Calyculins and Related Marine Natural Products as Serine- Threonine Protein Phosphatase PP1 and PP2A Inhibitors and Total Syntheses of Calyculin A, B, and C

Abstract
:
1. Introduction
2. Importance of Protein Phosphatases
3. Inhibition of Protein Phosphatases PP1 and PP2A by Naturally Occurring Toxins
4. Calyculins and Related Structures
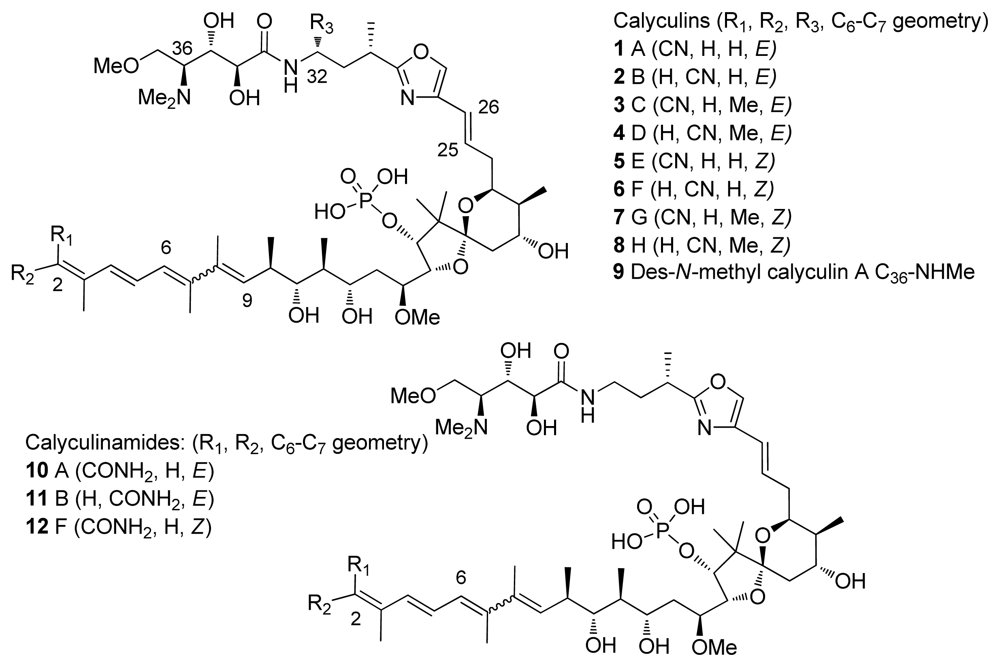
4.1. Origin
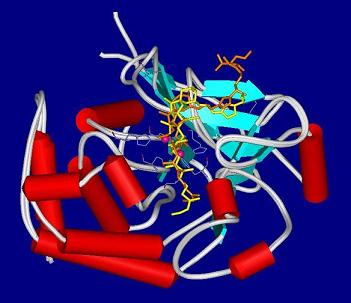
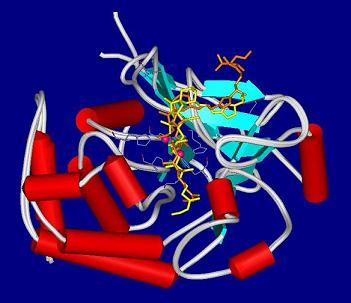
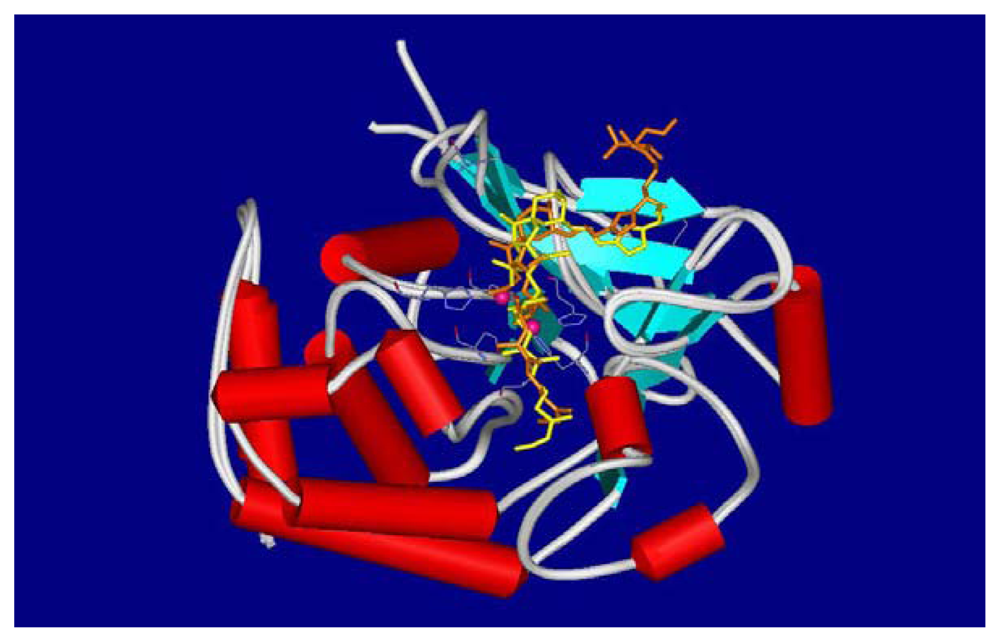
4.2. Crystal structures of calyculins and their binding to protein phosphatases
