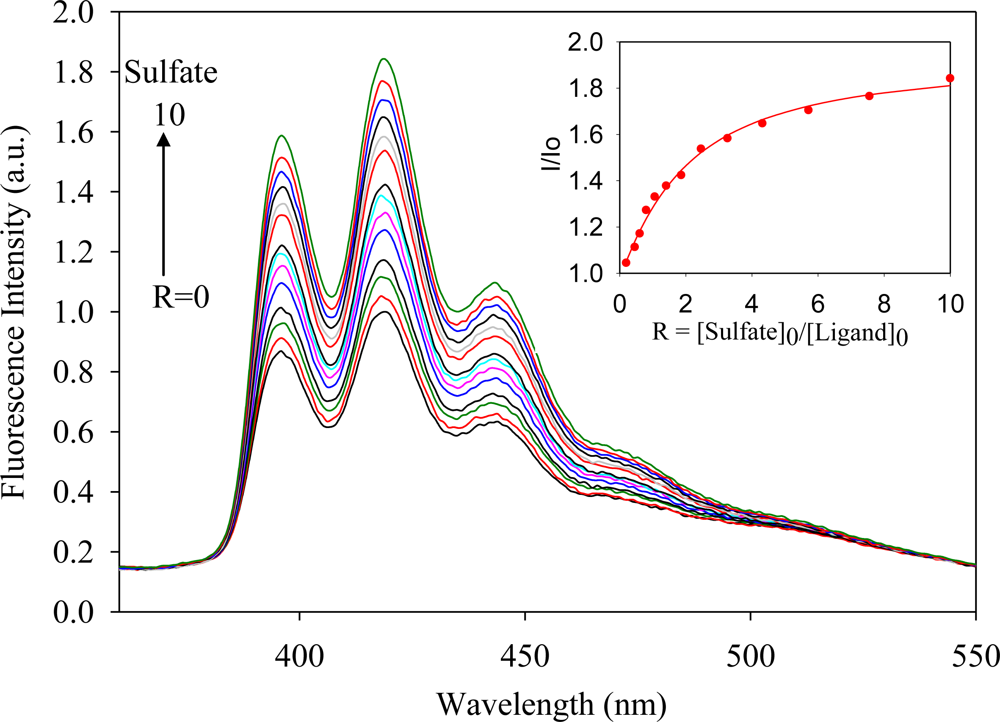
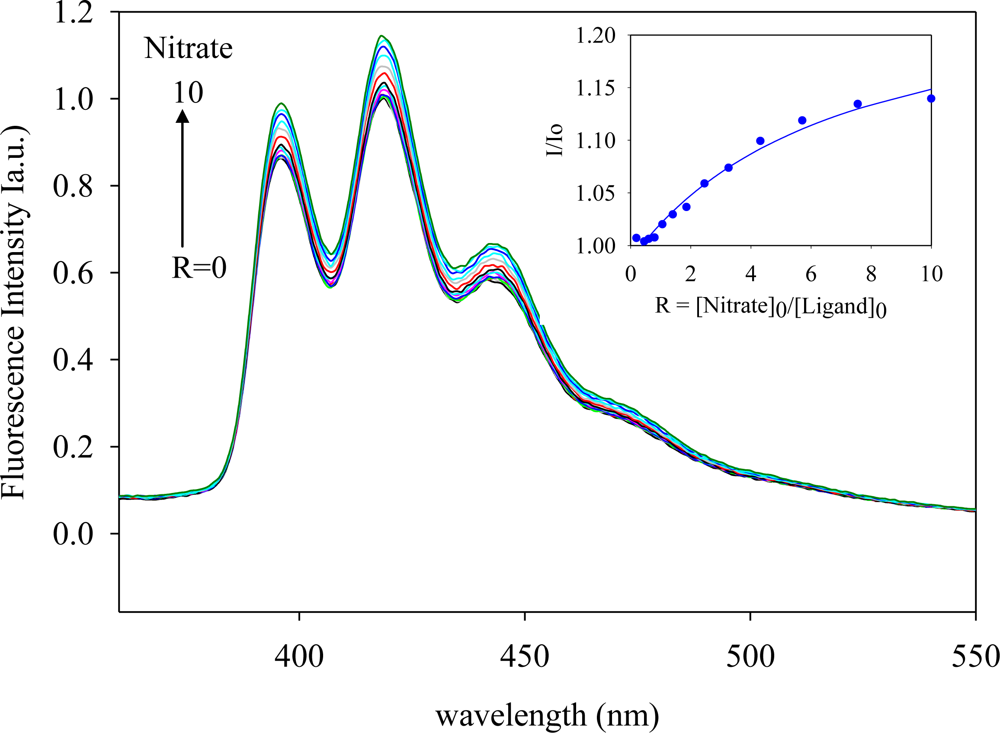
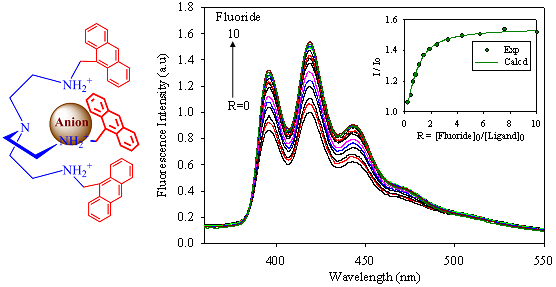
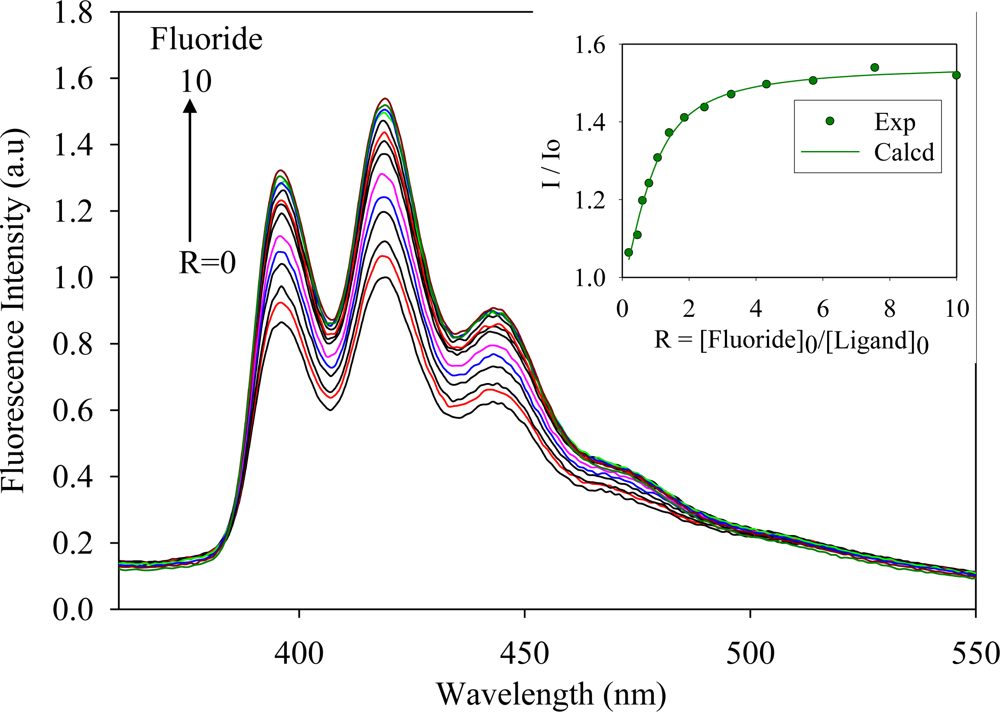
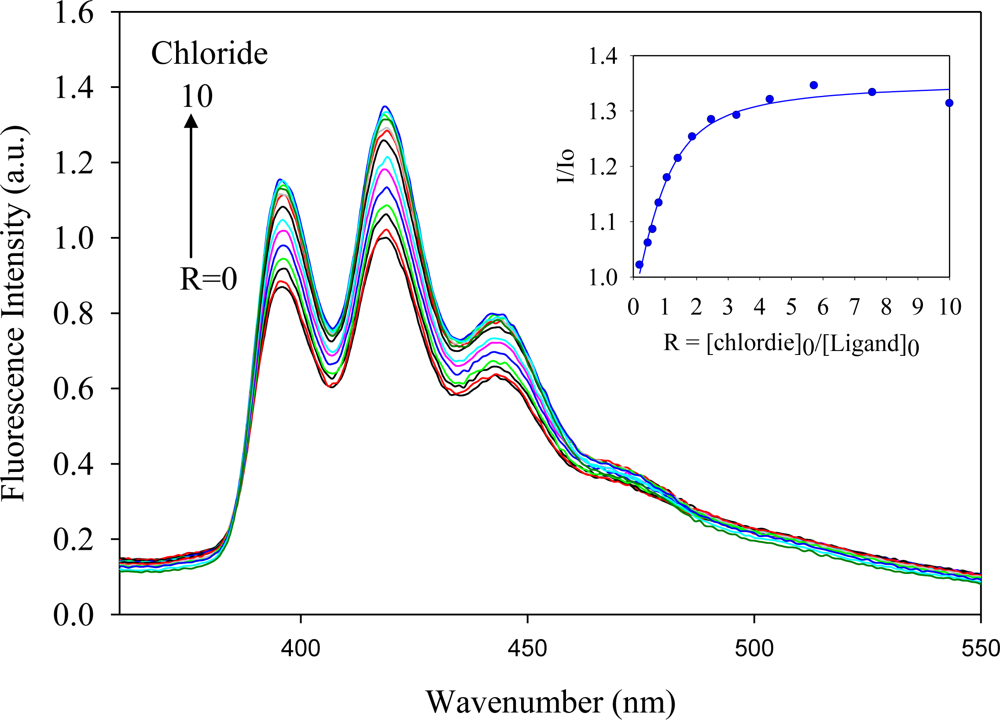
