Thimerosal Exposure and the Role of Sulfation Chemistry and Thiol Availability in Autism

{kind=link}
Abstract
:1. Introduction
2. Research Evidence
2.1. Use of TM in Vaccines
2.2. TM as a Toxin
2.3. TM Decreases GSH and Thiol Levels in General
2.4. TM Effects and the Importance of Thiols
2.5. Evidence of Abnormal Sulfation Chemistry in Autism
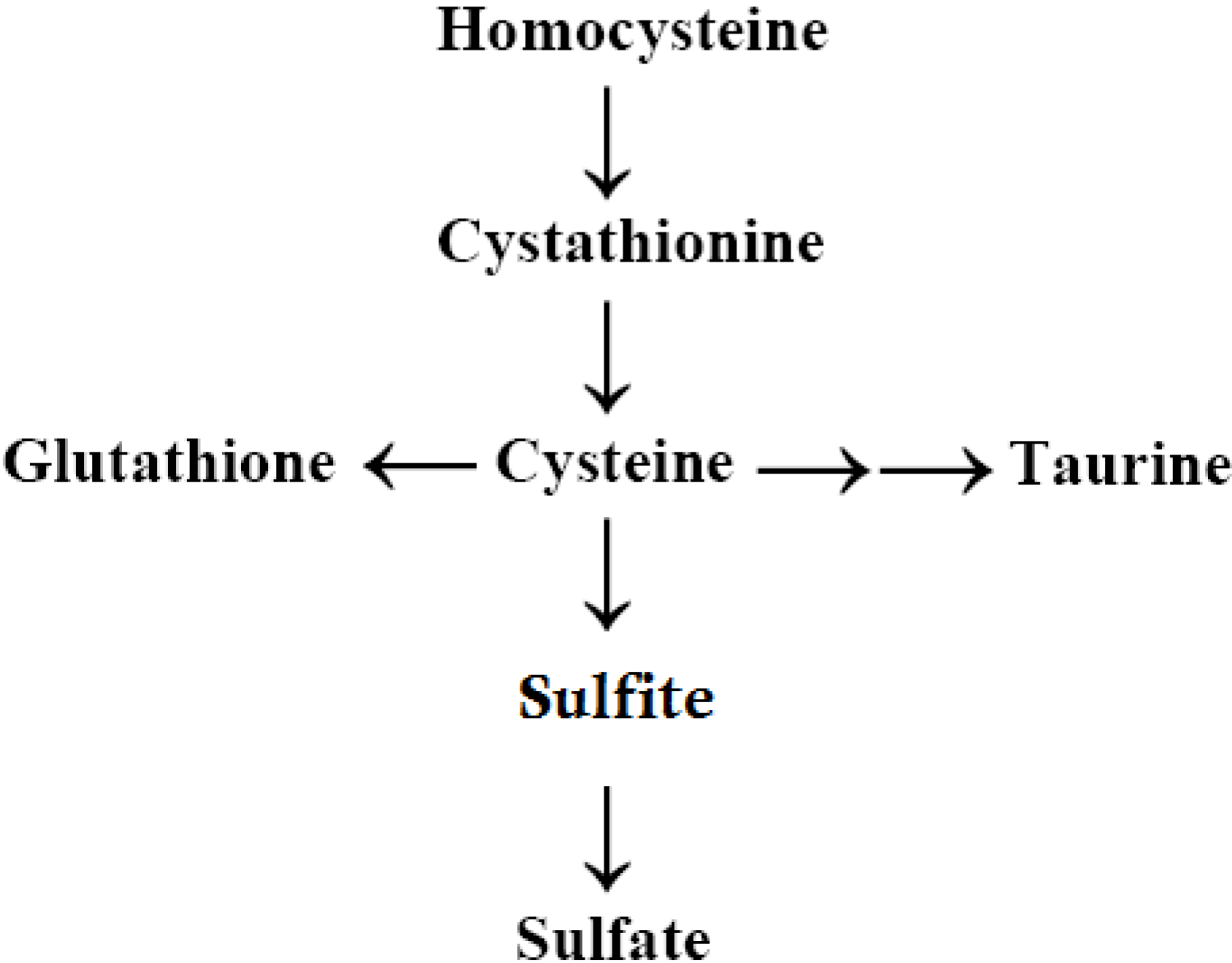
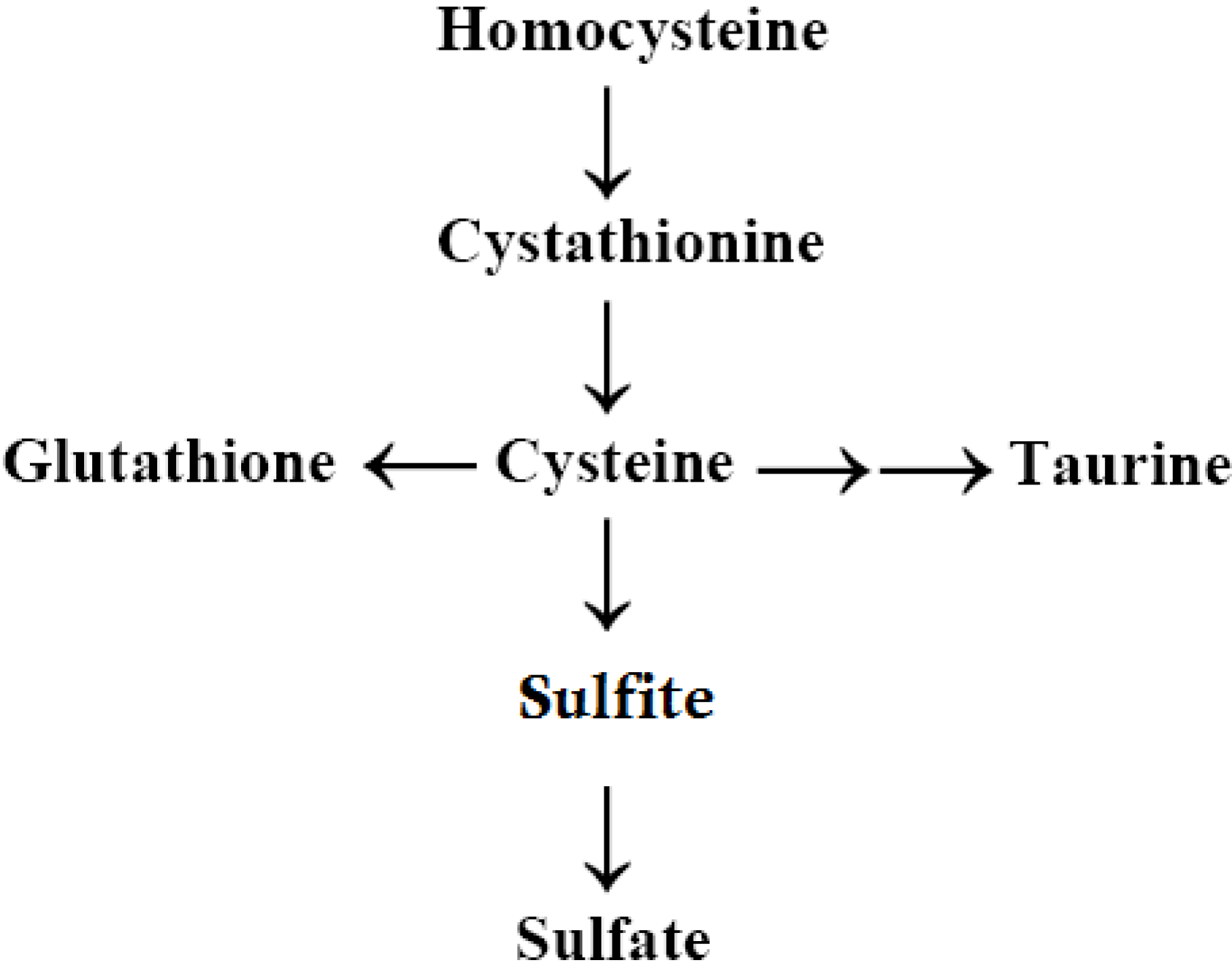
2.6. Autism, the Transsulfuration Pathway, and the Availability of Thiols
2.7. Availability of GSH in the Brain of Those with Autism
2.8. GSH: Complex Synthesis Process and the Potential for Excessive Demand
2.9. Direct Evidence of Decreased GSH Reserve Capacity and Increased Susceptibility in Autism
2.10. TM as a Source of Hg Exposure in Infants and Children
2.11. Neurotoxic Differences between Me- and Et-Hg
2.12. TM and Neurodevelopmental Periods
2.13. Hg and the Brain Pathology in Autism
2.14. TM as a Risk Factor in Other Developmental Issues
3. Conclusions
Conflicts of Interest
References
- American Psychiatric Association, Diagnostic Criteria for Autistic Disorder. In Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; American Psychiatric Association: Washington, DC, USA, 2013.
