The Association between Splenocyte Apoptosis and Alterations of Bax, Bcl-2 and Caspase-3 mRNA Expression, and Oxidative Stress Induced by Dietary Nickel Chloride in Broilers


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chickens and Diets
2.2. Detection of the Splenocyte Apoptosis by TUNEL and FCM
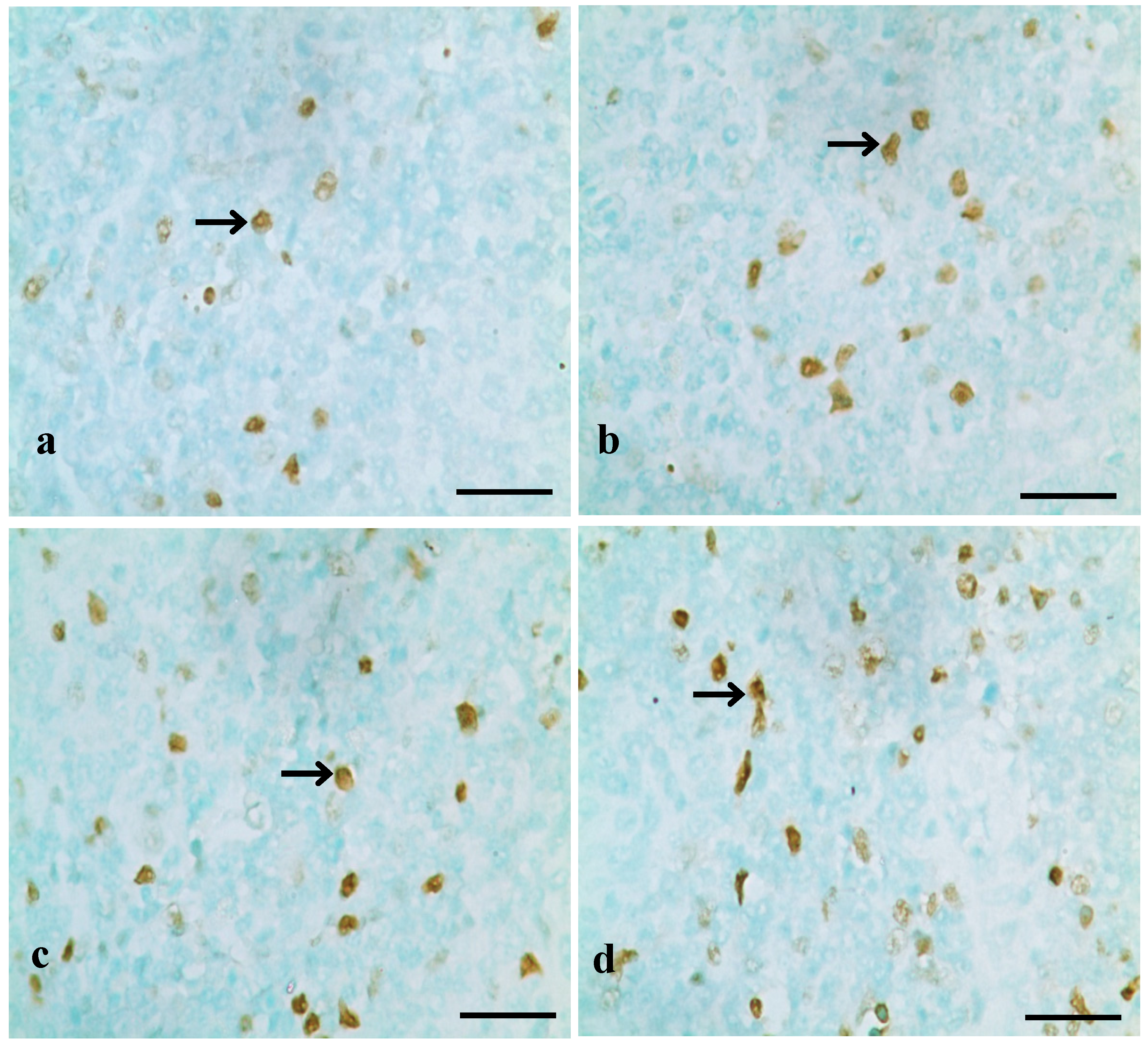
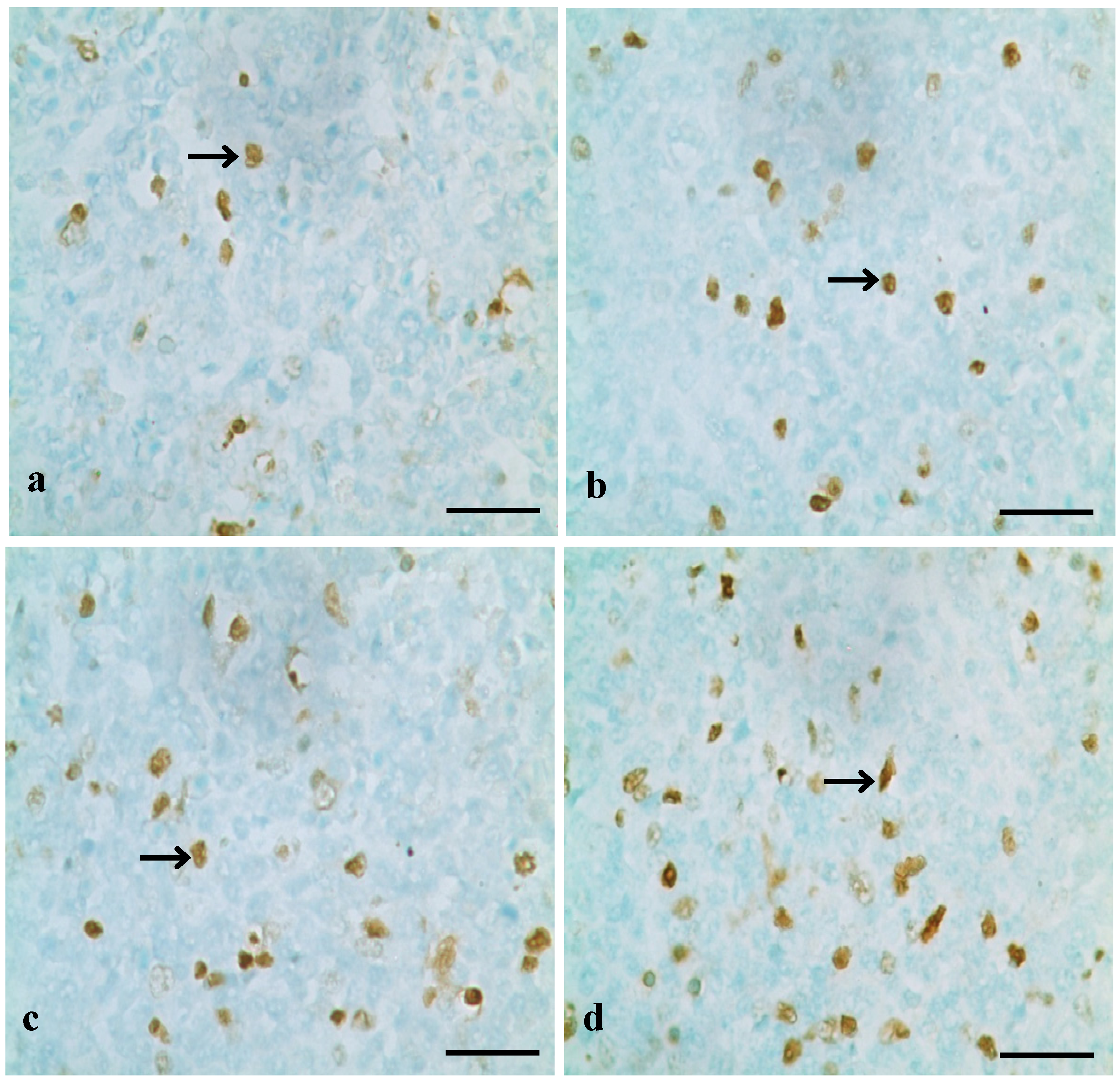
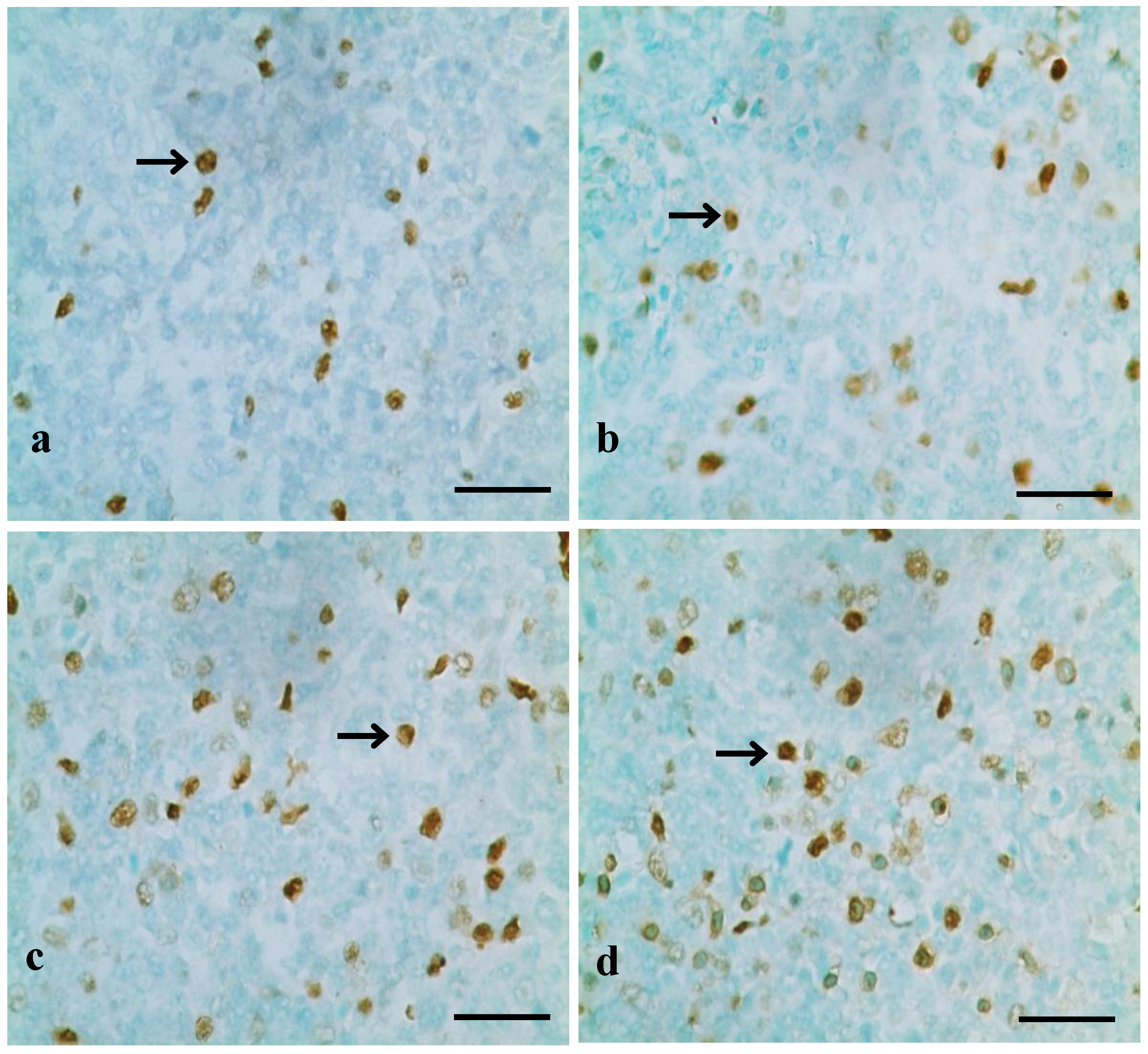
2.2.1. TUNEL Assay
2.2.2. Annexin V Apoptotic Detection by FCM
2.3. Determination of Bax, Bcl-2 and Caspase-3 in the Spleen
2.3.1. Detection of Bax, Bcl-2 and Caspase-3 mRNA Expression Levels by qRT-PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5’→3’) | Accession Number | |
|---|---|---|---|
| Bax | F | TCCTCATCGCCATGCTCAT | XM_422067 |
| R | CCTTGGTCTGGAAGCAGAAGA | XM_422067 | |
| Bcl-2 | F | GATGACCGAGTACCTGAACC | NM_205339 |
| R | CAGGAGAAATCGAACAAAGGC | NM_205339 | |
| Caspase-3 | F | TGGCCCTCTTGAACTGAAAG | NM_204725 |
| R | TCCACTGTCTGCTTCAATACC | NM_204725 | |
| β-actin | F | TGCTGTGTTCCCATCTATCG | L08165 |
| R | TTGGTGACAATACCGTGTTCA | L08165 | |
2.3.2. Detection of Bax, Bcl-2 and Caspase-3 Contents by ELISA
2.4. Detection of Oxidative Stress Parameters in the Spleen
2.5. Statistical Analysis
3. Results
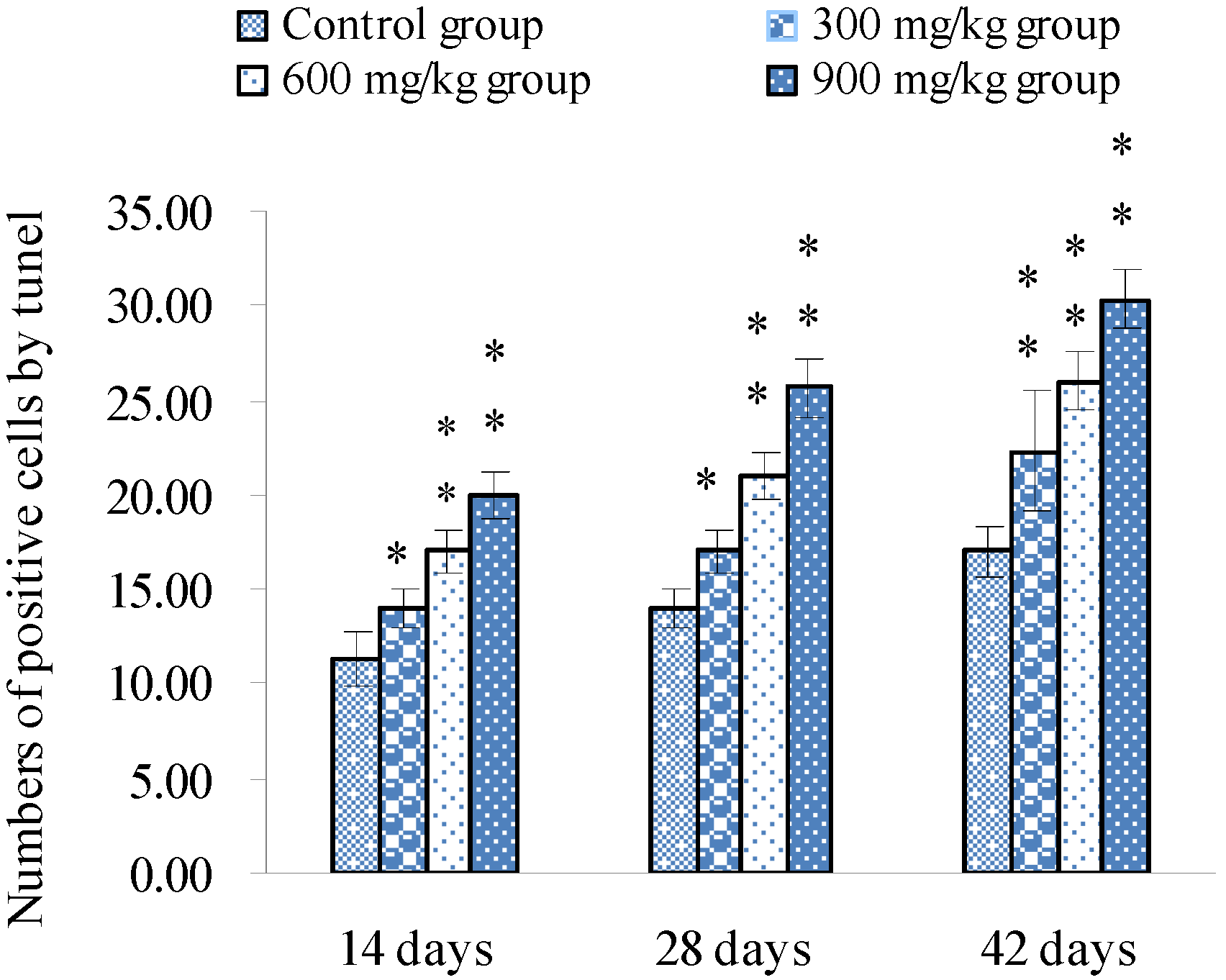
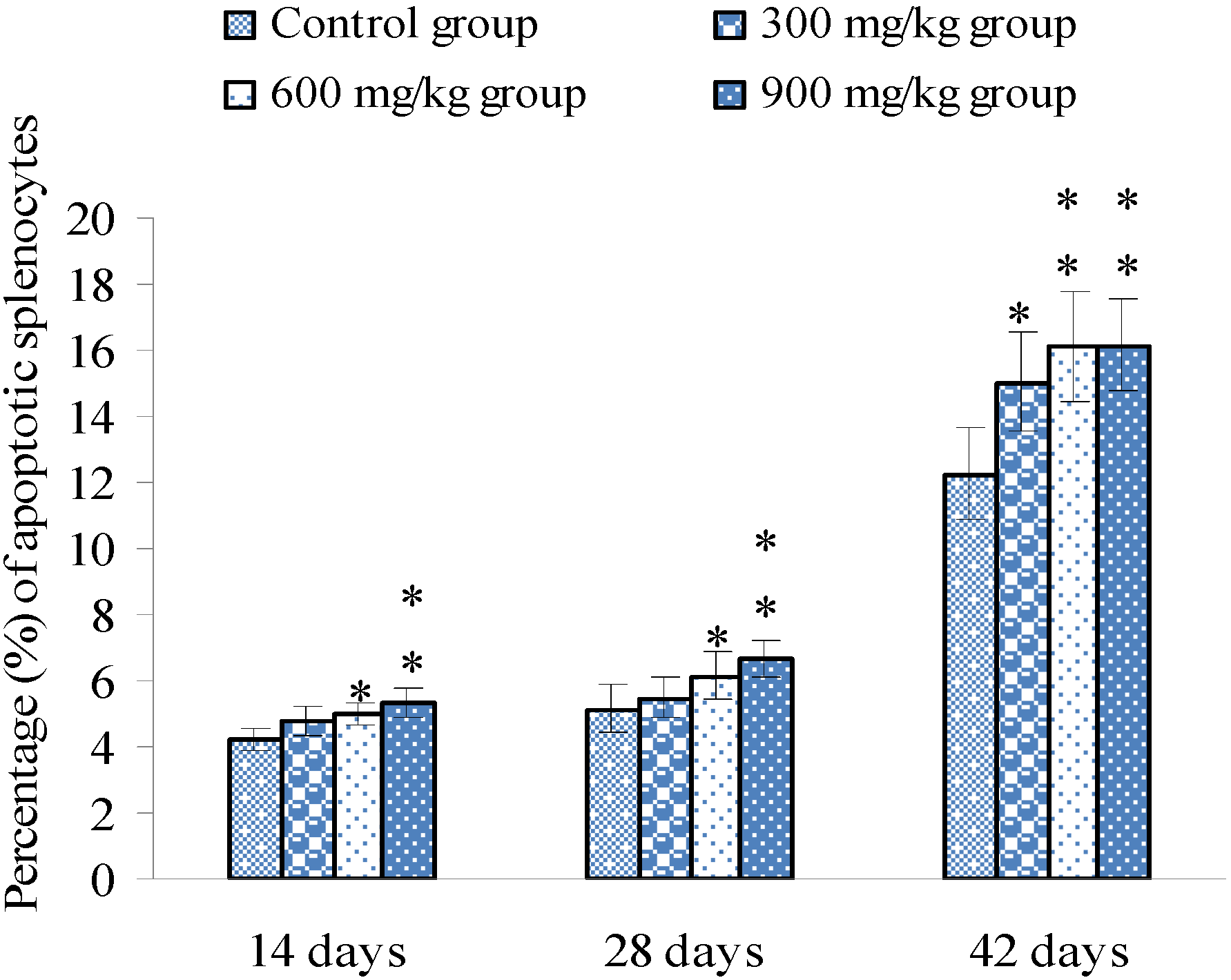
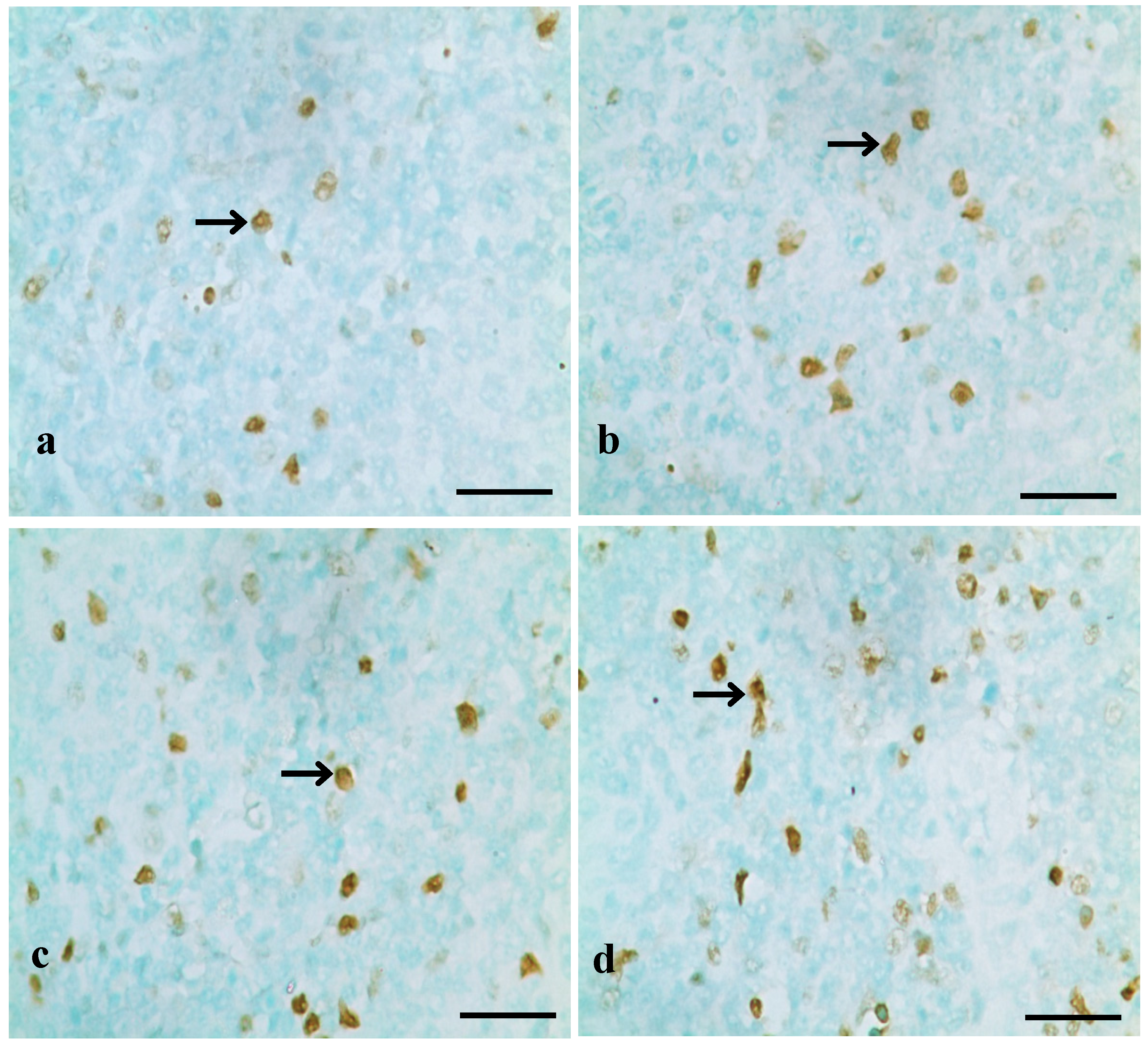
3.1. Changes of Splenocyte Apoptosis
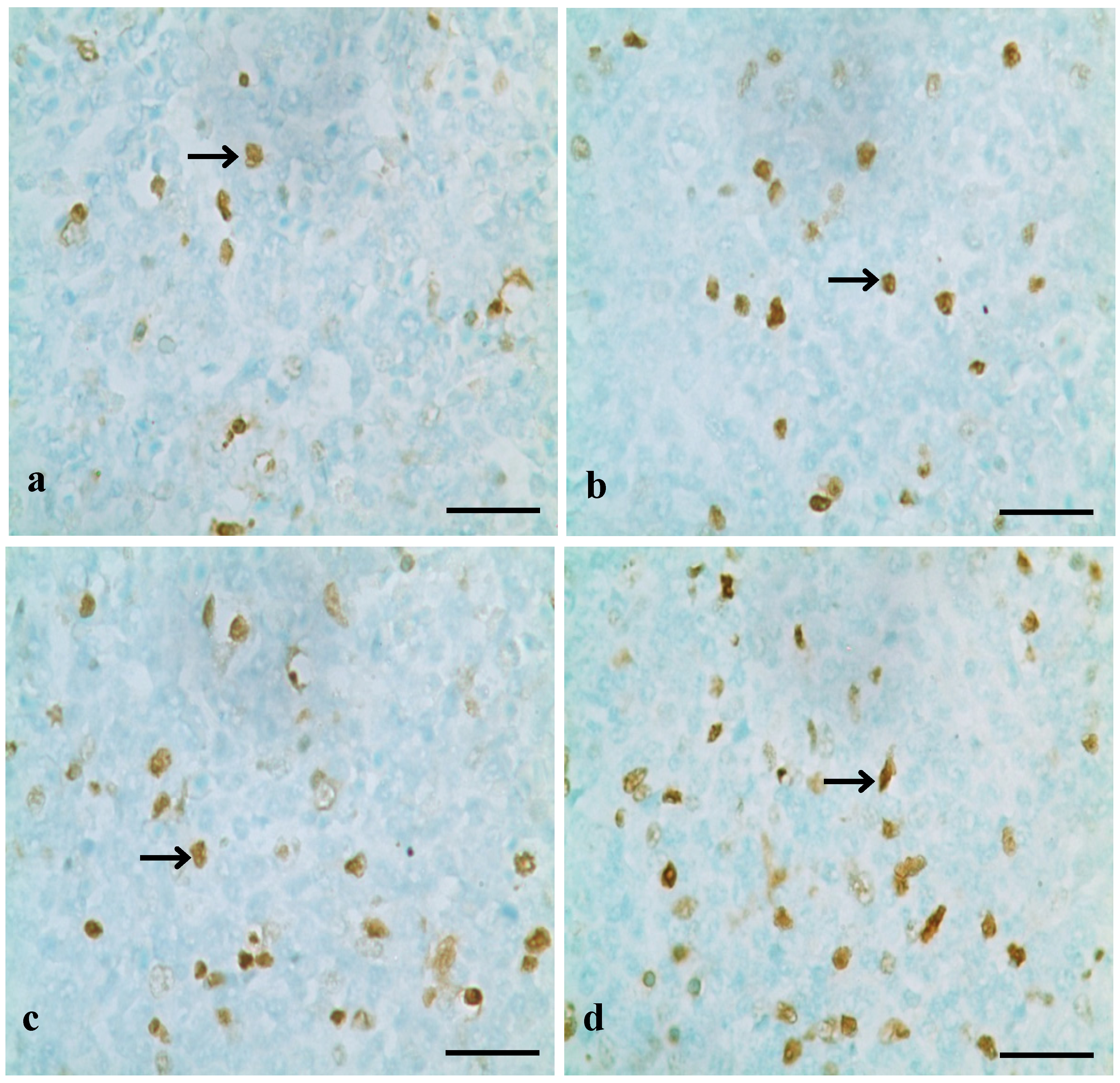
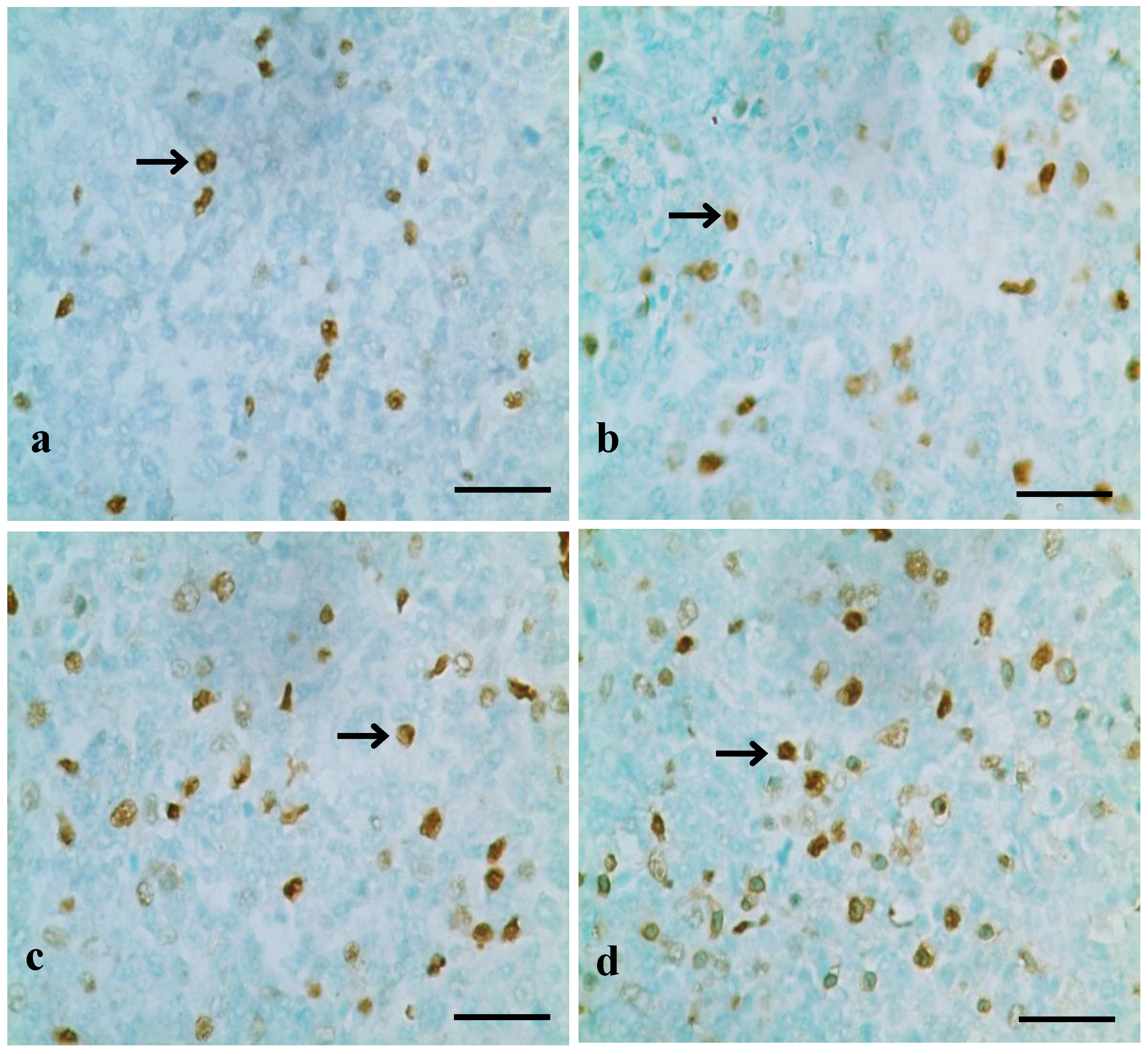
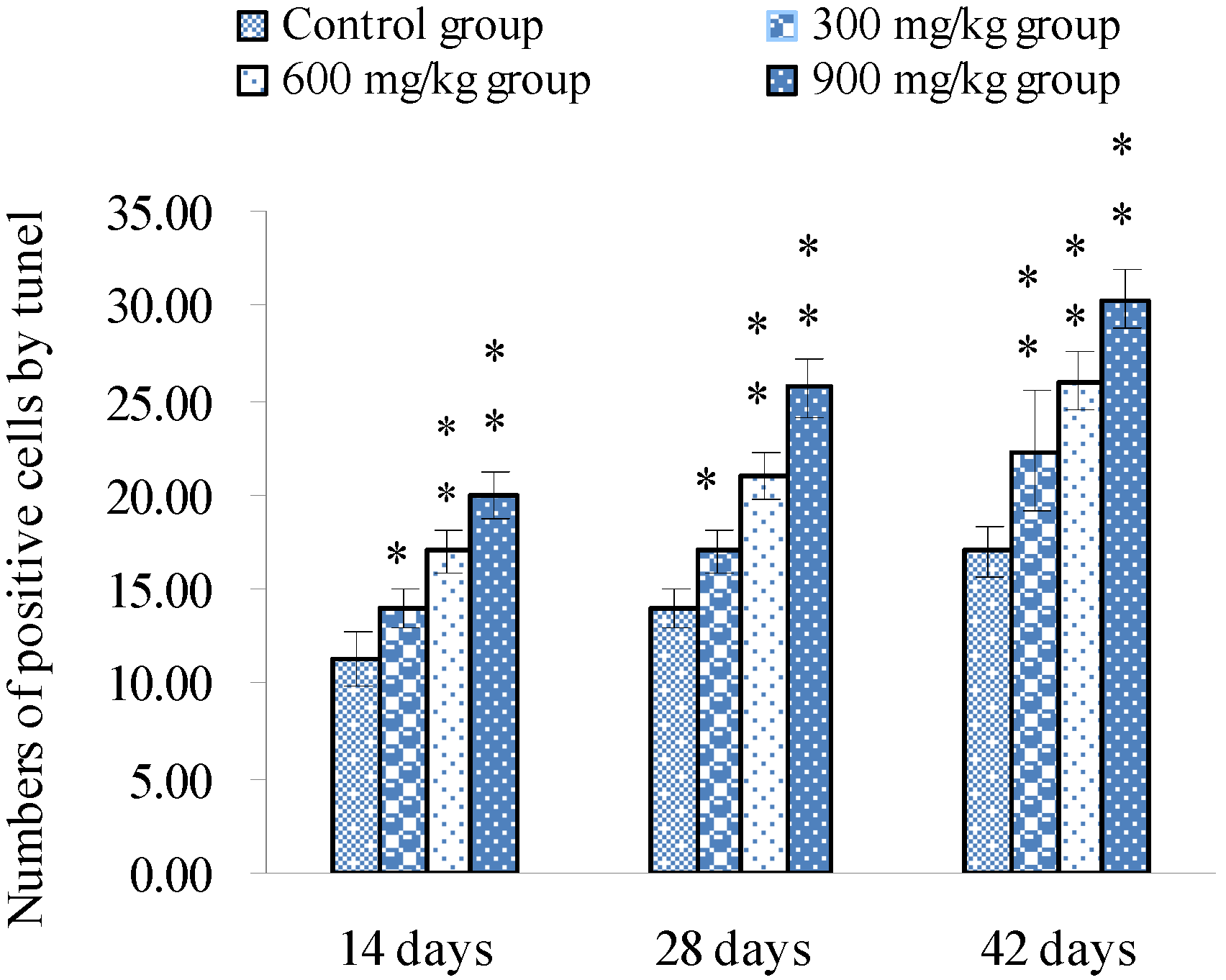
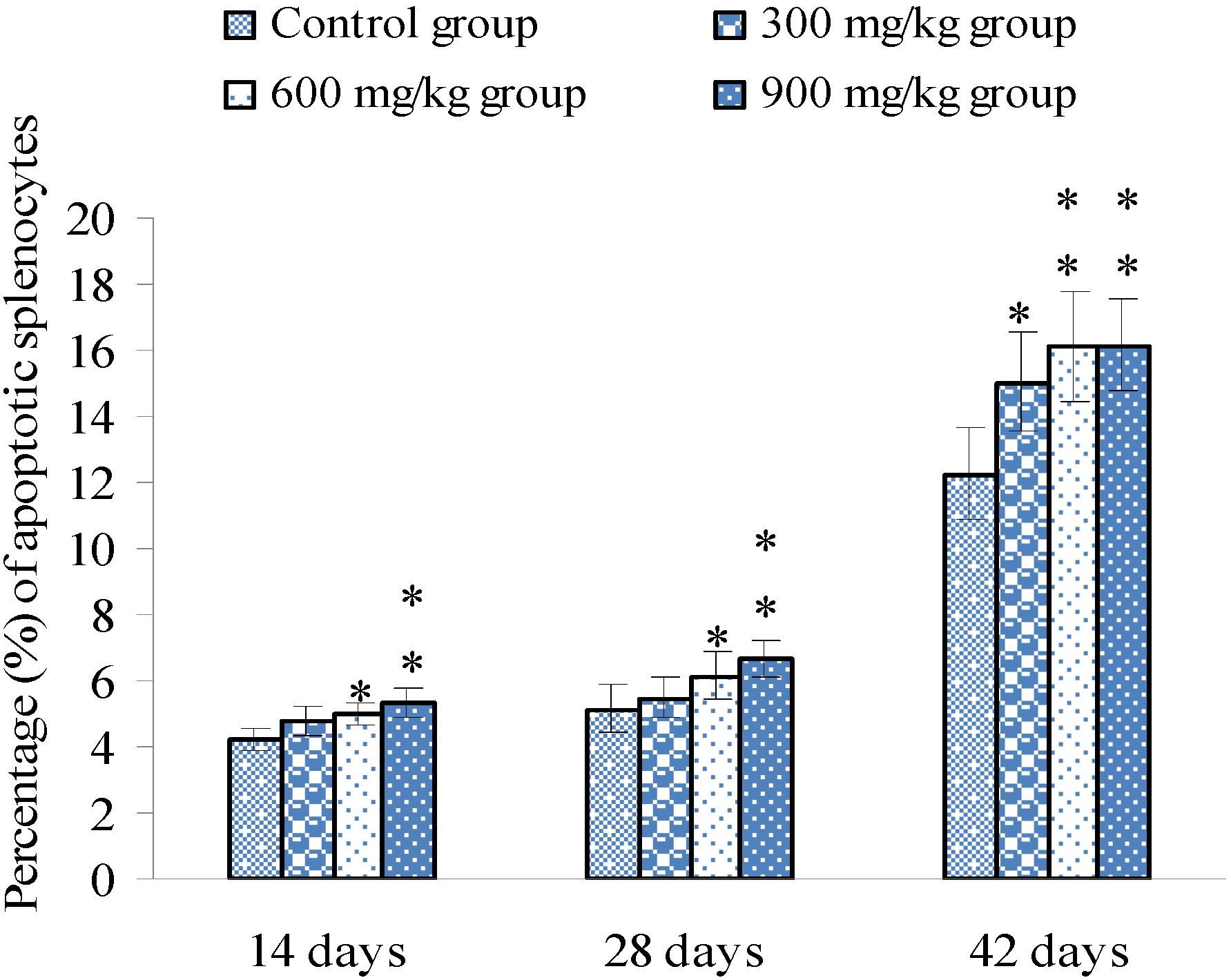
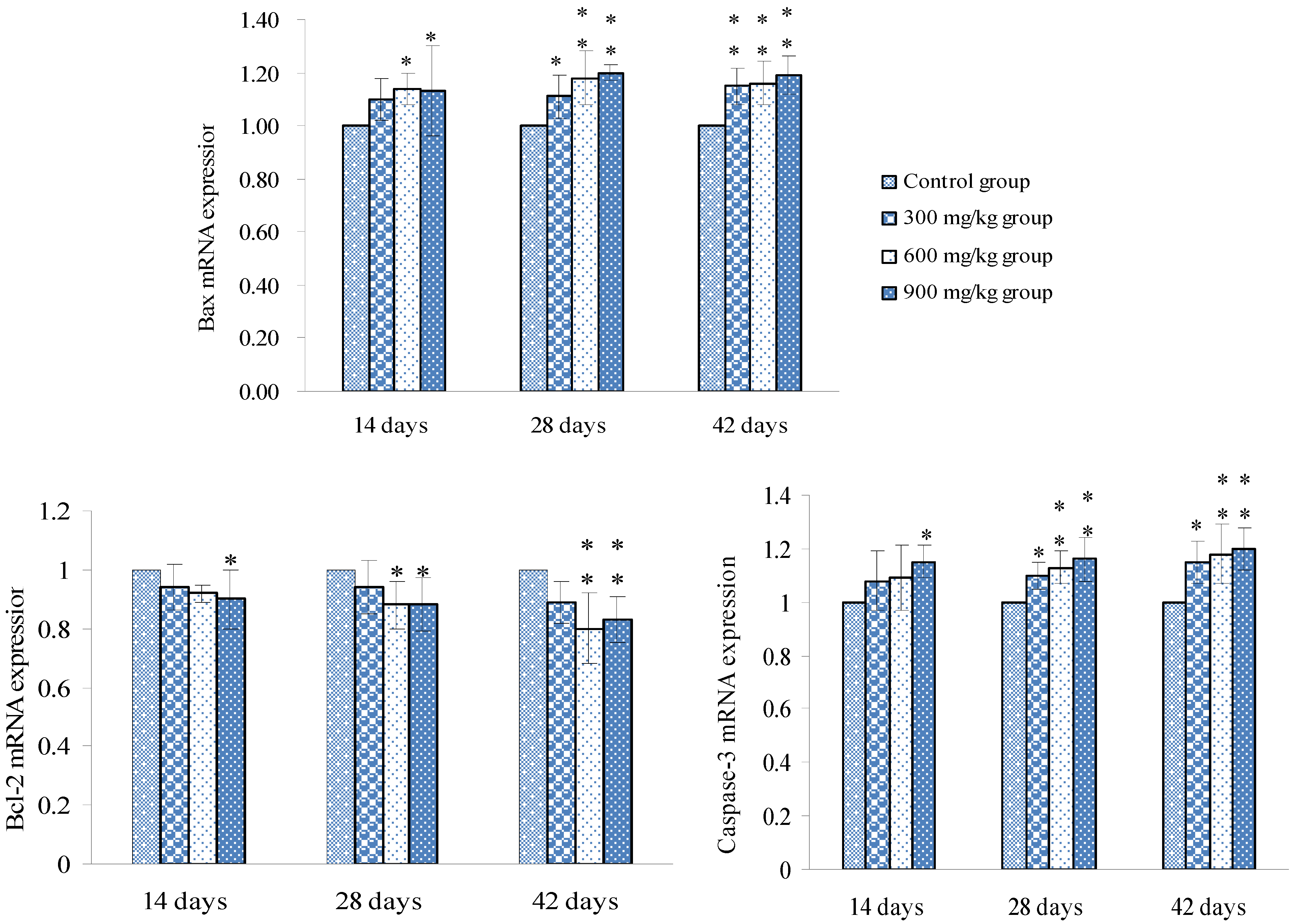
3.2. Changes of Bax, Bcl-2 and Caspase-3 mRNA Expression Levels and Contents in the Spleen
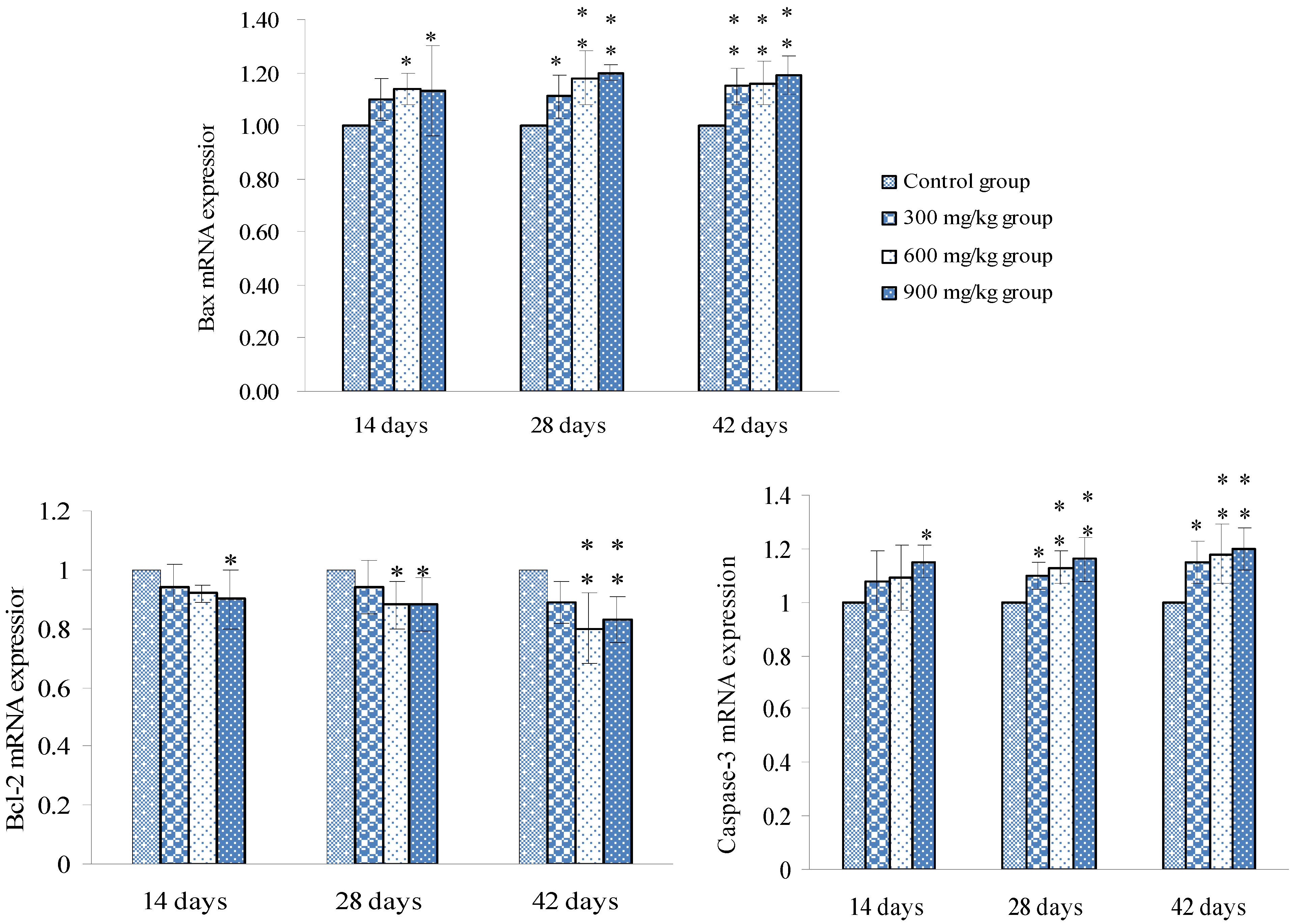
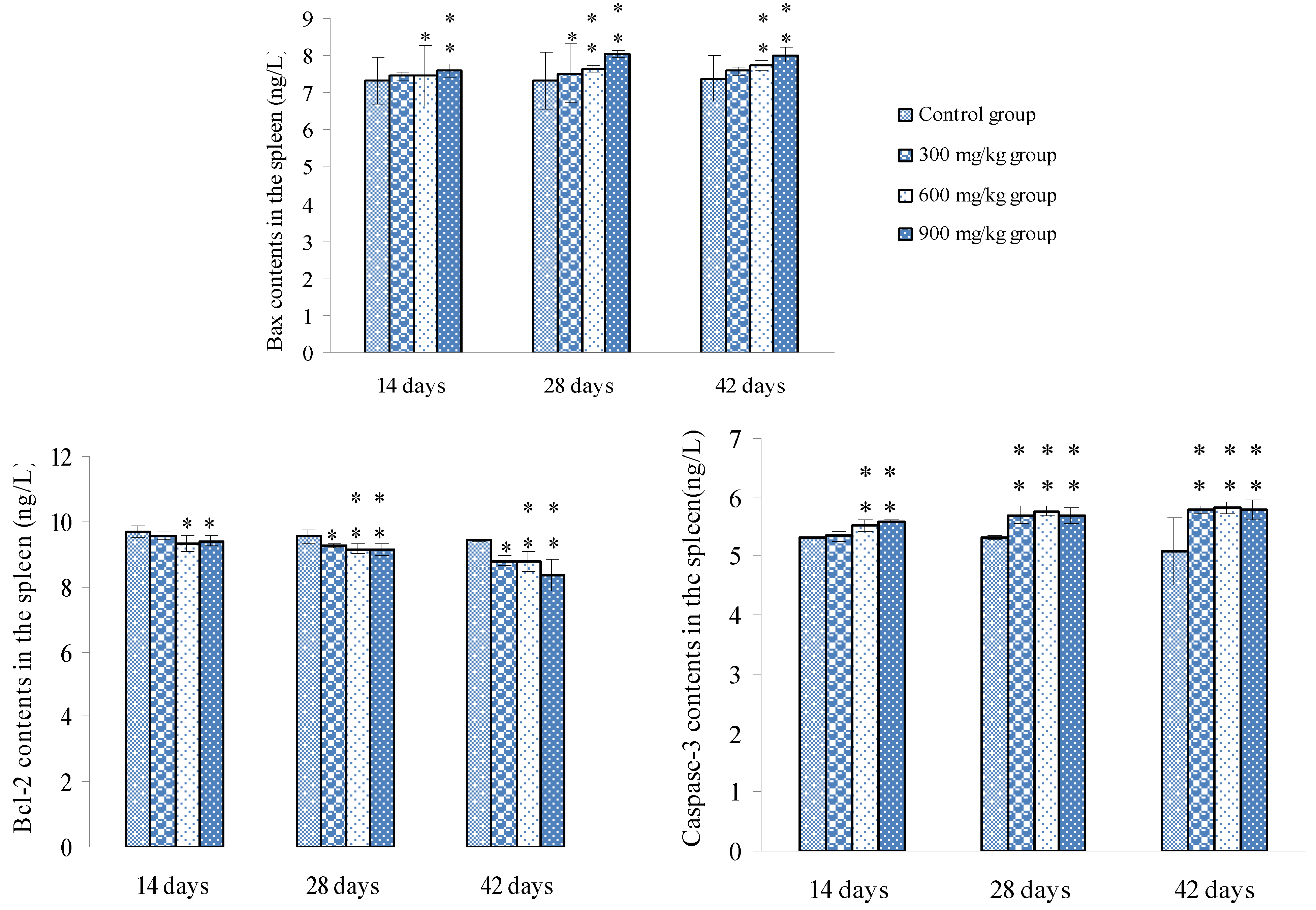
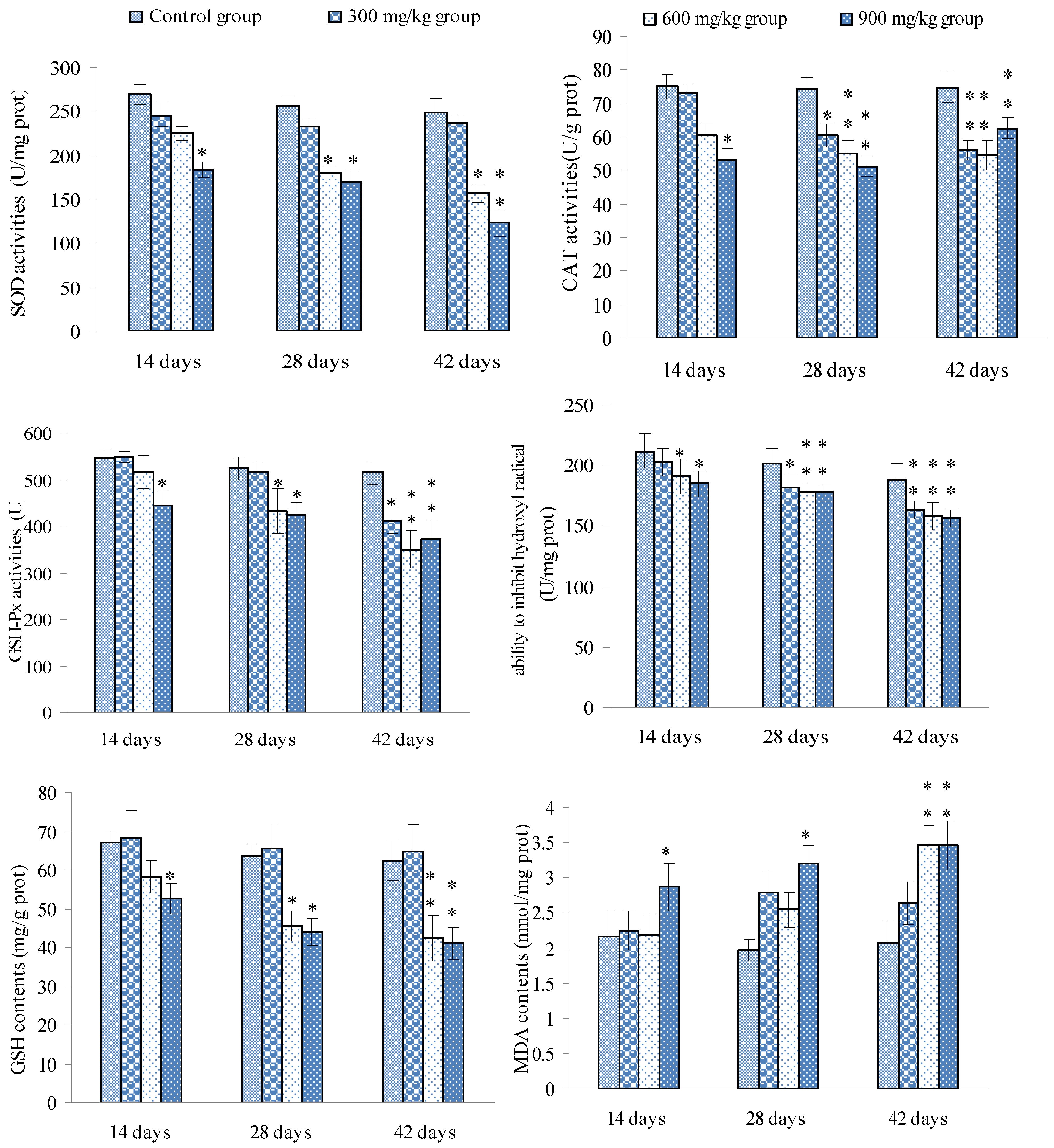
3.3. Changes of the Oxidative Stress Parameters in the Spleen

4. Discussion
5. Conclusions
Acknowledgment
Conflicts of Interest
References
- Scott-Fordsmand, J.J. Toxicity of nickel to soil organisms in Denmark. Rev. Environ. Contam. Toxicol. 1997, 148, 1–34. [Google Scholar]