5. Synthetic Approaches towards Calyculins
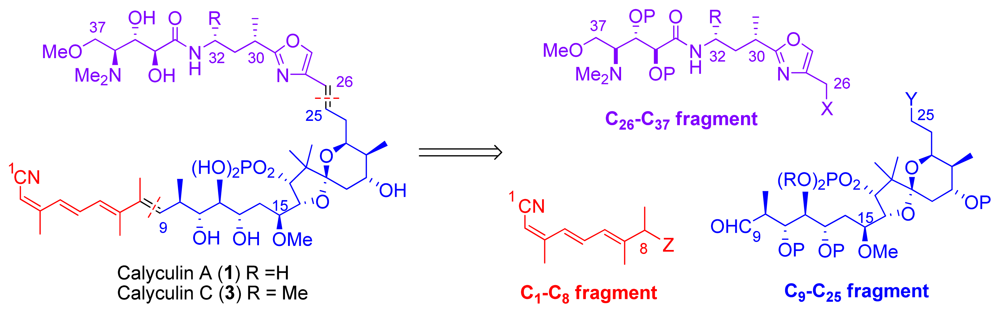
5.1. Retrosynthetic analysis
5.2. C1–C8 tetraene fragment
5.2.1. Evans [18]
5.2.2. Masamune [19]
5.2.3. Shioiri [20]
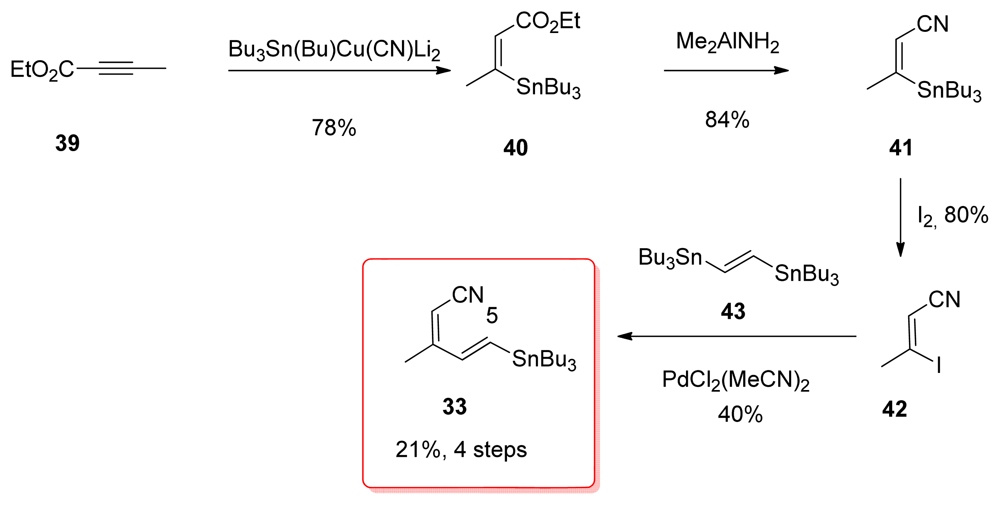