- Schieve, L.A.; Gonzalez, V.; Boulet, S.L.; Visser, S.N.; Rice, C.E.; van Naarden, B.K.; Boyle, C.A. Concurrent medical conditions and health care use and needs among children with learning and behavioral developmental disabilities, National Health Interview Survey, 2006–2010. Res. Dev. Disabil. 2011, 33, 467–476. [Google Scholar]
- Atladóttir, H.O.; Thorsen, P.; Schendel, D.E.; Østergaard, L.; pLemcke, S.; Parner, E.T. Association of hospitalization for infection in childhood with diagnosis of autism spectrum disorders, a Danish cohort study. Arch. Pediatr. Adolesc. Med. 2010, 164, 470–477. [Google Scholar] [CrossRef]
- Geier, D.A.; Kern, J.K.; Geier, M.R. A prospective cross-sectional cohort assessment of health, physical, and behavioral problems in autism spectrum disorders. Maedica (Buchar) 2012, 7, 193–200. [Google Scholar]
- Danielsson, S.; Gillberg, I.C.; Billstedt, E.; Gillberg, C.; Olsson, I. Epilepsy in young adults with autism: A prospective population-based follow-up study of 120 individuals diagnosed in childhood. Epilepsia 2005, 46, 918–923. [Google Scholar] [CrossRef]
- Hyman, S.E. Grouping diagnoses of mental disorders by their common risk factors. Am. J. Psychiatry 2011, 168, 1–3. [Google Scholar] [CrossRef]
- Stefanatos, G.A. Regression in autistic spectrum disorders. Neuropsych. Rev. 2008, 18, 305–319. [Google Scholar] [CrossRef]
- Ozonoff, S.; Williams, B.J.; Landa, R. Parental report of the early development of children with regressive autism, the delays-plus-regression phenotype. Autism 2005, 9, 461–486. [Google Scholar] [CrossRef]
- Filipek, P.A.; Accardo, P.J.; Baranek, G.T.; Cook, E.H., Jr.; Dawson, G.; Gordon, B.; Gravel, J.S.; Johnson, C.P.; Kallen, R.J.; Levy, S.E.; et al. The screening and diagnosis of autistic spectrum disorders. J. Autism Dev. Disord. 1999, 29, 439–484. [Google Scholar] [CrossRef]
- Davidovitch, M.; Glick, L.; Holtzman, G.; Tirosh, E.; Safir, M.P. Developmental regression in autism, maternal perception. J. Autism Dev. Disord. 2000, 30, 11311–11319. [Google Scholar]
- Tuchman, R. Pervasive developmental disorder, neurologic perspective. Acta Neuropediat. 1996, 2, 82–93. [Google Scholar]
- Kern, J.K.; Miller, V.S.; Evans, P.A.; Trivedi, M.H. Efficacy of porcine secretin in children with autism and pervasive developmental disorders. J. Autism Dev. Disord. 2002, 32, 153–160. [Google Scholar] [CrossRef]
- Goldberg, W.A.; Osann, K.; Filipek, P.A.; Laulhere, T.; Jarvis, K.; Modahl, C.; Flodman, P.; Spence, M.A. Language and other regression, assessment and timing. J. Autism Dev. Disord. 2003, 33, 607–616. [Google Scholar] [CrossRef]
- Malhi, P.; Singhi, P. Regression in children with autism spectrum disorders. Indian J. Pediatr. 2012, 27, 975–981. [Google Scholar]
- Ji, L.; Chauhan, V.; Flory, M.J.; Chauhan, A. Brain region-specific decrease in the activity and expression of protein kinase a in the frontal cortex of regressive autism. PLoS One 2011, 6, 23751. [Google Scholar] [CrossRef]
- Hansen, R.L.; Ozonoff, S.; Krakowiak, P.; Angkustsiri, K.; Jones, C.; Deprey, L.J.; Le, D.N.; Croen, L.A.; Hertz-Picciotto, I. Regression in autism, prevalence and associated factors in the CHARGE Study. Pediatrics 2008, 8, 25–31. [Google Scholar]
- Zappella, M. Autisticregression with and without EEG abnormalities followed by favourable outcome. Brain Dev. 2010, 32, 739–745. [Google Scholar] [CrossRef]
- Werner, E.; Dawson, G. Validation of the phenomenon of autistic regression using home videotapes. Arch. Gen. Psychiatry 2005, 62, 889–895. [Google Scholar] [CrossRef]
- Ozonoff, S.; Iosif, A.M.; Baguio, F.; Cook, I.C.; Hill, M.M.; Hutman, T.; Rogers, S.J.; Rozga, A.; Sangha, S.; Sigman, M.; Steinfeld, M.B.; Young, G.S. A prospective study of the emergence of early behavioral signs of autism. J. Am. Acad. Child Adolesc. Psychiatry 2010, 49, 256–266. [Google Scholar]
- Kern, J.K.; Waring, R.H.; Ramsden, D.B.; Grannemann, B.D.; Garver, C.R.; Trivedi, M.H. Abnormal Sulfation Chemistry in Autism. In Progress in Autism Research; Columbus, F., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2004. [Google Scholar]
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar]
- James, S.J.; Melnyk, S.; Jernigan, S.; Cleves, M.A.; Halsted, C.H.; Wong, D.H.; Cutler, P.; Bock, K.; Boris, M.; Bradstreet, J.J.; et al. Metabolic endophenotype and related genotypes are associated with oxidative stress in children with autism. A. J. Med. Genet. B Neuropsychiatr. Genet. 2006, 141B, 947–956. [Google Scholar] [CrossRef]
- James, S.J.; Rose, S.; Melnyk, S.; Jernigan, S.; Blossom, S.; Pavliv, O.; Gaylor, D.W. Cellular and mitochondrial glutathione redoximbalance in lymphoblastoid cells derived from children with autism. FASEB J. 2009, 23, 2374–2383. [Google Scholar] [CrossRef]
- Food and Drug Administration. Available online: http://www.fda.gov/BiologicsBloodVaccines/DevelopmentApprovalProcess/BiologicalApprovalsbyYear/default.htm (accessed on 14 June 2013).