- Haber, L.; Erdreicht, L.; Diamond, G.; Maier, A.; Ratney, R.; Zhao, Q.; Dourson, M. Hazard identification and dose response of inhaled nickel-soluble salts. Regul. Toxicol. Pharmacol. 2000, 31, 210–230. [Google Scholar] [CrossRef]
- Diagomanolin, V.; Farhang, M.; Ghazi-Khansari, M.; Jafarzadeh, N. Heavy metals (Ni, Cr, Cu) in the Karoon waterway river, Iran. Toxicol. Lett. 2004, 151, 63–67. [Google Scholar] [CrossRef]
- Coogan, T.P.; Latta, D.M.; Snow, E.T.; Costa, M.; Lawrence, A. Toxicity and carcinogenicity of nickel compounds. CRC Crit. Rev. Toxicol. 1989, 19, 341–384. [Google Scholar] [CrossRef]
- Grandjean, P. Human exposure to nickel. IARC Sci. Publ. 1984, 53, 469–485. [Google Scholar]
- Friberg, L.; Elinder, C.-G. Biological Monitoring of Toxic Metals. Scand. J. Work Environ. Health 1993, 19, 7–13. [Google Scholar]
- Von Burg, D.R. Toxicology update. J. Appl. Toxicol. 1999, 19, 379–386. [Google Scholar] [CrossRef]
- Anke, M.; Grun, M.; Dittrich, G.; Groppel, B.; Hennig, A. Low nickel rations for growth and reproduction in pigs. In International Symposium on Trace Element Metabolism Inanimal; University Park Press: Madison, WI, USA, 1974; Volume 2, pp. 715–718. [Google Scholar]
- Afridi, H.I.; Kazi, T.G.; Kazi, N.; Kandhro, G.A.; Baig, J.A.; Shah, A.Q.; Wadhwa, S.K.; Khan, S.; Kolachi, N.F.; Shah, F. Evaluation of status of cadmium, lead, and nickel levels in biological samples of normal and night blindness children of age groups 3–7 and 8–12 years. Biol. Trace Elem. Res. 2011, 142, 350–361. [Google Scholar] [CrossRef]
- Bencko, V. Nickel: A review of its occupational and environmental toxicology. J. Hyg. Epidemiol. Microbiol. Immunol. 1983, 27, 237–247. [Google Scholar]
- Yokoi, K.; Uthus, E.O.; Nielsen, F.H. Nickel deficiency diminishes sperm quantity and movement in rats. Biol. Trace Elem. Res. 2003, 93, 141–153. [Google Scholar] [CrossRef]
- Anke, M.; Hennig, A.; Grün, M.; Partschefeld, M.; Groppel, B.; Lüdke, H. Nickel—Ein essentielles Spurenelement. Arch. Anim. Nutr. 1977, 27, 25–38. [Google Scholar]
- Schnegg, A.; Kirchgessner, M. Ni deficiency and its effects on metabolism. Trace Elem. Metab. Man Anim. 1978, 3, 236–243. [Google Scholar]