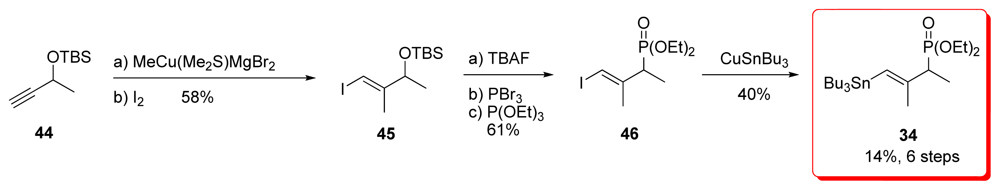
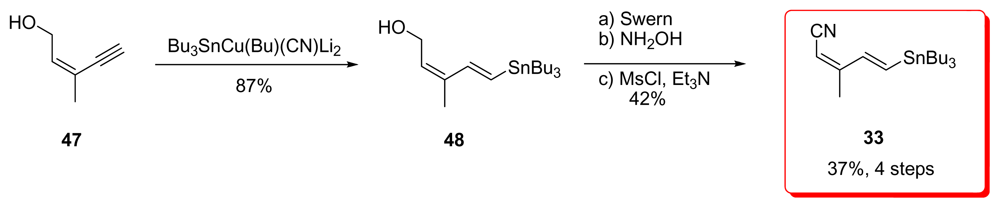
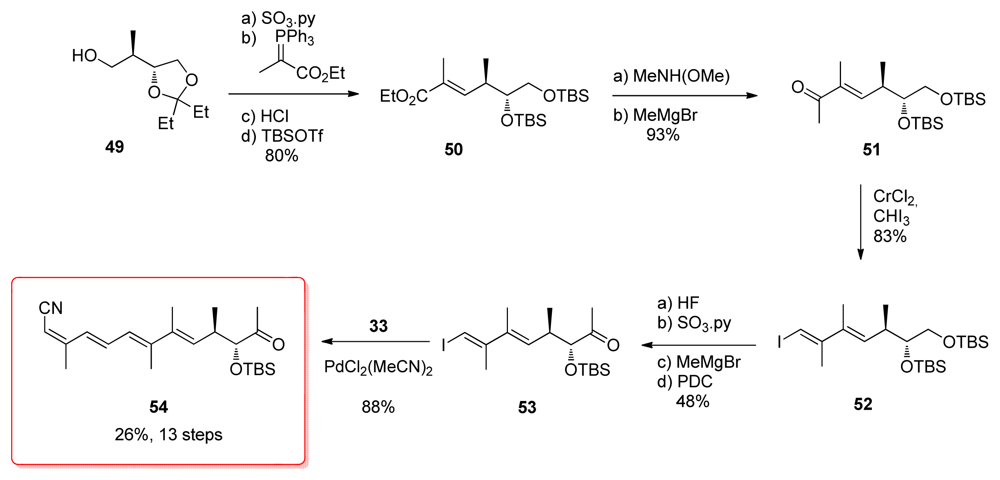
5.2.4. Smith [22,51]
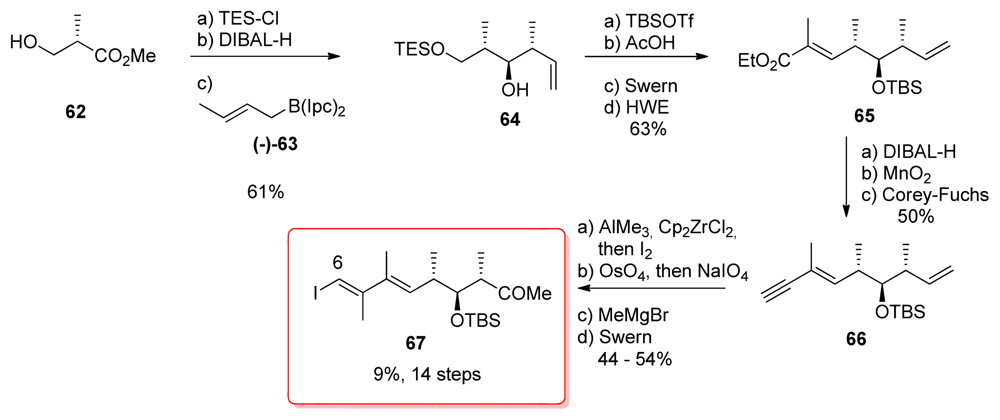
5.2.5. Armstrong [21,52]
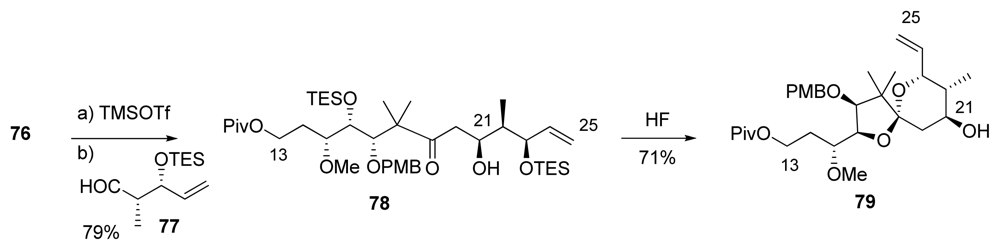
5.2.6. Barrett [23,53]
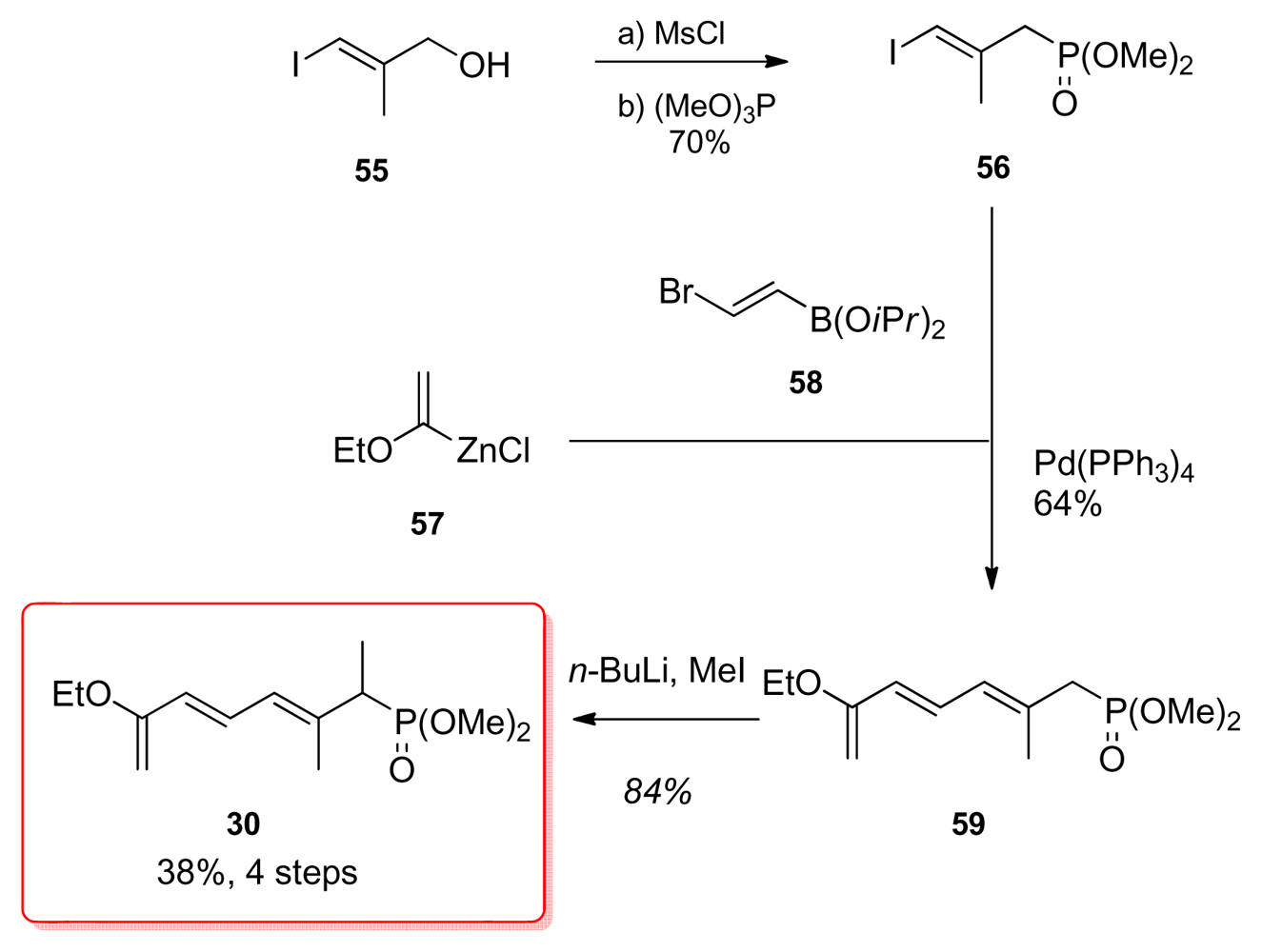