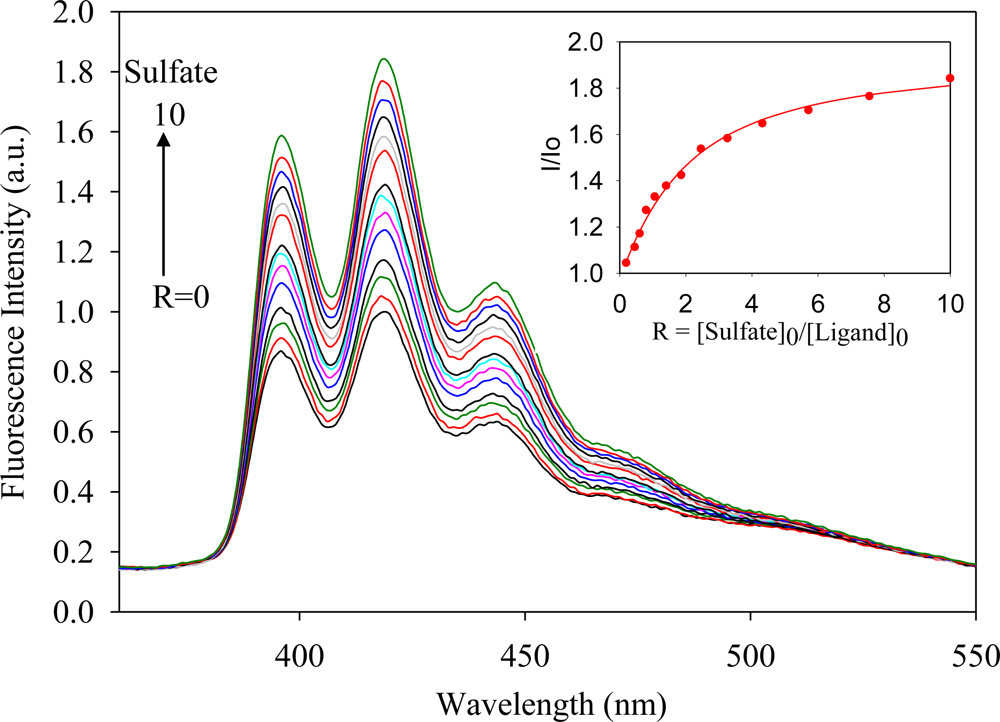
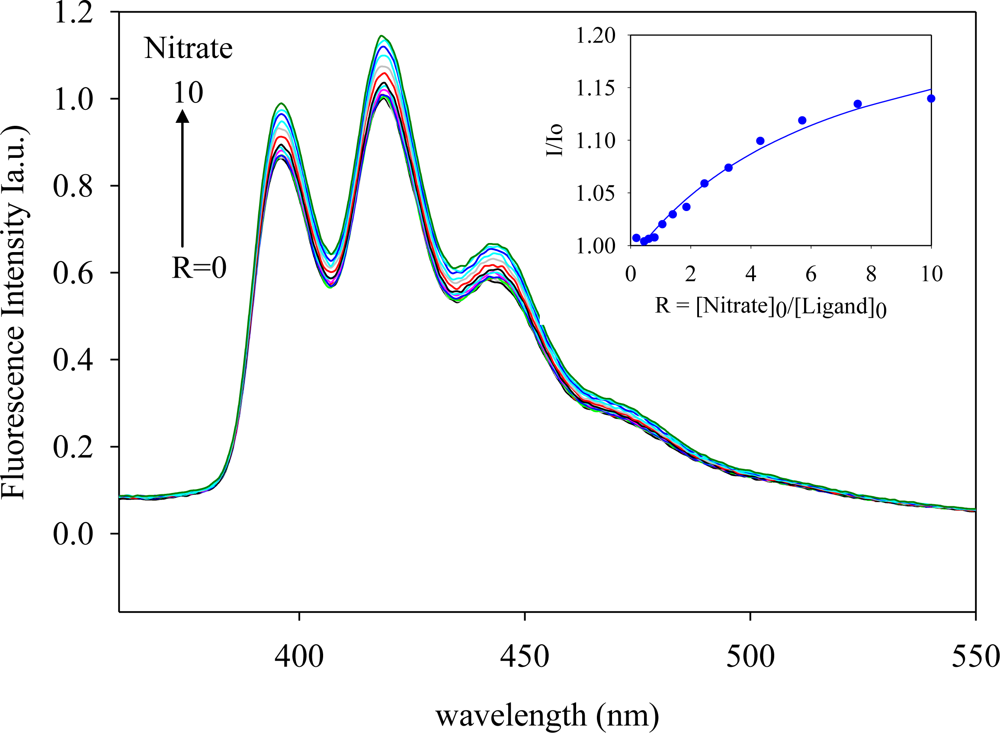
2.2. Fluorescence Studies
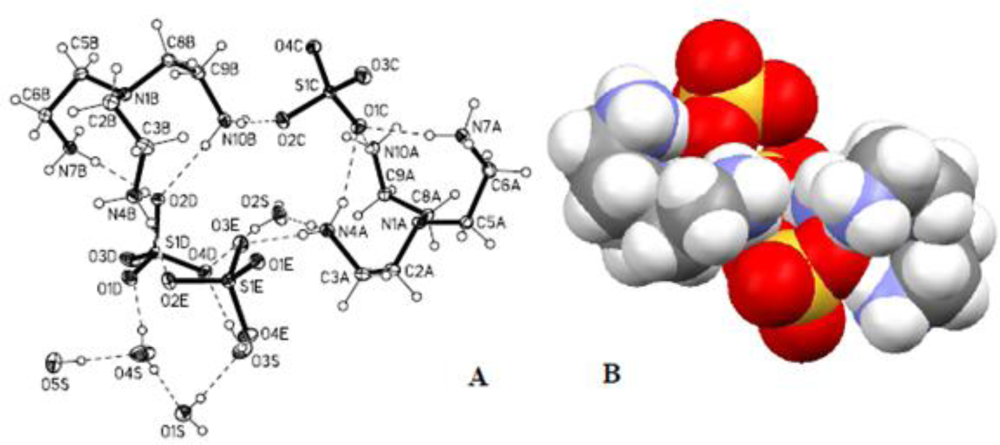