- Cempel, M.; Nikel, G. Nickel: A review of its sources and environmental toxicology. Pol. J. Environ. Studies 2006, 15, 375–382. [Google Scholar]
- Samal, L.; Mishra, C. Significance of Nickel in Livestock Health and Production. Inter. J. Agro Vet. Med. Sci. 2011, 5, 349–361. [Google Scholar] [CrossRef]
- Kasprzak, K.S.; Sunderman, F.W., Jr; Salnikow, K. Nickel carcinogenesis. Mutat. Res. 2003, 533, 67–97. [Google Scholar] [CrossRef]
- Haley, P.J.; Bice, D.E.; Muggenburg, B.A.; Hann, F.F.; Benjamin, S.A. Immunopathologic effects of nickel subsulfide on the primate pulmonary immune system. Toxicol. Appl. Pharmacol. 1987, 88, 1–12. [Google Scholar] [CrossRef]
- Donskoy, E.; Donskoy, M.; Forouhar, F.; Gillies, C.; Marzouk, A.; Reid, M.; Zaharia, O.; Sunderman, F. Hepatic toxicity of nickel chloride in rats. Ann. Clin. Lab. Sci. 1986, 16, 108–117. [Google Scholar]
- Doreswamy, K.; Shrilatha, B.; Rajeshkumar, T. Nickel-Induced oxidative stress in testis of mice: Evidence of DNA damage and genotoxic effects. J. Androl. 2004, 25, 996–1003. [Google Scholar]
- Sunderman, F.W., Jr. Mechanisms of nickel carcinogenesis. Scand. J. Work Environ. Health 1989, 15, 1–12. [Google Scholar] [CrossRef]
- Costa, M.; Klein, C.B. Nickel carcinogenesis, mutation, epigenetics, or selection. Environ. Health Persp. 1999, 107, A438–A439. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Sheu, J.-Y.; Lin, T.-H. Oxidative effects of nickel on bone marrow and blood of rats. J. Toxicol. Environ. Health 1999, 58, 475–483. [Google Scholar] [CrossRef]
- Stohs, S.; Bagchi, D. Oxidative mechanisms in the toxicity of metal ions. Free Radic. Biol. Med. 1995, 18, 321–336. [Google Scholar] [CrossRef]
- Chakrabarti, S.K.; Bai, C. Role of oxidative stress in nickel chloride-induced cell injury in rat renal cortical slices. Bioche. Pharm. 1999, 58, 1501–1510. [Google Scholar] [CrossRef]
- Misra, M.; Rodriguez, R.E.; Kasprzak, K.S. Nickel induced lipid peroxidation in the rat: Correlation with nickel effect on antioxidant defense systems. Toxicol. 1990, 64, 1–17. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Wang, Y.-F.; Lin, Y.-H.; Yen, S.-F. Nickel-induced oxidative stress and effect of antioxidants in human lymphocytes. Arch. Toxicol. 2003, 77, 123–130. [Google Scholar]