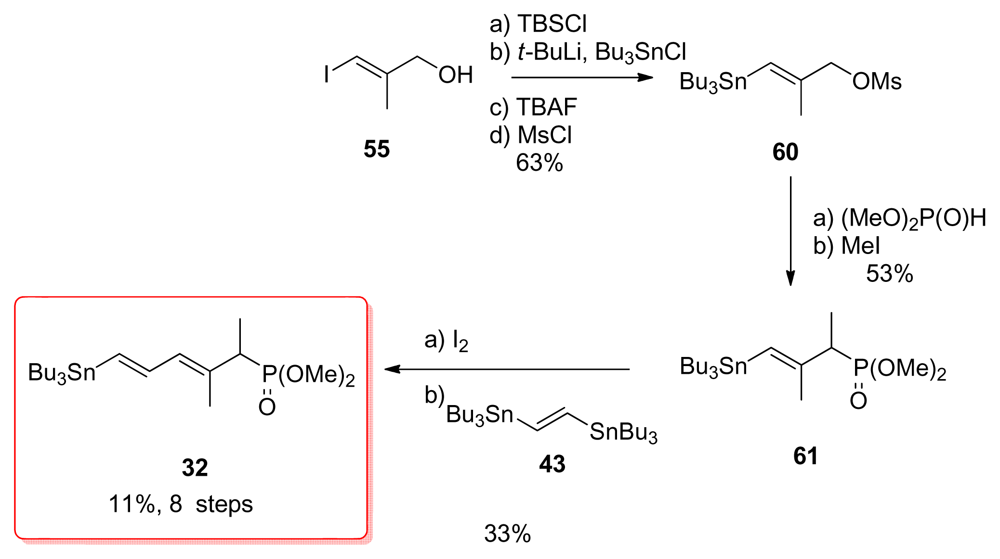
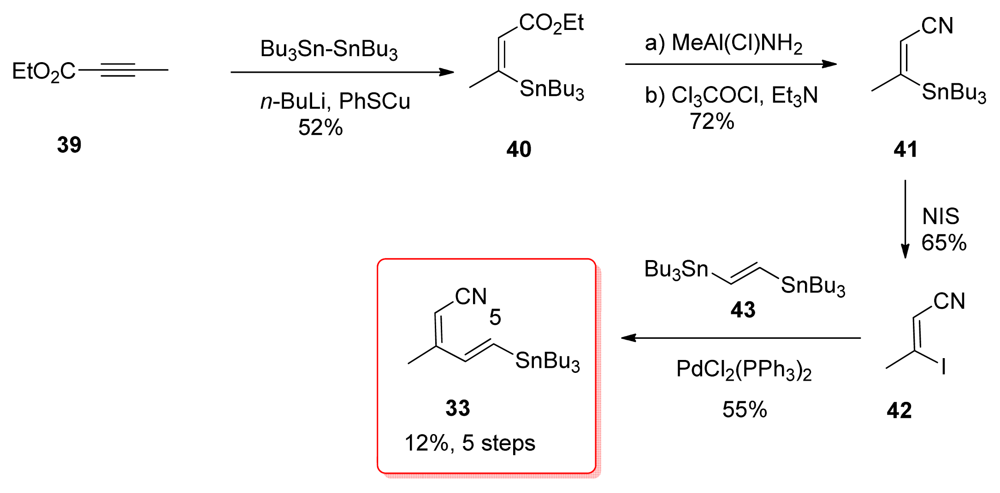
5.2.7. Koskinen [40]
5.3. Synthesis of the C9–C25 dipropionate-spiroketal subunit
5.3.1. Evans [18]
5.3.2. Masamune [19]
5.3.3. Shioiri [20,58]
5.3.4. Smith [22,59,60]
5.3.5. Armstrong [21,52]