- Centers for Disease Control. Prevention and Control of Influenza: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2002, 51, (RR-03). 1–31.
- Centers for Disease Control. Prevention and Control of Influenza: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2004, 53, (RR-06). 1–40.
- Centers for Disease Control. Prevention and Control of Influenza: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2006, 55, (RR10). 1–42.
- Centers for Disease Control. Prevention and Control of Seasonal Influenza with Vaccines Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 2009, 58, (RR08). 1–52.
- John Hopkin’s School of Public Health. Thimerosal Content in Some US Licensed Vaccines, 2012. Available online: http://www.vaccinesafety.edu/thi-table.htm (accessed on 7 March 2013).
- Centers for Disease Control. Prevention and Control of Influenza: Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR 1997, 46, (RR-9). 1–25.
- Goldman, G. Comparison of VAERS fetal-loss reports during three consecutive influenza seasons, was there a synergistic fetal toxicity associated with the two-vaccine 2009/2010 season? Hum. Exp. Toxicol. 2013, 32, 464–475. [Google Scholar] [CrossRef]
- Brown, I.A.; Austin, D.W. Maternal transfer of mercury to the developing embryo/fetus: Is there a safe level? Toxicol. Environ. Chem. 2012, 94, 1610–1627. [Google Scholar] [CrossRef]
- Muñoz, M.A.; Katia Abarca, V.K.; Jiménez, de la J.J.; Luchslnger, F.V.; O’Ryan, G.M.; Ripoll, M.E.; Valenzuela, B.M.T.; Vergara, F.R. Safety of thimerosal containing vaccines. Statement of the Consultive Committee of Immunizations on behalf of the Chilean Infectious Diseases Society. Rev. Chil. Infect. 2007, 24, 372–376. [Google Scholar]
- Dórea, J.G. Making sense of epidemiological studies of young children exposed to thimerosal in vaccines. Clin. Chim. Acta. 2010, 411, 1580–1586. [Google Scholar] [CrossRef]
- Dórea, J.G. Integrating experimental (in vitro and in vivo) neurotoxicity studies of low-dose thimerosal relevant to vaccines. Neurochem. Res. 2011, 36, 927–938. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Thimerosal in vaccines: A joint statement of the American Academy of Pediatrics and the Public Health Service. MMWR. 1999, 48, 563–565.
- Thimerosal. In The Merck Index, 12th ed.; Budavari, S. (Ed.) Merck & Co., Inc.: Whitehouse Station, NJ, USA, 1996; p. 1590.
- Environmental Protection Agency. Mercury Compounds; Hazard Summary-Created in April 1992; Revised in January 2000. Available online: http://www.epa.gov/ttnatw01/hlthef/mercury.html (accessed on 16 June 2013).
- Dirilgen, N. Mercury and lead, assessing the toxic effects on growth and metal accumulation by Lemna minor. Ecotoxicol. Environ. Saf. 2011, 74, 48–54. [Google Scholar] [CrossRef]
- Geier, D.A.; King, P.G.; Geier, M.R. Mitochondrial dysfunction, impaired oxidative-reduction activity, degeneration, and cell death in human neuronal and fetal cells induced by low-level exposure to Thimerosal and other metal compounds. Toxicol. Environ. Chem. 2009, 91, 735–749. [Google Scholar] [CrossRef]
- Farina, M.; Avila, D.S.; da Rocha, J.B.; Aschner, M. Metals, oxidative stress and neurodegeneration: A focus on iron, manganese and mercury. Neurochem. Int. 2013, 62, 575–594. [Google Scholar] [CrossRef]
- Stringari, J.; Nunes, A.K.; Franco, J.L.; Bohrer, D.; Garcia, S.C.; Dafre, A.L.; Milatovic, D.; Souza, D.O.; Rocha, J.B.; Aschner, M.; Farina, M. Prenatal methylmercury exposure hampers glutathione antioxidant system ontogenesis and causes long-lasting oxidative stress in the mouse brain. Toxicol. Appl. Pharmacol. 2008, 227, 147–154. [Google Scholar] [CrossRef]
- Sharpe, M.A.; Livingston, A.D.; Baskin, D.S. Thimerosal-derived ethylmercury is a mitochondrial toxin in human astrocytes, possible role of Fenton chemistry in the oxidation and breakage of mtDNA. J. Toxicol. 2012, 2012, 373678. [Google Scholar] [CrossRef]
- Ethylmercuric Chloride. In The Merck Index, 12th ed.; Budavari, S. (Ed.) Merck & Co., Inc.: Whitehouse Station, NJ, USA, 1996; p. 650.
- Qvarnström, J.; Lambertsson, L.; Havarinasab, S.; Hultman, P.; Frech, W. Determination of methylmercury, ethylmercury, and inorganic mercury in mouse tissues, following administration of thimerosal, by species-specific isotope dilution GC-inductively coupled plasma-MS. Anal. Chem. 2003, 75, 4120–4124. [Google Scholar] [CrossRef]
- Rodrigues, J.L.; Serpeloni, J.M.; Batista, B.L.; Souza, S.S.; Barbosa, F., Jr. Identification and distribution of mercury species in rat tissues following administration of thimerosal or methylmercury. Arch. Toxicol. 2010, 84, 891–896. [Google Scholar] [CrossRef]
- Sugita, M. The biological half-time of heavy metals. The existence of a third, “slowest” component. Int. Arch. Occup. Environ. Health 1978, 41, 25–40. [Google Scholar] [CrossRef]
- Secor, J.D.; Kotha, S.R.; Gurney, T.O.; Patel, R.B.; Kefauver, N.R.; Gupta, N.; Morris, A.J.; Haley, B.E.; Parinandi, N.L. Novel lipid-soluble thiol-redox antioxidant and heavy metal chelator, N,N'-bis(2-mercaptoethyl)isophthalamide (NBMI) and phospholipase D-specific inhibitor, 5-fluoro-2-indolyl des-chlorohalopemide (FIPI) attenuate mercury-induced lipid signaling leading to protection against cytotoxicity in aortic endothelial cells. Int. J. Toxicol. 2011, 30, 619–638. [Google Scholar] [CrossRef]
- Böhme, M.; Diener, M.; Mestres, P.; Rummel, W. Direct and indirect actions of HgCl2 and methyl mercury chloride on permeability and chloride secretion across the rat colonic mucosa. Toxicol. Appl. Pharmacol. 1992, 114, 285–294. [Google Scholar] [CrossRef]
- Lazo, J.S.; Kuo, S.M.; Woo, E.S.; Pitt, B.R. The protein thiol metallothionein as an antioxidant and protectant against antineoplastic drugs. Chem. Biol. Interact. 1998, 111–112, 255–262. [Google Scholar] [CrossRef]