- Wu, B.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Huang, J. Dietary nickel chloride induces oxidative intestinal damage in broilers. Int. J. Environ. Res. Public Health 2013, 10, 2109–2119. [Google Scholar] [CrossRef]
- Rodriguez, R.E.; Misra, M.; North, S.L.; Kasprzak, K.S. Nickel-Induced lipid peroxidation in the liver of different strains of mice and its relation to nickel effects on antioxidant systems. Toxicol. Lett. 1991, 57, 269–281. [Google Scholar] [CrossRef]
- Pereira, M.; Pereira, M.; Sousa, J. Evaluation of nickel toxicity on liver, spleen, and kidney of mice after administration of high-dose metal ion. J. Biomed. Mater. Res. 1998, 40, 40–47. [Google Scholar] [CrossRef]
- Dieter, M.; Jameson, C.; Tucker, A.; Luster, M.; French, J.; Hong, H.; Boorman, G. Evaluation of tissue disposition, myelopoietic, and immunologic responses in mice after long-term exposure to nickel sulfate in the drinking water. J. Toxicol. Environ. Health 1988, 24, 357–372. [Google Scholar] [CrossRef]
- Perminova, I.; Sinel’shchikova, T.; Alekhina, N.; Perminova, E.; Zasukhina, G. Individual sensitivity to genotoxic effects of nickel and antimutagenic activity of ascorbic acid. Bull. Exp. Biol. Med. 2001, 131, 367–370. [Google Scholar] [CrossRef]
- Lee-Chen, S.F.; Wang, M.C.; Yu, C.T.; Wu, D.R.; Jan, K.Y. Nickel chloride inhibits the DNA repair of UV-treated but not methyl methanesulfonate-treated Chinese hamster ovary cells. Biol. Trace Elem. Res. 1993, 37, 39–50. [Google Scholar] [CrossRef]
- Dally, H.; Hartwig, A. Induction and repair inhibition of oxidative DNA damage by nickel (II) and cadmium (II) in mammalian cells. Carcinog. 1997, 18, 1021–1026. [Google Scholar] [CrossRef]
- Au, A.; Ha, J.; Hernandez, M.; Polotsky, A.; Hungerford, D.S.; Frondoza, C.G. Nickel and vanadium metal ions induce apoptosis of T-lymphocyte Jurkat cells. J. Biomed. Mater. Res. 2006, 79, 512–521. [Google Scholar]
- Kim, K.; Lee, S.-H.; Seo, Y.-R.; Perkins, S.N.; Kasprzak, K.S. Nickel (II)-induced apoptosis in murine T cell hybridoma cells is associated with increased fas ligand expression. Toxicol. Appl. Pharmacol. 2002, 185, 41–47. [Google Scholar] [CrossRef]