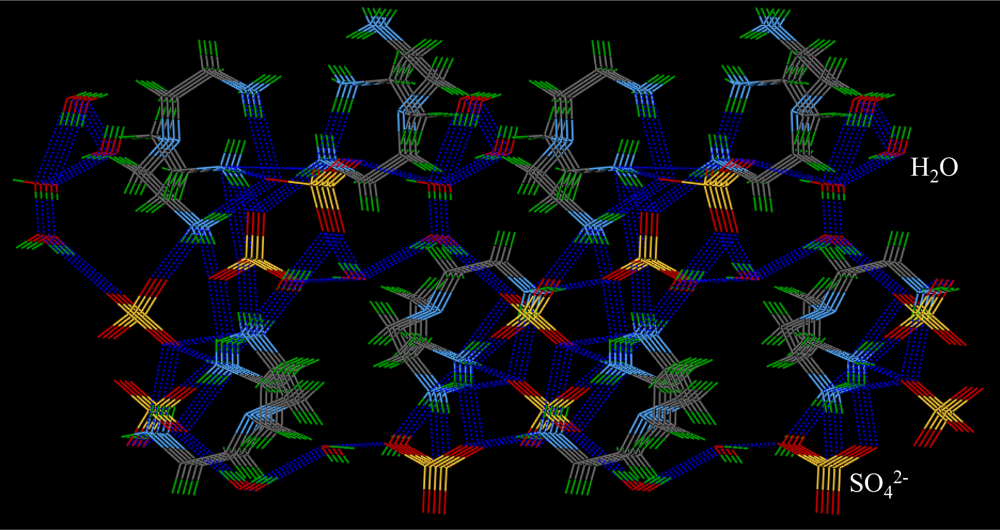
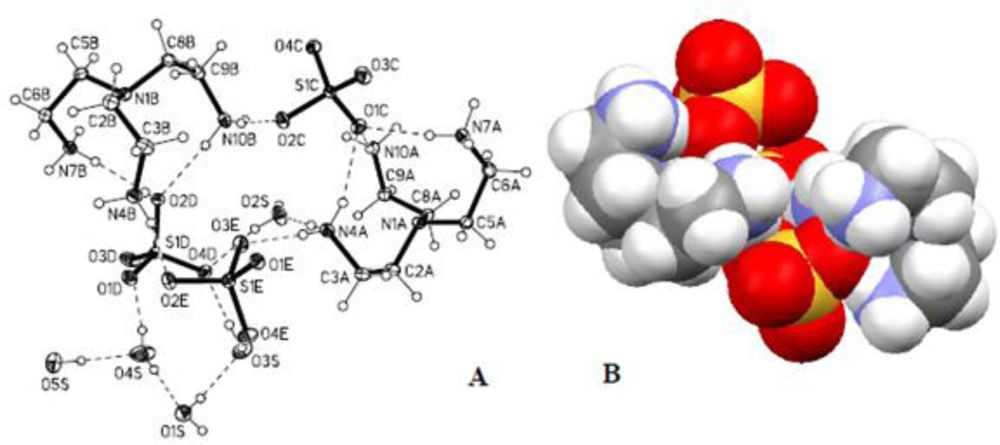
2.3. Crystallographic Studies
3. Experimental Section
3.1. General
3.2. Synthesis
3.3. X-ray Crystallography:
3.4. Fluorescence Studies
4. Conclusions
Acknowledgments
References
- Lehn, J-M. Supramolecular Chemistry: Concepts and Perspectives; VCH: Weinheim, Germany, 1995. [Google Scholar]
- Supramolecular Chemistry of Anions; Bianchi, A; Bowman-James, K; García-España, E (Eds.) Wiley-VCH: New York, NY, USA, 1997.
- Hirsch, AKH; Fischer, FR; Diederich, F. Angew. Chem. Int. Ed 2007, 46, 338–352.