5.3.6. Barrett [23,61]
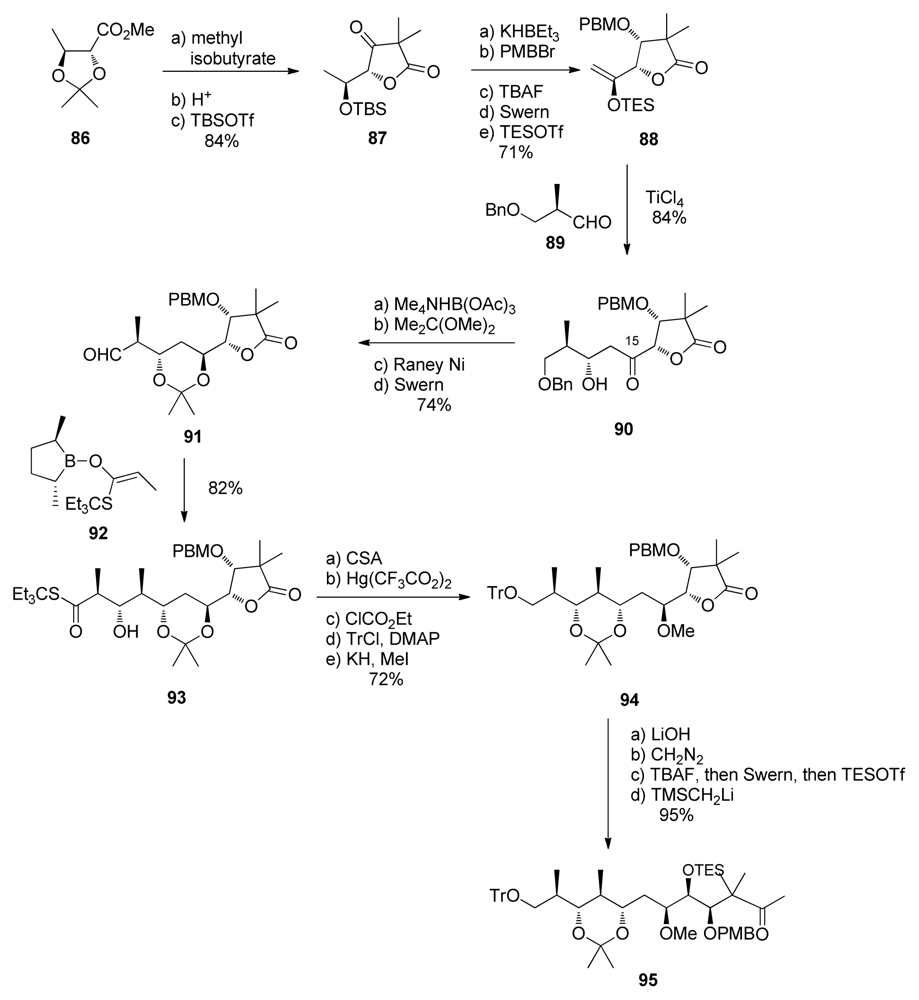
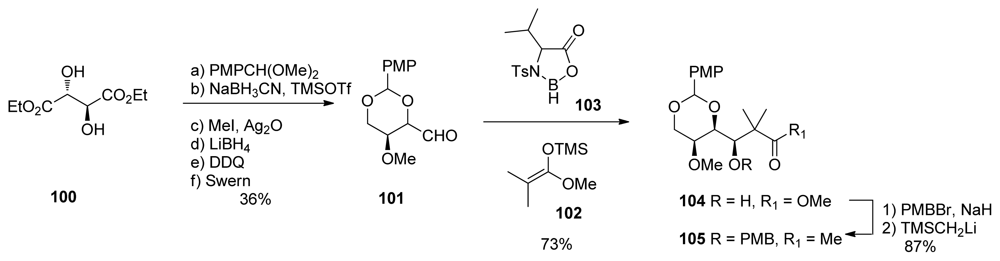
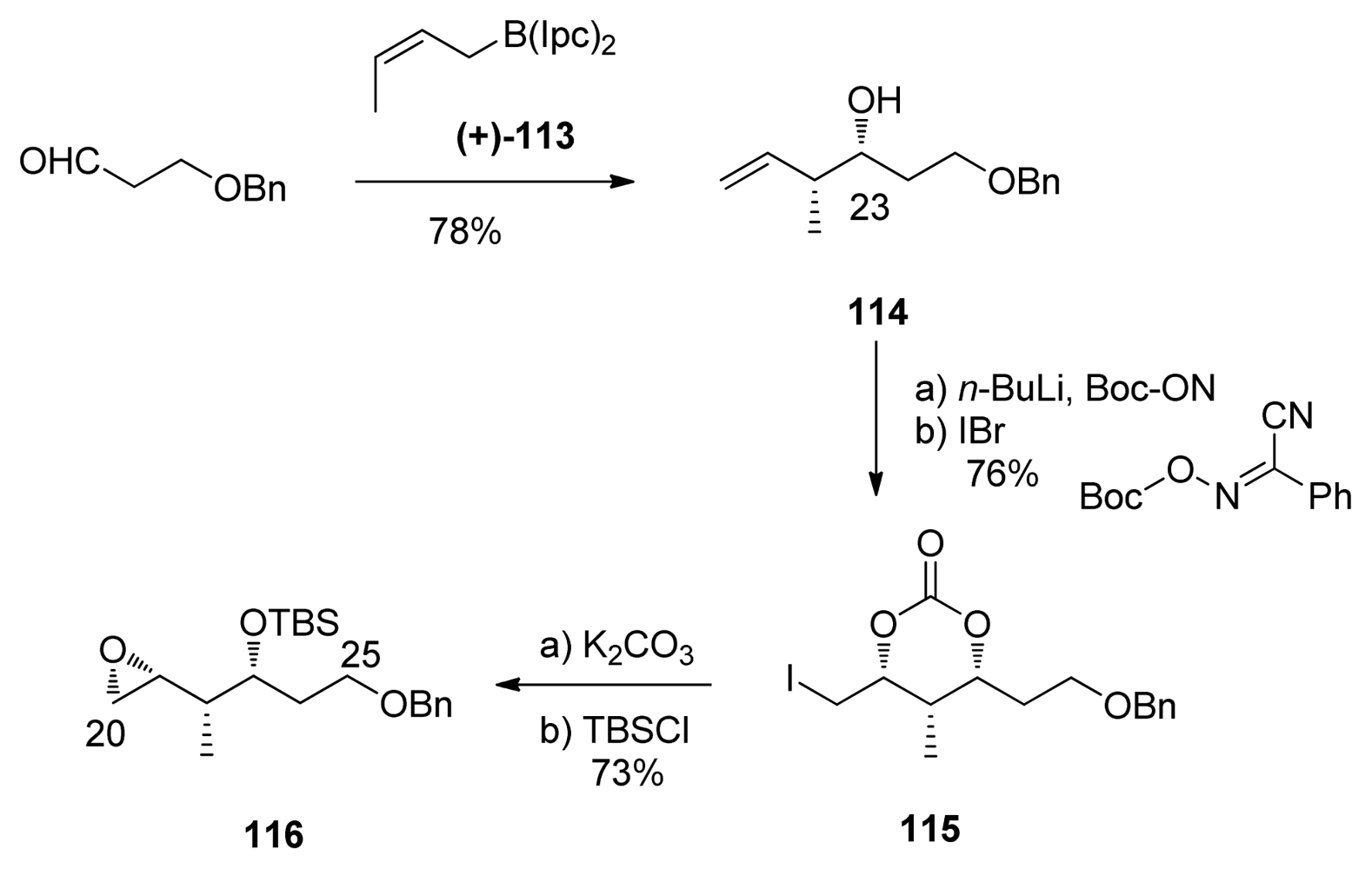
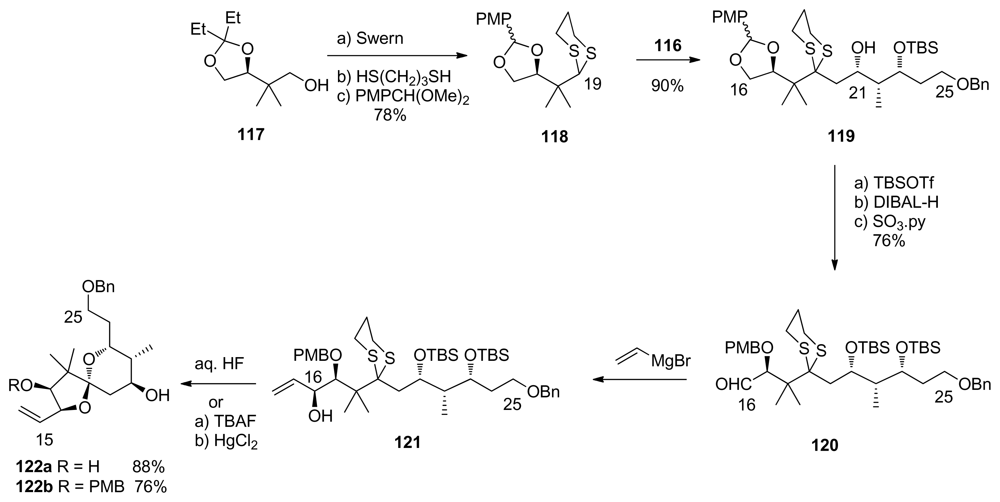
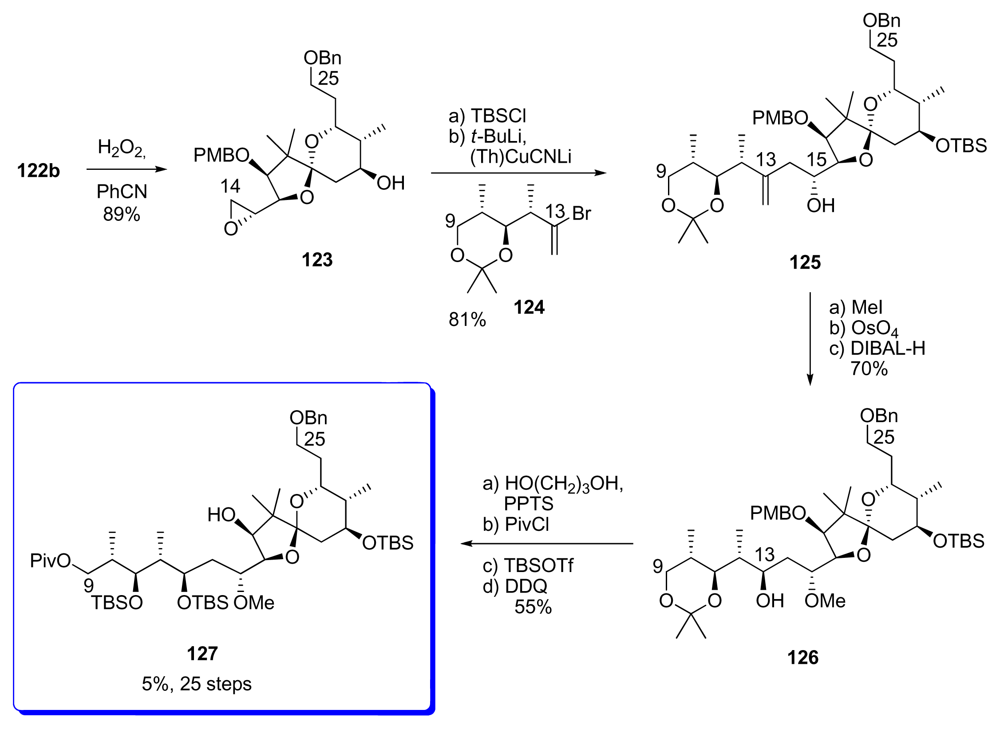
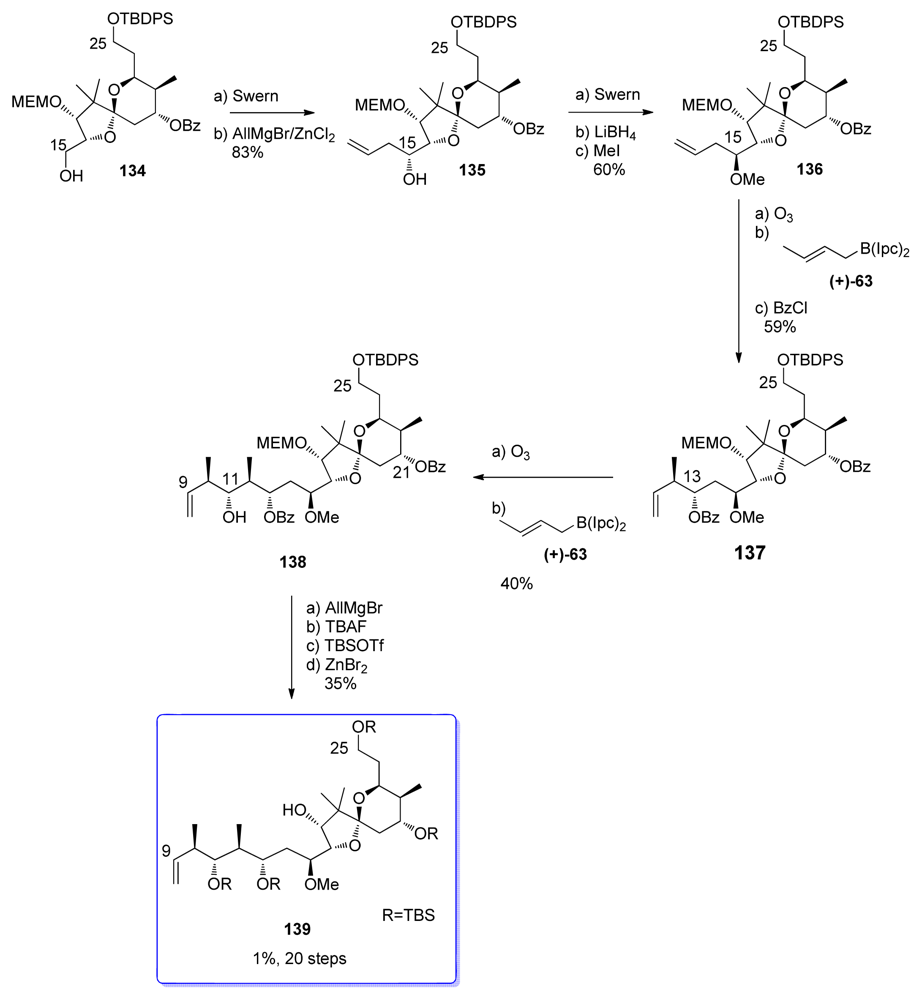