- Nishio, H.; Nezasa, K.; Hirano, J.; Nakata, Y. Effects of thimerosal, an organic sulfhydryl modifying agent, on serotonin transport activity into rabbit blood platelets. Neurochem. Int. 1996, 29, 391–396. [Google Scholar] [CrossRef]
- James, S.J.; Slikker, W., III; Melnyk, S.; New, E.; Pogribna, M.; Jernigan, S. Thimerosal neurotoxicity is associated with glutathione depletion, protection with glutathione precursors. Neurotoxicology 2005, 26, 1–8. [Google Scholar] [CrossRef]
- Anundi, I.; Högberg, J.; Stead, A.H. Glutathione depletion in isolated hepatocytes, its relation to lipid peroxidation and cell damage. Acta Pharmacol. Toxicol. (Copenh). 1979, 45, 45–51. [Google Scholar] [CrossRef]
- Agrawal, A.; Kaushal, P.; Agrawal, S.; Gollapudi, S.; Gupta, S. Thimerosal induces TH2 responses via influencing cytokine secretion by human dendritic cells. J. Leukoc. Biol. 2007, 81, 474–482. [Google Scholar] [CrossRef]
- Abdel-Rahman, M.; Mohamed, A.F.; Essam, N.; Moneim, A.E.A. Studies on H1N1 vaccine-induced monoamines alternations and oxidative stress on brain of adult mice. J. Appl. Pharm. Sci. 2013, 3, 48–53. [Google Scholar]
- Stringari, J.; Meotti, F.C.; Souza, D.O.; Santos, A.R.; Farina, M. Postnatal methylmercury exposure induces hyperlocomotor activity and cerebellar oxidative stress in mice, dependence on the neurodevelopmental period. Neurochem. Res. 2006, 31, 563–569. [Google Scholar] [CrossRef]
- Manfroi, C.B.; Schwalm, F.D.; Cereser, V.; Abreu, F.; Oliveira, A.; Bizarro, L.; Rocha, J.B.; Frizzo, M.E.; Souza, D.O.; Farina, M. Maternal milk as methylmercury source for suckling mice, neurotoxic effects involved with the cerebellar glutamatergic system. Toxicol. Sci. 2004, 81, 172–178. [Google Scholar] [CrossRef]
- Franco, J.L.; Teixeira, A.; Meotti, F.C.; Ribas, C.M.; Stringari, J.; Garcia Pomblum, S.C.; Moro, A.M.; Bohrer, D.; Bairros, A.V.; Dafre, A.L.; Santos, A.R.; Farina, M. Cerebellar thiol status and motor deficit after lactational exposure to methylmercury. Environ. Res. 2006, 102, 22–28. [Google Scholar] [CrossRef]
- Wu, X.; Liang, H.; O’Hara, K.A.; Yalowich, J.C.; Hasinoff, B.B. Thiol-modulated mechanisms of the cytotoxicity of thimerosal and inhibition of DNA topoisomerase II alpha. Chem. Res. Toxicol. 2008, 21, 483–493. [Google Scholar] [CrossRef]
- Nabemoto, M.; Ohsawa, K.; Nakamura, H.; Hirabayashi, T.; Saito, T.; Okuma, Y.; Nomura, Y.; Murayama, T. Reversible activation of secretory phospholipase A2 by sulfhydryl reagents. Arch. Biochem. Biophys. 2005, 436, 145–153. [Google Scholar] [CrossRef]
- Hagele, T.J.; Mazerik, J.N.; Gregory, A.; Kaufman, B.; Magalang, U.; Kuppusamy, M.L.; Marsh, C.B.; Kuppusamy, P.; Parinandi, N.L. Mercury activates vascular endothelial cell phospholipase D through thiols and oxidative stress. Int. J. Toxicol. 2007, 26, 57–69. [Google Scholar] [CrossRef]
- Zieminska, E.; Toczylowska, B.; Stafiej, A.; Lazarewicz, J.W. Low molecular weight thiols reduce thimerosal neurotoxicity in vitro, modulation by proteins. Toxicology 2010, 276, 154–163. [Google Scholar] [CrossRef]
- Olczak, M.; Duszczyk, M.; Mierzejewski, P.; Bobrowicz, T.; Majewska, M.D. Neonatal administration of thimerosal causes persistent changes in mu opioid receptors in the rat brain. Neurochem. Res. 2010, 35, 1840–1847. [Google Scholar] [CrossRef]
- Makani, S.; Gollapudi, S.; Yel, L.; Chiplunkar, S.; Gupta, S. Biochemical and molecular basis of thimerosal-induced apoptosis in T cells, a major role of mitochondrial pathway. Genes Immun. 2002, 3, 270–278. [Google Scholar] [CrossRef]
- Vas, J.; Monestier, M. Immunology of mercury. Ann. N.Y. Acad. Sci. 2008, 1143, 240–267. [Google Scholar] [CrossRef]
- Shenker, B.J.; Mayro, J.S.; Rooney, C.; Vitale, L.; Shapiro, I.M. Immunotoxic effects of mercuric compounds on human lymphocytes and monocytes. IV. Alterations in cellular glutathione content. Immunopharmacol. Immunotoxicol. 1993, 15, 273–290. [Google Scholar] [CrossRef]
- Migdal, C.; Tailhardat, M.; Courtellemont, P.; Haftek, M.; Serres, M. Responsiveness of human monocyte-derived dendritic cells to thimerosal and mercury derivatives. Toxicol. Appl. Pharmacol. 2010, 246, 66–73. [Google Scholar] [CrossRef]
- Mian, M.F.; Kang, C.; Lee, S.; Choi, J.H.; Bae, S.S.; Kim, S.H.; Kim, Y.H.; Ryu, S.H.; Suh, P.G.; Kim, J.S.; Kim, E. Cleavage of focal adhesion kinase is an early marker and modulator of oxidative stress-induced apoptosis. Chem. Biol. Interact. 2008, 171, 57–66. [Google Scholar] [CrossRef]
- Liu, S.I.; Huang, C.C.; Huang, C.J.; Wang, B.W.; Chang, P.M.; Fang, Y.C.; Chen, W.C.; Wang, J.L.; Lu, Y.C.; Chu, S.T.; et al. Thimerosal-induced apoptosis in human SCM1 gastric cancer cells, activation of p38 MAP kinase and caspase-3 pathways without involvement of [Ca2+]i elevation. Toxicol. Sci. 2007, 100, 109–117. [Google Scholar] [CrossRef]
- Migdal, C.; Foggia, L.; Tailhardat, M.; Courtellemont, P.; Haftek, M.; Serres, M. Sensitization effect of thimerosal is mediated in vitro via reactive oxygen species and calcium signaling. Toxicology 2010, 274, 1–9. [Google Scholar] [CrossRef]
- Ueha-Ishibashi, T.; Tatsuishi, T.; Iwase, K.; Umebayashi, C.; Hirama, S.; Sakai, Y.; Ishida, S.; Okano, Y. Property of thimerosal-induced decrease in cellular content of glutathione in rat thymocytes, a flow cytometric study with 5-chloromethylfluorescein diacetate. Toxicol. in Vitro 2004, 18, 563–569. [Google Scholar] [CrossRef]
- Montero, M.; Barrero, M.J.; Torrecilla, F.; Lobatón, C.D.; Moreno, A.; Alvarez, J. Stimulation by thimerosal of histamine-induced Ca(2+) release in intact HeLa cells seen with aequorin targeted to the endoplasmic reticulum. Cell Calcium 2001, 30, 181–190. [Google Scholar] [CrossRef]
- Bootman, M.D.; Taylor, C.W.; Berridge, M.J. The thiol reagent, thimerosal, evokes Ca2+ spikes in HeLa cells by sensitizing the inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 1992, 267, 25113–25119. [Google Scholar]