- National Academy of Sciences. The Health Effects of Nitrate, Nitrite and N-Nitroso Compounds; National Academy Press: Washington, DC, USA, 1981. [Google Scholar]
- Scragg, RK. Birth defects and Household water supply. Med. J. Aust 1982, 2, 577–579. [Google Scholar]
- Morton, WE. Hypertension and drinking constituents in Colorado. Am. J. Pubi. Hlth 1971, 61, 1371–1378. [Google Scholar]
- Van-Maanen, JM; Van- Dijk, A; Mulder, K; De-Baets, MH; Kleinjans, JC. Consumption of drinking water with high nitrate levels cause hypertrophy of the thyroid. Toxicol. Lett 1994, 72, 365–374. [Google Scholar]
- Waldron, TT; Modestou, MA; Murphy, KP. Anion binding to a protein-protein complex lacks dependence on net charge. Protein Sci 2003, 12, 871–874. [Google Scholar]
- Hossain, MA. Inclusion complexes of halide anions with macrocyclic receptors. Curr. Org. Chem 2008, 12, 1231–1256. [Google Scholar]
- Jahansouz, H; Jiang, Z; Himes, RH; Mertes, MP; Bowman Mertes, K. Formate activation in neutral aqueous solution mediated by a polyammonium macrocycle. J. Am. Chem. Soc 1989, 111, 1409–1413. [Google Scholar]
- Boudon, S; Decian, A; Fischer, J; Hosseini, MW; Lehn, J-M; Wipff, G. Structural and anion coordination features of macrocyclic polyammonium cations in the solid, solution, and computational phases. J. Coord. Chem 1991, 23, 113–123. [Google Scholar]
- Morgan, G; McKee, V; Nelson, J. Caged anions: perchlorate and perfluoroanion cryptates. J Chem Soc Chem Commun 1995, 1649–1652. [Google Scholar]
- Ichikawa, K; Hossain, MA. Structure of encapsulated chloride anion with the preorganization of a macrotricyclic ammonium cage host. J Chem Soc Chem Commun 1996, 1721–1722. [Google Scholar]
- Mason, S; Seib, L; Bowman-James, K. Unusual encapsulation of two nitrates in a single bicyclic cage. J. Am. Chem. Soc 1998, 120, 8899–8900. [Google Scholar]
- Hossain, MA; Llinares, JM; Miller, C; Seib, L; Bowman-James, K. Further insight to selectivity issues in halide binding in the tiny octaaza cryptand. Chem Commun 2000, 2269–2270. [Google Scholar]
- Hossain, MA; Llinares, J; Mason, MS; Morehouse, P; Powell, D; Bowman-James, K. Parallels in cation and anion coordination: A new class of cascade complexes. Angew. Chem. Int. Ed 2002, 41, 2335–2338. [Google Scholar]
- Bondy, CR; Loeb, SJ. Amide based receptors for anions. Coord. Chem. Rev 2003, 240, 77–99. [Google Scholar]
- Sessler, JL; Pantos, GD; Gale, PA; Light, ME. Synthesis and anion binding properties of N,N′-bispyrrol-2-yl-2,5-diamidopyrrole. Org. Lett 2006, 8, 1593–159. [Google Scholar]
- Gale, PA; García-Garrido, SE; Garric, J. Anion receptors based on organic frameworks: highlights from 2005 and 2006. Chem. Soc. Rev 2008, 37, 151–190. [Google Scholar]
- Saeed, MA; Fronczek, FR; Hossain, MA. Encapsulated chloride coordinating with two in-in protons of bridgehead amines in an octaprotonated azacryptand. Chem Commun 2009, 6409–6411. [Google Scholar]
- Gibson, D; Dey, KR; Fronczek, FR; Hossain, MA. A new hexaaminomacrocycle for ditopic binding of bromide. Tetrahedron Letters 2009, 50, 6537–6539. [Google Scholar]
- Kang, SO; Day, VW; Bowman-James, K. Fluoride: solution- and solid-state structural binding probe. J. Org. Chem 2010, 75, 277–283. [Google Scholar]
- Dey, KR; Wong, BW; Hossain, MA. Rational design of a macrocycle-based chemosensor for anions. Tetrahedron Letters 2010, 23, 1329–1332. [Google Scholar]