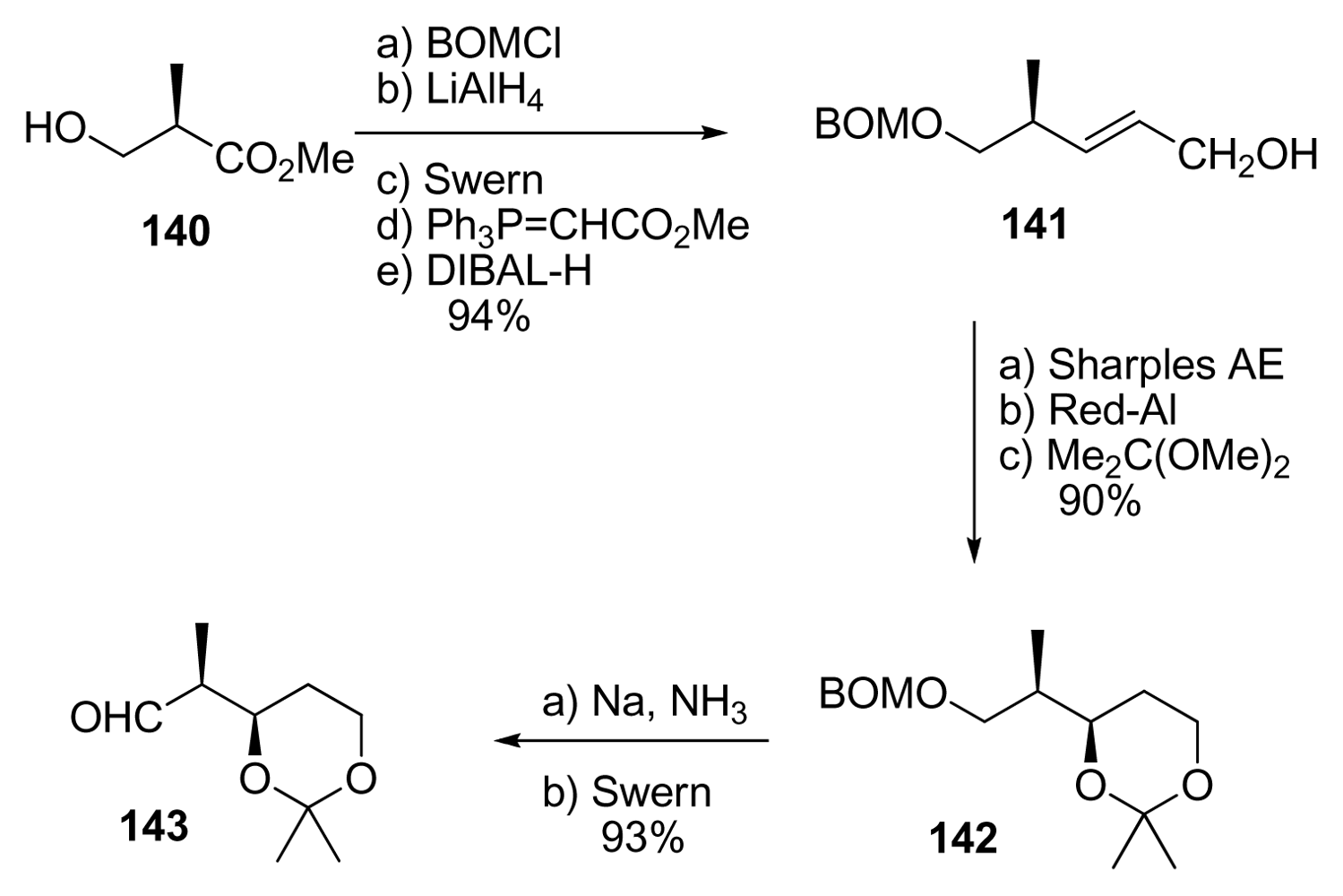
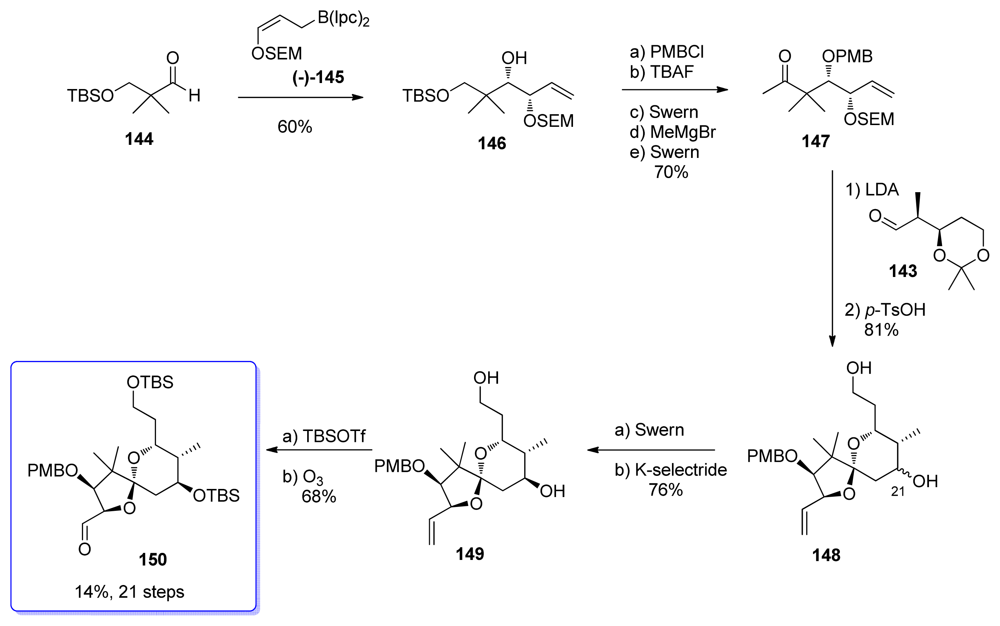
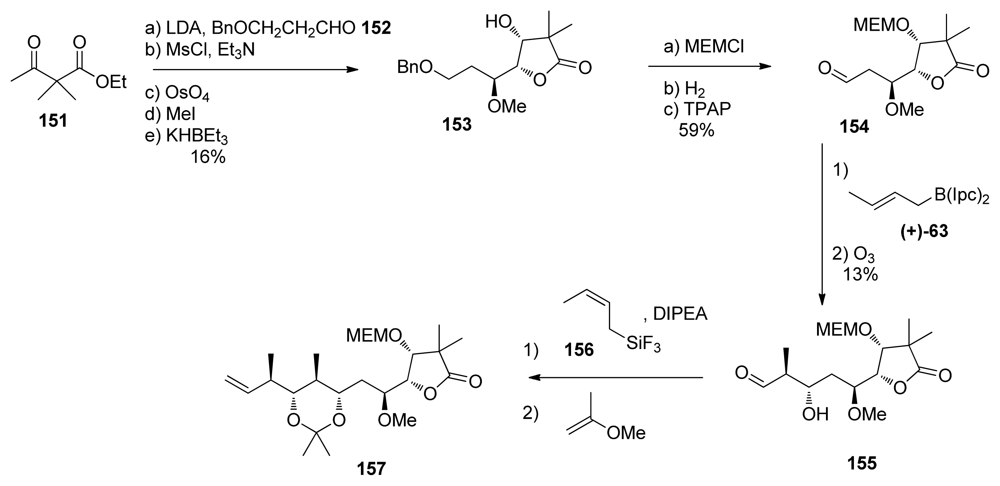
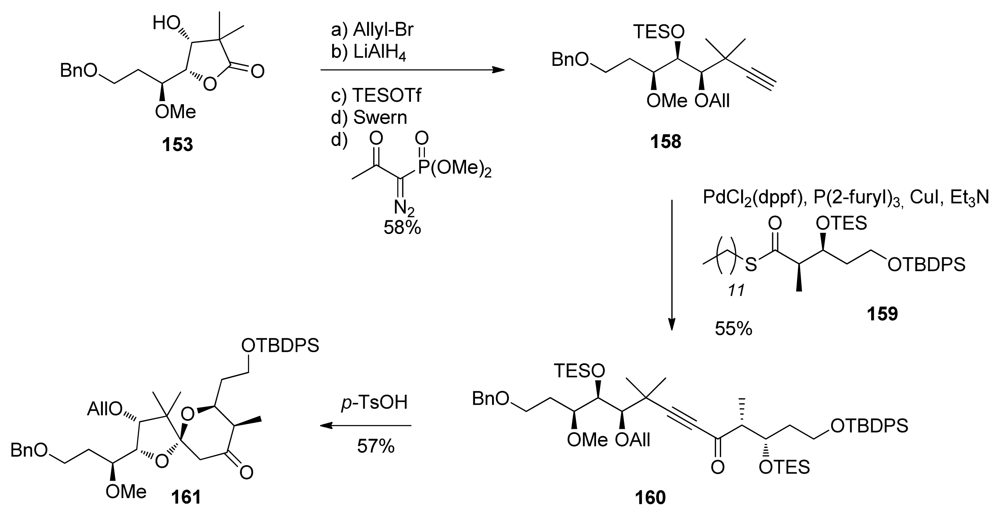
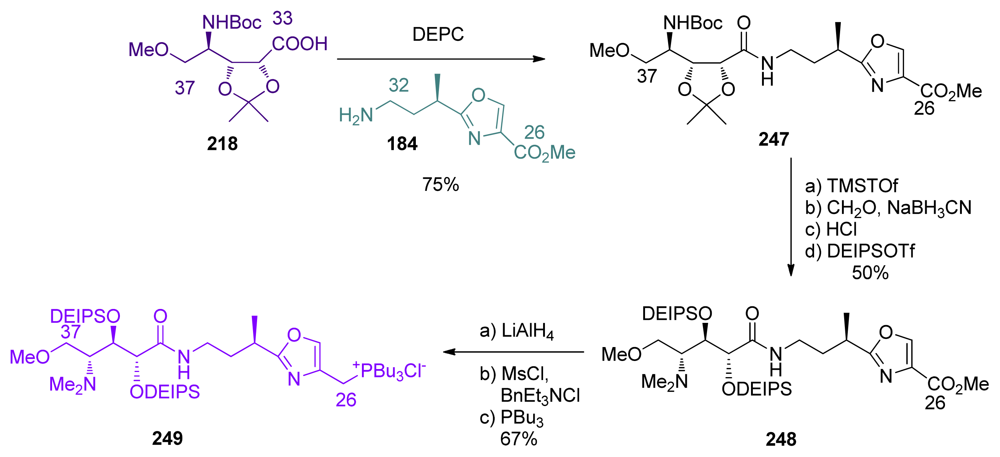
5.3.7. Koskinen [42–46]
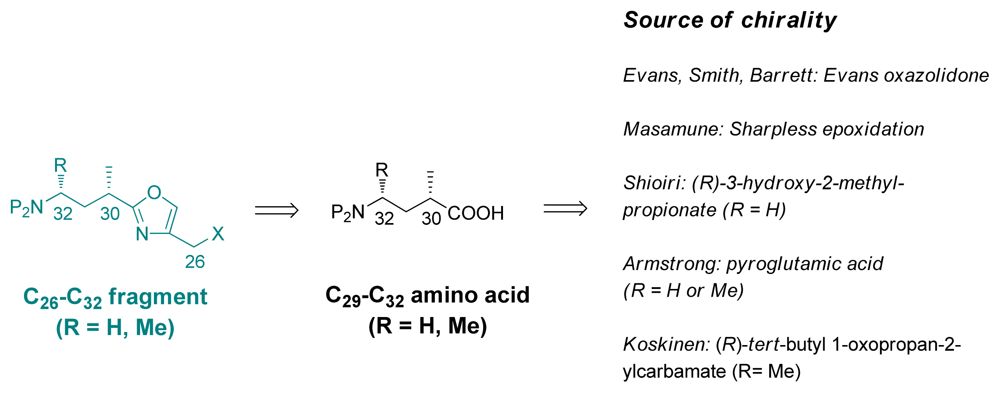