- Ahamed, M.; Akhtar, M.J.; Siddiqui, M.A.; Ahmad, J.; Musarrat, J.; Al-Khedhairy, A.A.; AlSalhi, M.S.; Alrokayan, S.A. Oxidative stress mediated apoptosis induced by nickel ferrite nanoparticles in cultured A549 cells. Toxicol. 2011, 283, 101–108. [Google Scholar] [CrossRef]
- Graham, J.; Miller, F.; Daniels, M.; Payne, E.; Gardner, D. Influence of cadmium, nickel, and chromium on primary immunity in mice. Environ. Res. 1978, 16, 77–87. [Google Scholar] [CrossRef]
- Jaramillo, A.; Sonnenfeld, G. Potentiation of lymphocyte proliferative responses by nickel sulfide. Oncol. 1992, 49, 396–406. [Google Scholar] [CrossRef]
- Warner, G.L.; Lawrence, D.A. Stimulation of murine lymphocyte responses by cations. Cell. Immunol. 1986, 101, 425–439. [Google Scholar] [CrossRef]
- Smialowicz, R.; Rogers, R.R.; Riddle, M.M.; Garner, R.; Rowe, D.; Luebke, R. Immunologic effects of nickel: II. Suppression of natural killer cell activity. Environ. Res. 1985, 36, 56–66. [Google Scholar] [CrossRef]
- Gaça, M.D.; Pickering, J.A.; Arthur, M.J.; Benyon, R.C. Human and rat hepatic stellate cells produce stem cell factor: a possible mechanism for mast cell recruitment in liver fibrosis. J. Hepatol. 1999, 30, 850–858. [Google Scholar] [CrossRef]
- Lee, S.-H. Early gene expression in mouse spleen cells after exposure to nickel acetate. J. Toxicol. Public Health 2006, 22, 95–102. [Google Scholar]
- Shirkey, R.; Chakraborty, J.; Bridges, J. An improved method for preparing rat small intestine microsomal fractions for studying drug metabolism. Anal. Biochem. 1979, 93, 73–81. [Google Scholar] [CrossRef]
- Gagliano, N.; Donne, I.D.; Torri, C.; Migliori, M.; Grizzi, F.; Milzani, A.; Filippi, C.; Annoni, G.; Colombo, P.; Costa, F. Early cytotoxic effects of ochratoxin A in rat liver: A morphological, biochemical and molecular study. Toxicol. 2006, 225, 214–224. [Google Scholar] [CrossRef]
- Bersényi, A.; Fekete, S.G.; Szilágyi, M.; Berta, E.; Zöldág, L.; Glávits, R. Effects of nickel supply on the fattening performance and several biochemical parameters of broiler chickens and rabbits. Acta Vet. Hung. 2004, 52, 185–197. [Google Scholar] [CrossRef]