- Saeed, MA; Fronczek, FR; Huang, MJ; Hossain, MA. Unusual bridging of three nitrates with two bridgehead protons in an octaprotonated azacryptand. Chem. Commun 2010, 46, 404–406. [Google Scholar]
- Saeed, MA; Thompson, JJ; Fronczek, FR; Hossain, MA. Ditopic binding of perchlorate anion to hexaazamacrocyclic hosts. CrystEngComm 2010, 12, 674–676. [Google Scholar]
- Gale, PA. Structural and molecular recognition studies with acyclic anion receptors. Acc. Chem. Res 2006, 39, 465–475. [Google Scholar]
- Valiyaveettil, SK; Engbersen, JFJ; Verboom, W; Reinhoudt, DN. Synthesis and complexation studies of neutral anion receptors. Angew. Chem. Int. Ed. Engl 1993, 32, 900–901. [Google Scholar]
- Ravikumar, I; Lakshminarayanan, PS; Arunachalam, M; Suresh, E; Ghosh, P. Anion complexation of a pentafluorophenyl-substituted tripodal urea receptor in solution and the solid state: selectivity toward phosphate. Dalton Trans 2009, 4160–4168. [Google Scholar]
- Lakshminarayanan, PS; Ravikumar, I; Suresh, E; Ghosh, P. Trapped inorganic phosphate dimmer. Chem Commun 2007, 5214–5216. [Google Scholar]
- Hossain, MA; Liljegren, JA; Powell, D; Bowman-James, K. Anion binding with a tripodal amine. Inorg. Chem 2004, 43, 3751–3755. [Google Scholar]
- Shao, J; Qiao, Y; Lin, H; Lin, H. Rational design of novel benzimidazole-based sensor molecules that display positive and negative fluorescence responses to anions. J. Fluoresc 2009, 19, 183–188. [Google Scholar]
- Schneider, H; Kramer, R; Simova, S; Schneider, U. Host-guest chemistry. 14. Solvent and salt effects on binding constants of organic substrates in macrocyclic host compounds. A general equation measuring hydrophobic binding contributions. J. Am. Chem. Soc 1988, 110, 6442–6448. [Google Scholar]
- Bazzicalupi, C; Bencini, A; Bianchi, A; Danesi, A; Giorgi, C; Valtancoli, B. Anion binding by protonated forms of the tripodal ligand tren. Inorg. Chem 2009, 48, 2391–2398. [Google Scholar]
- Collins, KD; Washabaugh, MW. The Hofmeister effect and the behaviour of water at interfaces. Q. Rev. Biophys 1985, 18, 323–422. [Google Scholar]
- Ilioudis, CA; Georganopoulou, DG; Steed, JW. Insights into supramolecular design 2: Analysis of anion coordination geometry of oxoanions in a protonatedpolyamine matrix. CrystEngComm 2002, 4, 26–36. [Google Scholar]
- Kang, SO; Hossain, MA; Powell, D; Bowman-James, K. Sulphate encapsulated in two cryptands: Insight to binding aspects. Chem Comm 2005, 328–330. [Google Scholar]
- (a) Data Collection: SMART Software Reference Manual. 1998, Bruker-AXS, 5465 E. Cheryl Parkway, Madison, WI 53711-5373, USA. (b) Data Reduction: SAINT Software Reference Manual, 1998. Bruker-AXS, 5465 E. Cheryl Parkway, Madison, WI 53711-5373, USA.
- Sheldrick, GM. SADABS. Program for Empirical Absorption Correction of Area Detector Data; University of Göttingen: Göttingen, Germany, 2007. [Google Scholar]
- Sheldrick, GM. Acta Cryst 2008, A64, 112–122.(b) International Tables for Crystallography; , Vol C, Tables 6114, 4268, and 4242, Kluwer: Boston, MA, USA, 1995.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Anion | Log Ka | −ΔG/Kcal mol−1 |
|---|---|---|
| Fluoride | 5.8 | 7.9 |
| Chloride | 5.5 | 7.5 |
| Bromide | 3.8 | 5.2 |
| Sulfate | 4.8 | 6.5 |
| Phosphate | 4.6 | 6.3 |
| Nitrate | 3.9 | 5.3 |
| Complex | 2(C6 H21 N)3+ 3(SO4)2−·4.5(H2O) |
|---|---|
| Empirical formula | C12 H51 N8 O16.5 S3 |