- Waring, R.H.; O’Reilly, B.A. Enzyme and sulphur oxidation deficiencies in autistic children with known food/chemical intolerances. Xenobiotica 1990, 20, 117–122. [Google Scholar] [CrossRef]
- Alberti, A.; Pirrone, P.; Elia, M.; Waring, R.H.; Romano, C. Sulphation deficit in low-functioning autistic children, a pilot study. Biol. Psychiatr. 1999, 46, 420–424. [Google Scholar] [CrossRef]
- Waring, R.H.; Klovrza, L.V. Sulphur metabolism in autism. J. Nutr. Environ. Med. 2000, 10, 25–32. [Google Scholar] [CrossRef]
- Dawson, P.A.; Markovich, D. Genetic polymorphisms of human sulfate transporters. Curr. Pharmacogenom. 2007, 5, 262–274. [Google Scholar] [CrossRef]
- Markovich, D.; Mure, H.; Biber, J.; Sakhaee, K.; Pak, C.; Levi, M. Dietary sulfate regulates the expression of the renal brush border Na/Si cotransporter, NaSi-1. J. Am. Soc. Nephrol. 1998, 9, 1568–1573. [Google Scholar]
- Blaurock-Busch, E.; Amin, O.R.; Dessoki, H.H.; Rabah, T. Toxic metals and essential elements in hair and severity of symptoms among children with autism. Maedica (Buchar) 2012, 7, 38–48. [Google Scholar]
- Geier, D.A.; Geier, M.R. A clinical and laboratory evaluation of methionine cycle-transsulfuration and androgen pathway markers in children with autistic disorders. Horm. Res. 2006, 66, 182–188. [Google Scholar] [CrossRef]
- Paşca, S.P.; Dronca, E.; Kaucsár, T.; Craciun, E.C.; Endreffy, E.; Ferencz, B.K.; Iftene, F.; Benga, I.; Cornean, R.; Banerjee, R.; Dronca, M. One carbon metabolism disturbances and the C677T MTHFR gene polymorphism in children with autism spectrum disorders. J. Cell Mol. Med. 2009, 13, 4229–4238. [Google Scholar] [CrossRef]
- Al-Yafee, Y.A.; Al-Ayadhi, L.Y.; Haq, S.H.; El-Ansary, A.K. Novel metabolic biomarkers related to sulfur-dependent detoxification pathways in autistic patients of Saudi Arabia. BMC Neurol. 2011, 11, 139. [Google Scholar] [CrossRef]
- Al-Gadani, Y.; El-Ansary, A.; Attas, O.; Al-Ayadhi, L. Metabolic biomarkers related to oxidative stress and antioxidant status in Saudi autistic children. Clin. Biochem. 2009, 42, 1032–1040. [Google Scholar] [CrossRef]
- Geier, D.A.; Kern, J.K.; Adams, J.B.; Garver, C.R.; Geier, M.R. A prospective study of oxidative stress biomarkers in autistic disorders. J. Appl. Psychol. 2009, 5, 2–10. [Google Scholar] [CrossRef]
- Adams, J.B.; Audhya, T.; McDonough-Means, S.; Rubin, R.A.; Quig, D.; Geis, E.; Gehn, E.; Loresto, M.; Mitchell, J.; Atwood, S.; Barnhouse, S.; Lee, W. Nutritional and metabolic status of children with autism vs. neurotypical children, and the association with autism severity. Nutr. Metab. (Lond). 2011, 8, 34. [Google Scholar] [CrossRef]
- Main, P.A.; Angley, M.T.; O'Doherty, C.E.; Thomas, P.; Fenech, M. The potential role of the antioxidant and detoxification properties of glutathione in autism spectrum disorders: A systematic review and meta-analysis. Nutr. Metab. (Lond). 2012, 9, 35. [Google Scholar] [CrossRef]
- Chauhan, A.; Audhya, T.; Chauhan, V. Brain region-specific glutathione redox imbalance in autism. Neurochem. Res. 2012, 37, 1681–1689. [Google Scholar] [CrossRef]
- Rose, S.; Melnyk, S.; Pavliv, O.; Bai, S.; Nick, T.G.; Frye, R.E.; James, S.J. Evidence of oxidative damage and inflammation associated with low glutathione redox status in the autism brain. Transl. Psychiatry 2012, 2, e134. [Google Scholar] [CrossRef]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar]
- Bounous, G.; Molson, J. Competition for glutathione precursors between the immune system and the skeletal muscle, pathogenesis of chronic fatigue syndrome. Med. Hypotheses 1999, 53, 347–349. [Google Scholar] [CrossRef]
- Ho, W.Z.; Douglas, S.D. Glutathione and N-acetylcysteine suppression of human immunodeficiency virus replication in human monocyte/macrophages in vitro. AIDS Res. Hum. Retrovir. 1992, 8, 1249–1253. [Google Scholar] [CrossRef]
- Sen, C.K. Nutritional biochemistry of cellular glutathione. J. Nutr. Biochem. 1997, 8, 660–672. [Google Scholar] [CrossRef]
- Baruchel, S.; Viau, G.; Olive, R.; Bounous, G. Nutraceutical Modulation with a Humanized Native Milk Serum Protein Isolate, Immunocal®, Application in AIDS and Cancer. In Oxidative Stress in Cancer, AIDS and Neurodegenerative Diseases; Montagnier, L., Olivier, R., Pasquier, C., Eds.; Marcel Dekker, Inc.: New York, NY, USA, 1998; pp. 447–461. [Google Scholar]
- Gutman, J. Glutathione—Your Bodies Most Powerful Protector, 3rd ed.; Communications Kudo.ca Inc.: Montreal, Canada, 2002. [Google Scholar]
- Takahashi, T.; Kimura, T.; Sato, Y.; Shiraki, H.; Ukita, T. Time-dependent distribution of [203] Hg-mercury compounds in rat and monkey as studied by whole body autoradiography. J. Hygenic. Chem. (Japan) 1971, 17, 93–107. [Google Scholar]
- Geier, D.A.; Kern, J.K.; Adams, J.B.; Geier, M.R. Biomarkers of environmental toxicity and susceptibility in autism. J. Neurolog. Sci. 2009, 280, 101–108. [Google Scholar] [CrossRef]
- Geier, D.A.; Kern, J.K.; Garver, C.R.; Adams, J.B.; Audhya, T.; Geier, M.R. A prospective study of transsulfuration biomarkers in autistic disorders. Neurochem. Res. 2009, 34, 386–393. [Google Scholar] [CrossRef]
- Nkabyo, Y.S.; Gu, L.H.; Jones, D.P.; Ziegler, T.R. Thiol/disulfide redox status is oxidized in plasma and small intestinal and colonic mucosa of rats with inadequate sulfur amino acid intake. J. Nutr. 2006, 136, 1242–1248. [Google Scholar]
- Sharpe, M.A.; Taylor, L.; Gist, T.L.; Baskin, D.S. B-Lymphocytes from a population of children with autism spectrum disorder and their unaffected siblings exhibit hypersensitivity to Thimerosal. J. Toxicol. 2013, 2013, 801517:1–801517:11. [Google Scholar]
- Bigham, M.; Copes, R. Thiomersal in vaccines, balancing the risk of adverse effects with the risk of vaccine-preventable disease. Drug Saf. 2005, 28, 89–101. [Google Scholar] [CrossRef]