- Martinez, D.; Diaz, G. Effect of graded levels of dietary nickel and manganese on blood haemoglobin content and pulmonary hypertension in broiler chickens. Avian Pathol. 1996, 25, 537–549. [Google Scholar] [CrossRef]
- Wilson, J.H.; Wilson, E.J.; Ruszler, P.L. Dietary nickel improves male broiler (Gallus domesticus) bone strength. Biol. Trace Elem. Res. 2001, 83, 239–249. [Google Scholar] [CrossRef]
- Ling, J.R.; Leach, R.M. Studies on nickel metabolism: Interaction with other elements. Poult. Sci. 1979, 58, 591–596. [Google Scholar] [CrossRef]
- Kremling, K. The distribution of cadmium, copper, nickel, manganese, and aluminium in surface waters of the open Atlantic and European shelf area. Deep Sea Research Part A. Oceanogr. Res. Pap. 1985, 32, 531–555. [Google Scholar] [CrossRef]
- National Research Council (NRC). Nutrient Requirements of Poultry; National Academy Press: Washington, DC, USA, 1994. [Google Scholar]
- Sun, L.; Lam, W.; Wong, Y.; Lam, L.; Tang, H.; Wai, M.; Mak, Y.; Pan, F.; Yew, D. Permanent deficits in brain functions caused by long-term ketamine treatment in mice. Hum. Exp. Toxicol. 2011, 30, 1287–1296. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Wei, M.C.; Zong, W.-X.; Cheng, E.H.-Y.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Sci. 2001, 292, 727–730. [Google Scholar] [CrossRef]
- Bratton, S.B.; MacFarlane, M.; Cain, K.; Cohen, G.M. Protein complexes activate distinct caspase cascades in death receptor and stress-induced apoptosis. Exp. Cell Res. 2000, 256, 27–33. [Google Scholar] [CrossRef]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar] [CrossRef]
- Martin, L.J.; Liu, Z.; Pipino, J.; Chestnut, B.; Landek, M.A. Molecular regulation of DNA damage-induced apoptosis in neurons of cerebral cortex. Cereb. Cortex 2009, 19, 1273–1293. [Google Scholar]
- Sun, X.-M.; MacFarlane, M.; Zhuang, J.; Wolf, B.B.; Green, D.R.; Cohen, G.M. Distinct caspase cascades are initiated in receptor-mediated and chemical-induced apoptosis. J. Biol. Chem. 1999, 274, 5053–5060. [Google Scholar] [CrossRef]
- Marnett, L.J. Lipid peroxidation—DNA damage by malondialdehyde. Mutat. Res. 1999, 424, 83–95. [Google Scholar] [CrossRef]