| Formula weight | 667.79 |
| Crystal size (mm3) | 0.50 × 0.21 × 0.11 |
| Crystal system | Triclinic |
| Space group | P1̅ |
| a, Å | 8.6555(16) |
| b, Å | 12.247(2) |
| c, Å | 14.919(3) |
| α, deg | 75.836(5) |
| β, deg | 82.716(5) |
| γ, deg | 76.736(6) |
| Volume (Å3) | 1,488.3(5) |
| Z, Z' | 2, 1 |
| dcalc g cm−3 | 1.490 |
| λ (Å) | 0.71073 |
| T (K) | 100(2) |
| F(000) | 718 |
| abs coeff (mm−1) | 0.330 |
| Absorption correction | Semi-empirical from equivalents |
| Max. and min. transmission | 0.967 and 0.850 |
| θ range (°) | 1.75 to 28.35 |
| Reflections collected | 20,882 |
| Independent reflections | 7,427 |
| R(int) | 0.0312 |
| wR(F2 all data) | wR2 = 0.0896 |
| R(F obsd data) | R1 = 0.0322 |
| Goodness-of-fit on F2 | 1.010 |
| Observed data [I > 2σ(I)] | 6,666 |
| Largest and mean shift / s.u. | 0.001 and 0.000 |
| Δρmax , Δρmin (e Å−3) | 0.380 and −0.456 |
| D-H...A | d(D-H) | d(H...A) | d(D...A) | <(DHA) |
|---|---|---|---|---|
| N(4A)-H(4AA)...O(2S) | 0.898(18) | 1.930(18) | 2.8277(17) | 179.0(17) |
| N(4A)-H(4AB)...O(3E) | 0.925(18) | 1.876(18) | 2.7840(16) | 166.4(16) |
| N(4A)-H(4AC)...O(1C) | 0.848(19) | 2.121(19) | 2.8920(16) | 151.0(16) |
| N(4A)-H(4AC)...O(3C)i | 0.848(19) | 2.583(17) | 3.0014(16) | 111.7(14) |
| N(4A)-H(4AC)...O(3C) i | 0.848(19) | 2.583(17) | 3.0014(16) | 111.7(14) |
| N(7A)-H(7AA)...O(1E) i | 0.898(18) | 1.959(18) | 2.8559(15) | 175.9(16) |
| N(7A)-H(7AB)...O(1C) | 0.874(18) | 1.925(19) | 2.7957(16) | 174.1(16) |
| N(7A)-H(7AC)...O(2C) i | 0.889(18) | 1.911(18) | 2.7981(16) | 175.6(16) |
| N(10A)-H(10A)...O(1C) | 0.847(18) | 2.008(18) | 2.8238(15) | 161.5(16) |
| N(10A)-H(10B)...O(1E) ii | 0.906(18) | 1.926(18) | 2.7961(16) | 160.4(15) |
| N(10A)-H(10C)...O(4C) iii | 0.896(18) | 1.904(19) | 2.7862(15) | 167.9(16) |
| N(4B)-H(4BA)...O(1D) iv | 0.914(19) | 1.838(19) | 2.7463(16) | 172.2(16) |
| N(4B)-H(4BB)...O(2D) | 0.878(19) | 2.022(19) | 2.8838(17) | 166.9(16) |
| N(4B)-H(4BC)...O(2E) | 0.882(19) | 2.056(19) | 2.8821(16) | 155.5(16) |
| N(4B)-H(4BC)...O(3E) | 0.882(19) | 2.330(18) | 2.9970(17) | 132.5(15) |
| N(7B)-H(7BA)...O(2D) | 0.860(18) | 2.029(19) | 2.8748(16) | 167.5(16) |
| N(7B)-H(7BB)...O(3D) v | 0.911(18) | 1.911(19) | 2.8006(16) | 165.0(16) |
| N(7B)-H(7BC)...O(2E) iv | 0.886(18) | 1.891(19) | 2.7748(15) | 175.1(16) |
| N(10B)-H(10D)...O(2C) | 0.898(18) | 1.964(19) | 2.8569(16) | 172.3(16) |
| N(10B)-H(10E)...O(2D) | 0.872(18) | 1.981(19) | 2.8494(15) | 174.0(16) |
| N(10B)-H(10F)...O(1E) ii | 0.899(18) | 1.981(18) | 2.8686(15) | 169.0(16) |
| N(10B)-H(10F)...O(2E) ii | 0.899(18) | 2.612(17) | 3.1698(16) | 121.0(13) |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Quinn, W.A.; Saeed, M.A.; Powell, D.R.; Hossain, M.A. An Anthracene-Based Tripodal Chemosensor for Anion Sensing. Int. J. Environ. Res. Public Health 2010, 7, 2057-2070. https://doi.org/10.3390/ijerph7052057
Quinn WA, Saeed MA, Powell DR, Hossain MA. An Anthracene-Based Tripodal Chemosensor for Anion Sensing. International Journal of Environmental Research and Public Health. 2010; 7(5):2057-2070. https://doi.org/10.3390/ijerph7052057
Chicago/Turabian StyleQuinn, Whitney A., Musabbir A. Saeed, Douglas R. Powell, and Md. Alamgir Hossain. 2010. "An Anthracene-Based Tripodal Chemosensor for Anion Sensing" International Journal of Environmental Research and Public Health 7, no. 5: 2057-2070. https://doi.org/10.3390/ijerph7052057