- Dórea, J.G.; Bezerra, V.L.; Fajon, V.; Horvat, M. Speciation of methyl- and ethyl-mercury in hair of breastfed infants acutely exposed to thimerosal-containing vaccines. Clin. Chim. Acta 2011, 412, 1563–1566. [Google Scholar] [CrossRef]
- Dórea, J.G.; Marques, R.C.; Isejima, C. Neurodevelopment of Amazonian infants, antenatal and postnatal exposure to methyl- and ethylmercury. J. Biomed. Biotechnol. 2012, 2012, 132876. [Google Scholar] [CrossRef]
- Redwood, L.; Bernard, S.; Brown, D. Predicted mercury concentrations in hair from infant immunizations, cause for concern. Neurotoxicology 2001, 22, 691–677. [Google Scholar] [CrossRef]
- El-baz, F.; Elhossiny, E.M.; Elsayed, A.B.; Gaber, G.M. Hair mercury measurement in Egyptian autistic children. Egyptian J. Med. Hum. Genet. 2010, 11, 135–141. [Google Scholar] [CrossRef]
- Majewska, M.D.; Urbanowicz, E.; Rok-Bujko, P.; Namyslowska, I.; Mierzejewski, P. Age-dependent lower or higher levels of hair mercury in autistic children than in healthy controls. Acta Neurobiol. Exp. (Wars) 2010, 70, 96–208. [Google Scholar]
- Jedrychowski, W.; Jankowski, J.; Flak, E.; Skarupa, A.; Mroz, E.; Sochacka-Tatara, E.; Lisowska-Miszczyk, I.; Szpanowska-Wohn, A.; Rauh, V.; Skolicki, Z.; et al. Effects of prenatal exposure to mercury on cognitive and psychomotor function in one-year-old infants, epidemiologic cohort study in Poland. Ann. Epidemiol. 2006, 16, 439–447. [Google Scholar] [CrossRef]
- Lederman, S.A.; Jones, R.L.; Caldwell, K.L.; Rauh, V.; Sheets, S.E.; Tang, D.; Viswanathan, S.; Becker, M.; Stein, J.L.; Wang, R.Y.; Perera, F.P. Relation between cord blood mercury levels and early child development in a World Trade Center cohort. Environ. Health Perspect. 2008, 116, 1085–1091. [Google Scholar] [CrossRef]
- Björnberg, K.A.; Vahter, M.; Petersson-Grawé, K.; Glynn, A.; Cnattingius, S.; Darnerud, P.O.; Atuma, S.; Aune, M.; Becker, W.; Berglund, M. Methyl mercury and inorganic mercury in Swedish pregnant women and in cord blood, influence of fish consumption. Environ. Health Perspect. 2003, 111, 637–641. [Google Scholar] [CrossRef]
- Ouédraogo, O.; Amyot, M. Effects of various cookingmethods and foodcomponents on bioaccessibility of mercury from fish. Environ. Res. 2011, 111, 1064–1069. [Google Scholar] [CrossRef]
- Burbacher, T.M.; Shen, D.D.; Liberato, N.; Grant, K.S.; Cernichiari, E.; Clarkson, T. Comparison of blood and brain mercury levels in infant monkeys exposed to methylmercury or vaccines containing thimerosal. Environ. Health Perspect. 2005, 113, 1015–1021. [Google Scholar] [CrossRef]
- World Health Organization. Mercury Intoxication. Available online: http://www.paho.org/hq/index.php?option=com_content&view=article&id=8158&Itemid=39767&lang=en (accessed 7 February 2013).
- Pichichero, M.E.; Gentile, A.; Giglio, N.; Umido, V.; Clarkson, T.; Cernichiari, E.; Zareba, G.; Gotelli, C.; Gotelli, M.; Yan, L.; Treanor, J. Mercury levels in newborns and infants after receipt of thimerosal-containing vaccines. Pediatrics 2008, 121, e208–e214. [Google Scholar] [CrossRef]
- Takeda, Y.; Kunugi, T.; Hoshino, O.; Ukita, T. Distribution of inorganic, aryl, and alkyl mercury compounds in rats. Toxicol. Appl. Pharmacol. 1968, 13, 156–164. [Google Scholar] [CrossRef]
- Zimmer, B.; Lee, G.; Balmer, N.V.; Meganathan, K.; Sachinidis, A.; Studer, L.; Leist, M. Evaluation of developmental toxicants and signaling pathways in a functional test based on the migration of human neural crest cells. Environ. Health Perspect. 2012, 120, 1116–1122. [Google Scholar] [CrossRef]
- Ueha-Ishibashi, T.; Oyama, Y.; Nakao, H.; Umebayashi, C.; Nishizaki, Y.; Tatsuishi, T.; Iwase, K.; Murao, K.; Seo, H. Effect of thimerosal, a preservative in vaccines, on intracellular Ca2+ concentration of rat cerebellar neurons. Toxicology 2004, 195, 77–84. [Google Scholar] [CrossRef]
- Zimmermann, L.T.; Santos, D.B.; Naime, A.A.; Leal, R.B.; Dórea, J.G.; Barbosa, F., Jr.; Aschner, M.; Rocha, J.B.; Farina, M. Comparative study on methyl- and ethylmercury-induced toxicity in C6 glioma cells and the potential role of LAT-1 in mediating mercurial-thiol complexes uptake. Neurotoxicology 2013. [Google Scholar] [CrossRef]
- Peltz, A.; Sherwani, S.I.; Kotha, S.R.; Mazerik, J.N.; O'Connor Butler, E.S.; Kuppusamy, M.L.; Hagele, T.; Magalang, U.J.; Kuppusamy, P.; Marsh, C.B.; Parinandi, N.L. Calcium and calmodulin regulate mercury-induced phospholipase D activation in vascular endothelial cells. Int. J. Toxicol. 2009, 28, 190–206. [Google Scholar] [CrossRef]
- Olczak, M.; Duszczyk, M.; Mierzejewski, P.; Wierzba-Bobrowicz, T.; Majewska, M.D. Lasting neuropathological changes in rat brain after intermittent neonatal administration of thimerosal. Folia Neuropathol. 2010, 48, 258–269. [Google Scholar]
- Hargreaves, R.J.; Eley, B.P.; Moorhouse, S.R.; Pelling, D. Regional cerebral glucose metabolism and blood flow during the silent phase of methylmercury neurotoxicity in rats. J. Neurochem. 1988, 51, 1350–1355. [Google Scholar] [CrossRef]
- Nelson, K.B.; Bauman, M.L. Thimerosal and autism? Pediatrics 2003, 111, 674–679. [Google Scholar] [CrossRef]
- Dórea, J.G.; Farina, M.; Rocha, J.B. Toxicity of ethylmercury (and Thimerosal): A comparison with methylmercury. J. Appl. Toxicol. 2013, 33, 700–711. [Google Scholar] [CrossRef]
- Korbas, M.; O’Donoghue, J.L.; Watson, G.E.; Pickering, I.J.; Singh, S.P.; Myers, G.J.; Clarkson, T.W.; George, G.N. The chemical nature of mercury in human brain following poisoning or environmental exposure. ACS Chem. Neurosci. 2010, 1, 810–818. [Google Scholar] [CrossRef]
- U. S. Food and Drug Administration. Vaccines, Blood & Biologics. Thimerosal in Vaccines. Available online: http://www.fda.gov/BiologicsBloodVaccines/SafetyAvailability/VaccineSafety/UCM096228 (accessed on 3 July 2013).