- Desagher, S.; Osen-Sand, A.; Nichols, A.; Eskes, R.; Montessuit, S.; Lauper, S.; Maundrell, K.; Antonsson, B.; Martinou, J.-C. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 1999, 144, 891–901. [Google Scholar] [CrossRef]
- Saito, M.; Korsmeyer, S.J.; Schlesinger, P.H. BAX-dependent transport of cytochrome c reconstituted in pure liposomes. Nat. Cell Biol. 2000, 2, 553–555. [Google Scholar]
- Wei, M.C.; Lindsten, T.; Mootha, V.K.; Weiler, S.; Gross, A.; Ashiya, M.; Thompson, C.B.; Korsmeyer, S.J. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000, 14, 2060–2071. [Google Scholar]
- Szegezdi, E.; MacDonald, D.C.; Chonghaile, T.N.; Gupta, S.; Samali, A. Bcl-2 family on guard at the ER. Am. J. Physiol. Cell Physiol. 2009, 296, C941–C953. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, X. Cytochrome C-mediated apoptosis. Annu. Rev. Biochem. 2004, 73, 87–106. [Google Scholar] [CrossRef]
- Chen, J.J.; Yu, B.P. Alterations in mitochondrial membrane fluidity by lipid peroxidation products. Free Radic. Biol. Med. 1994, 17, 411–418. [Google Scholar] [CrossRef]
- Thornberry, N. Caspases: a decade of death research. Cell Death Differ. 1999, 6, 1023–1027. [Google Scholar]
- Johansson, A.-C.; Appelqvist, H.; Nilsson, C.; Kågedal, K.; Roberg, K.; Öllinger, K. Regulation of apoptosis-associated lysosomal membrane permeabilization. Apoptosis 2010, 15, 527–540. [Google Scholar] [CrossRef]
- Streit, W.J.; Kincaid-Colton, C.A. The brain’s immune system. Sci. Am. 1995, 273, 54–55. [Google Scholar] [CrossRef]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef]
- Matés, J.M.; Sánchez-Jiménez, F.M. Role of reactive oxygen species in apoptosis: Implications for cancer therapy. Int. J. Biochem. Cell Biol. 2000, 32, 157–170. [Google Scholar] [CrossRef]
- Lynn, S.; Yew, F.; Chen, K.; Jan, K. Reactive oxygen species are involved in nickel inhibition of DNA repair. Environ. Mol. Mutagen. 1997, 29, 208–216. [Google Scholar]
- Muller, F.L.; Lustgarten, M.S.; Jang, Y.; Richardson, A.; Van Remmen, H. Trends in oxidative aging theories. Free Radic. Biol. Med. 2007, 43, 477–503. [Google Scholar] [CrossRef]
- Halliwell, B.; Chirico, S. Lipid peroxidation: its mechanism, measurement, and significance. Am. J. Clin. Nutr. 1993, 57, 715S–724S. [Google Scholar]
- Reiter, R.J.; Melchiorri, D.; Sewerynek, E.; Poeggeler, B.; Barlow-Walden, L.; Chuang, J.; Ortiz, G.G.; AcuñaCastroviejo, D. A review of the evidence supporting melatonin’s role as an antioxidant. J. Pineal Res. 1995, 18, 1–11. [Google Scholar] [CrossRef]
- Fletcher, J.I.; Meusburger, S.; Hawkins, C.J.; Riglar, D.T.; Lee, E.F.; Fairlie, W.D.; Huang, D.C.; Adams, J.M. Apoptosis is triggered when prosurvival Bcl-2 proteins cannot restrain Bax. Proc. Natl. Acad. Sci. 2008, 105, 18081–18087. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, J.; Cui, H.; Peng, X.; Fang, J.; Zuo, Z.; Deng, J.; Wu, B. The Association between Splenocyte Apoptosis and Alterations of Bax, Bcl-2 and Caspase-3 mRNA Expression, and Oxidative Stress Induced by Dietary Nickel Chloride in Broilers. Int. J. Environ. Res. Public Health 2013, 10, 7310-7326. https://doi.org/10.3390/ijerph10127310
Huang J, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wu B. The Association between Splenocyte Apoptosis and Alterations of Bax, Bcl-2 and Caspase-3 mRNA Expression, and Oxidative Stress Induced by Dietary Nickel Chloride in Broilers. International Journal of Environmental Research and Public Health. 2013; 10(12):7310-7326. https://doi.org/10.3390/ijerph10127310
Chicago/Turabian StyleHuang, Jianying, Hengmin Cui, Xi Peng, Jing Fang, Zhicai Zuo, Junliang Deng, and Bangyuan Wu. 2013. "The Association between Splenocyte Apoptosis and Alterations of Bax, Bcl-2 and Caspase-3 mRNA Expression, and Oxidative Stress Induced by Dietary Nickel Chloride in Broilers" International Journal of Environmental Research and Public Health 10, no. 12: 7310-7326. https://doi.org/10.3390/ijerph10127310
APA StyleHuang, J., Cui, H., Peng, X., Fang, J., Zuo, Z., Deng, J., & Wu, B. (2013). The Association between Splenocyte Apoptosis and Alterations of Bax, Bcl-2 and Caspase-3 mRNA Expression, and Oxidative Stress Induced by Dietary Nickel Chloride in Broilers. International Journal of Environmental Research and Public Health, 10(12), 7310-7326. https://doi.org/10.3390/ijerph10127310