- World Health Organization. WHO Statement on Thiomersal; WHO Global Advisory Committee on Vaccine Safety: Geneva, Switzerland, 2006. Available online: http://www.who.int/vaccine_safety/topics/thiomersal (accessed on 3 July 2013).
- Geier, D.A.; Sykes, L.K.; Geier, M.R. A review of Thimerosal (Merthiolate) and its ethylmercury breakdown product: Specific historical considerations regarding safety and effectiveness. J. Toxicol. Environ. Health B Crit. Rev. 2007, 10, 575–596. [Google Scholar] [CrossRef]
- Migliarini, S.; Pacini, G.; Pelosi, B.; Lunardi, G.; Pasqualetti, M. Lack of brain serotonin affects postnatal development and serotonergic neuronal circuitry formation. Mol. Psychiatry 2012. [Google Scholar] [CrossRef]
- LeBlanc, J.J.; Fagiolini, M. Autism, a “critical period” disorder? Neural Plast. 2011, 2011, 921680. [Google Scholar] [CrossRef]
- Makri, A.; Goveia, M.; Balbus, J.; Parkin, R. Children’s susceptibility to chemicals, a review by developmental stage. J. Toxicol. Environ. Health B 2004, 7, 417–435. [Google Scholar] [CrossRef]
- Graeter, L.J.; Mortensen, M.E. Kids are different, developmental variability in toxicology. Toxicology 1996, 111, 15–20. [Google Scholar] [CrossRef]
- Ballatori, N.; Clarkson, T.W. Developmental changes in the biliary excretion of methylmercury and glutathione. Science 1982, 216, 61–63. [Google Scholar]
- Blackburn, S.T. Renal function in the neonate. J. Perinat. Neonatal Nurs. 1994, 8, 37–47. [Google Scholar] [CrossRef]
- Ida-Eto, M.; Oyabu, A.; Ohkawara, T.; Tashiro, Y.; Narita, N.; Narita, M. Prenatal exposure to organomercury, thimerosal, persistently impairs the serotonergic and dopaminergic systems in the rat brain: implications for association with developmental disorders. Brain Dev. 2013, 35, 261–264. [Google Scholar] [CrossRef]
- Palmer, R.F.; Banchard, S.; Stein, Z.; Mandell, D.; Miller, C. Environmental mercury release, special education rates, and autistic disorder, an ecological study of Texas. Health Place 2006, 12, 203–209. [Google Scholar] [CrossRef]
- Palmer, R.F.; Blanchard, S.; Wood, R. Proximity to point sources of environmental mercury release as a predictor of autism prevalence. Health Place 2009, 15, 18–24. [Google Scholar] [CrossRef]
- Windham, G.C.; Zhang, L.; Gunier, R.; Croen, L.A.; Grether, J.K. Autism spectrum disorders in relation to distribution of hazardous air pollutants in the San Francisco Bay area. Environ. Health Perspect. 2006, 114, 1438–1444. [Google Scholar] [CrossRef]
- Young, H.A.; Geier, D.A.; Geier, M.R. Thimerosal exposure in infants and neurodevelopmental disorders, an assessment of computerized medical records in the Vaccine Safety Datalink. J. Neurol. Sci. 2008, 271, 110–118. [Google Scholar] [CrossRef]
- Geier, D.A.; Geier, M.R. A prospective study of mercury toxicity biomarkers in autistic spectrum disorders. J. Toxicol. Environ. Health 2007, 70, 1723–1730. [Google Scholar] [CrossRef]
- Geier, D.A.; Mumper, E.; Gladfelter, B.; Coleman, L.; Geier, M.R. Neurodevelopmental disorders, maternal Rh-negativity, and Rho(D) immune globulins, a multi-center assessment. Neuro. Endocrinol. Lett. 2008, 29, 272–280. [Google Scholar]
- Geier, D.A.; Kern, J.K.; King, P.G.; Sykes, L.K.; Geier, M.R. Hair toxic metal concentrations and autism spectrum disorder severity in young children. Int. J. Environ. Res. Public Health 2012, 9, 4486–4497. [Google Scholar] [CrossRef]
- Adams, J.B.; Baral, M.; Geis, E.; Mitchell, J.; Ingram, J.; Hensley, A.; Zappia, I.; Newmark, S.; Gehn, E.; Rubin, R.A.; Mitchell, K.; Bradstreet, J.; El-Dahr, J. Safety and efficacy of oral DMSA therapy for children with autism spectrum disorders, part A–Medical results. BMC Clin. Pharmacol. 2009, 9, 1–22. [Google Scholar]
- Blanchard, K.S.; Palmer, R.F.; Stein, Z. The value of ecologic studies, mercury concentration in ambient air and the risk of autism. Rev. Environ. Health 2011, 26, 111–118. [Google Scholar]
- Elshenshtawy, E.; Tobar, S.; Sherra, K.; Atallah, S.; Elkasaby, R. Study of some biomarkers in hair of children with autism. Middle East Curr. Psychiatry 2011, 18, 6–10. [Google Scholar] [CrossRef]
- Lakshmi Priya, M.D.; Geetha, A. Level of trace elements (copper, zinc, magnesium and selenium) and toxic elements (lead and mercury) in the hair and nail of children with autism. Biol. Trace Elem. Res. 2011, 142, 148–158. [Google Scholar] [CrossRef]
- Sajdel-Sulkowska, E.M.; Lipinsk, B.; Windom, H.; Audhya, T.; McGinnis, W. Oxidative stress in autism, elevated cerebellar 3-nitrotyrosine levels. Am. J. Biochem. Biotechnol. 2008, 4, 73–84. [Google Scholar] [CrossRef]
- Kern, J.K.; Geier, D.A.; Adams, J.B.; Grannemann, B.D.; Mehta, J.A.; Geier, M.R. Toxicity biomarkers related to autism spectrum disorder, a blinded study of urinary porphyrins. Pediatr. Int. 2011, 53, 147–153. [Google Scholar] [CrossRef]
- Kern, J.K.; Geier, D.A.; Audhya, T.; King, P.G.; Sykes, L.; Geier, M. Evidence of parallels between mercury intoxication and the brain pathology in autism. Acta Neurobiol. Exp. (Warsz) 2012, 72, 113–153. [Google Scholar]
- Geier, D.A.; Audhya, T.; Kern, J.K.; Geier, M.R. Differences in blood mercury levels in autism spectrum disorders, is there a threshold level? Acta Neurobiol. Exp. 2010, 70, 177–186. [Google Scholar]
- Kern, J.K.; Geier, D.A.; Adams, J.B.; Geier, M.R. A Biomarker of mercury body-burden correlated with diagnostic domain specific clinical symptoms of autistic disorders. Biometals. 2010, 23, 1043–1051. [Google Scholar] [CrossRef]
- Nataf, R.; Skorupka, C.; Amet, L.; Lam, A.; Springbett, A.; Lathe, R. Porphyinuria in childhood autistic disorder, implications for environmental toxicity. Toxicol. Applied Pharmacol. 2006, 14, 99–108. [Google Scholar]
- Holmes, A.S.; Blaxill, M.F.; Haley, B.E. Reduced levels of mercury in first baby haircuts of autistic children. Int. J. Toxicol. 2003, 22, 277–285. [Google Scholar] [CrossRef]
- Ida-Eto, M.; Oyabu, A.; Ohkawara, T.; Tashiro, Y.; Narita, N.; Narita, M. Embryonic exposure to thimerosal, an organomercury compound, causes abnormal early development of serotonergic neurons. Neuropharmacology 2011, 60, 1347–1354. [Google Scholar] [CrossRef]
- Azmitia, E.C.; Singh, J.S.; Whitaker-Azmitia, P.M. Increased serotonin axons (immunoreactive to 5-HT transporter) in postmortem brains from young autism donors. Neurosci. Lett. 2011, 505, 61–64. [Google Scholar] [CrossRef]
- Mrozek-Budzyn, D.; Majewska, R.; Kieltyka, A.; Augustyniak, M. Neonatal exposure to Thimerosal from vaccines and child development in the first 3 years of life. Neurotoxicol. Teratol. 2012, 34, 592–597. [Google Scholar] [CrossRef]
- Thompson, W.W.; Price, C.; Goodson, B.; Shay, D.K.; Benson, P.; Hinrichsen, V.L.; Lewis, E.; Eriksen, E.; Ray, P.; Marcy, S.M.; et al. Early thimerosal exposure and neuropsychological outcomes at 7 to 10 years. Vaccine Safety Datalink Team. N. Engl. J. Med. 2007, 357, 1281–1292. [Google Scholar]
- Verstraeten, T.; Davis, R.L.; DeStefano, F.; Lieu, T.A.; Rhodes, P.H.; Black, S.B.; Shinefield, H.; Chen, R.T.; Vaccine Safety Datalink Team. Safety of thimerosal-containing vaccines, a two-phased study of computerized health maintenance organization databases. Pediatrics 2003, 112, 1039–1048. [Google Scholar]
- Andrews, N.; Miller, E.; Grant, A.; Stowe, J.; Osborne, V.; Taylor, B. Thimerosal exposure in infants and developmental disorders, a retrospective cohort study in the United Kingdom does not support a causal association. Pediatrics 2004, 114, 584–591. [Google Scholar] [CrossRef]
- Barile, J.P.; Kuperminc, G.P.; Weintraub, E.S.; Mink, J.W.; Thompson, W.W. Thimerosal exposure in early life and neuropsychological outcomes 7–10 years later. J. Pediatr. Psychol. 2012, 37, 106–118. [Google Scholar] [CrossRef]
- Geier, D.A.; Geier, M.R. A two-phased population epidemiological study of the safety of thimerosal-containing vaccines, a follow-up analysis. Med. Sci. Monit. 2005, 11, CR160–CR170. [Google Scholar]
- Moro, P.L.; Broder, K.; Zheteyeva, Y.; Revzina, N.; Tepper, N.; Kissin, D.; Barash, F.; Arana, J.; Brantley, M.D.; Ding, H.; et al. Adverse events following administration to pregnant women of influenza A (H1N1) 2009 monovalent vaccine reported to the Vaccine Adverse Event Reporting System. Am. J. Obstet. Gynecol. 2011, 205, 473. [Google Scholar] [CrossRef]
- Geier, D.A.; Young, H.A.; Geier, M.R. Thimerosal exposure & increasing trends of premature puberty in the vaccine safety datalink. Indian J. Med. Res. 2010, 131, 500–507. [Google Scholar]
- Balabanič, D.; Rupnik, M.; Klemenčič, A.K. Negative impact of endocrine-disrupting compounds on human reproductive health. Reprod. Fertil. Dev. 2011, 23, 403–416. [Google Scholar]
- Hotchkiss, A.K.; Rider, C.V.; Blystone, C.R.; Wilson, V.S.; Hartig, P.C.; Ankley, G.T.; Foster, P.M.; Gray, C.L.; Gray, L.E. Fifteen years after “Wingspread”—Environmental endocrine disrupters and human and wildlife health: Where we are today and where we need to go. Toxicol. Sci. 2008, 105, 235–259. [Google Scholar] [CrossRef]
- Tan, S.W.; Meiller, J.C.; Mahaffey, K.R. The endocrine effects of mercury in humans and wildlife. Crit. Rev. Toxicol. 2009, 39, 228–269. [Google Scholar] [CrossRef]
- Fagan, D.G.; Pritchard, J.S.; Clarkson, T.W.; Greenwood, M.R. Organ mercury levels in infants with omphaloceles treated with organic mercurial antiseptic. Arch. Dis. Child 1977, 52, 962–964. [Google Scholar] [CrossRef]
- Barcelos, G.R.; Grotto, D.; de Marco, K.C.; Valentini, J.; Lengert, A.V.; Oliveira, A.A.; Garcia, S.C.; Braga, G.U.; Schläwicke Engström, K.; Cólus, I.M.; et al. Polymorphisms in glutathione-related genes modify mercury concentrations and antioxidant status in subjects environmentally exposed to methylmercury. Sci. Total Environ. 2013, 463–464, 319–325. [Google Scholar] [CrossRef]
- Blumberg, S.J.; Bramlett, M.D.; Kogan, M.D.; Schieve, L.A.; Jones, J.R.; Lu, M.C. Changes in Prevalence of Parent-Reported Autism Spectrum Disorder in School-Aged U.S. Children, 2007 to 2011–2012. Available online: http://www.cdc.gov/nchs/data/nhsr/nhsr065.pdf (accessed on 21 March 2013).
- Boyle, C.A.; Boulet, S.; Schieve, L.A.; Cohen, R.A.; Blumberg, S.J.; Yeargin-Allsopp, M.; Visser, S.; Kogan, M.D. Trends in the prevalence of developmental disabilities in US children, 1997–2008. Pediatrics 2011, 127, 1034–1042. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kern, J.K.; Haley, B.E.; Geier, D.A.; Sykes, L.K.; King, P.G.; Geier, M.R. Thimerosal Exposure and the Role of Sulfation Chemistry and Thiol Availability in Autism. Int. J. Environ. Res. Public Health 2013, 10, 3771-3800. https://doi.org/10.3390/ijerph10083771
Kern JK, Haley BE, Geier DA, Sykes LK, King PG, Geier MR. Thimerosal Exposure and the Role of Sulfation Chemistry and Thiol Availability in Autism. International Journal of Environmental Research and Public Health. 2013; 10(8):3771-3800. https://doi.org/10.3390/ijerph10083771
Chicago/Turabian StyleKern, Janet K., Boyd E. Haley, David A. Geier, Lisa K. Sykes, Paul G. King, and Mark R. Geier. 2013. "Thimerosal Exposure and the Role of Sulfation Chemistry and Thiol Availability in Autism" International Journal of Environmental Research and Public Health 10, no. 8: 3771-3800. https://doi.org/10.3390/ijerph10083771
APA StyleKern, J. K., Haley, B. E., Geier, D. A., Sykes, L. K., King, P. G., & Geier, M. R. (2013). Thimerosal Exposure and the Role of Sulfation Chemistry and Thiol Availability in Autism. International Journal of Environmental Research and Public Health, 10(8), 3771-3800. https://doi.org/10.3390/ijerph10083771