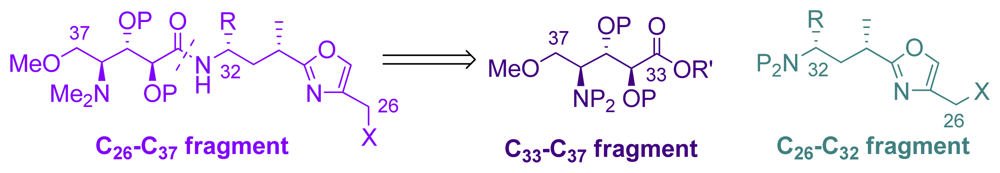
5.4. Syntheses of the C26-C32 oxazole fragment
5.4.1. Evans [18]
5.4.2. Masamune [19,63]
5.4.3. Shioiri [20,64]
5.4.4. Smith [22,51]
5.4.5. Armstrong [21,65]
5.4.6. Barrett [23,66]
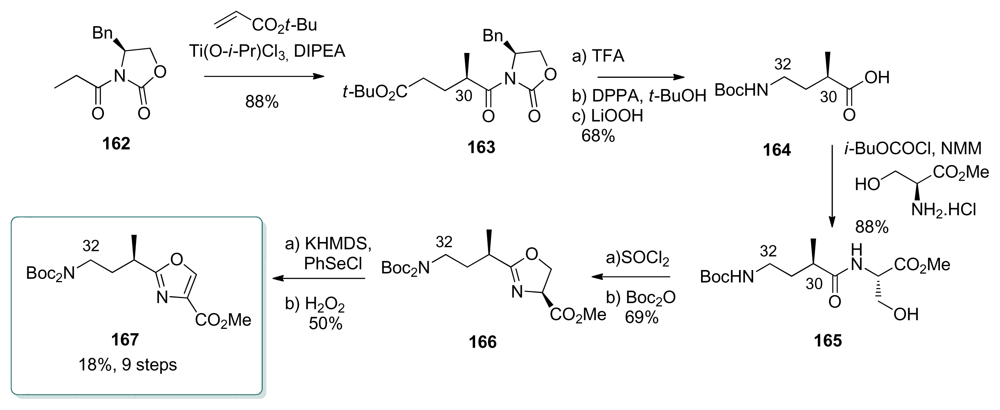
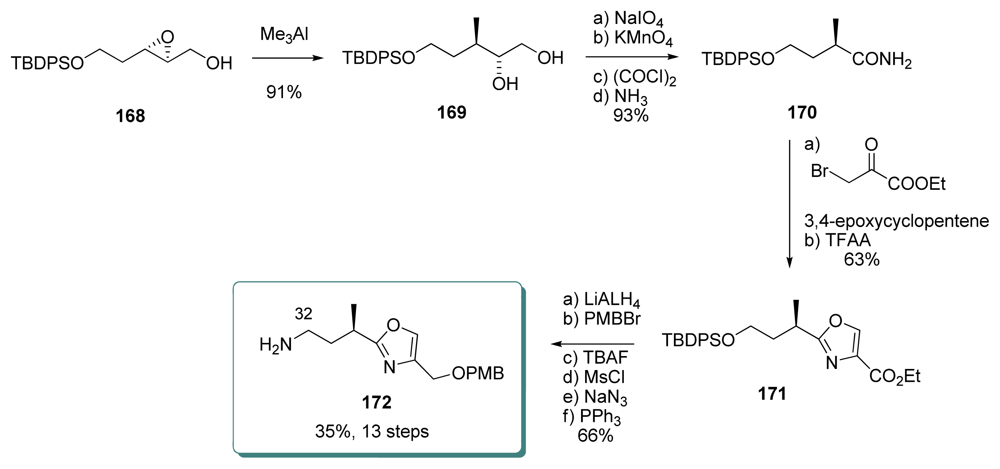
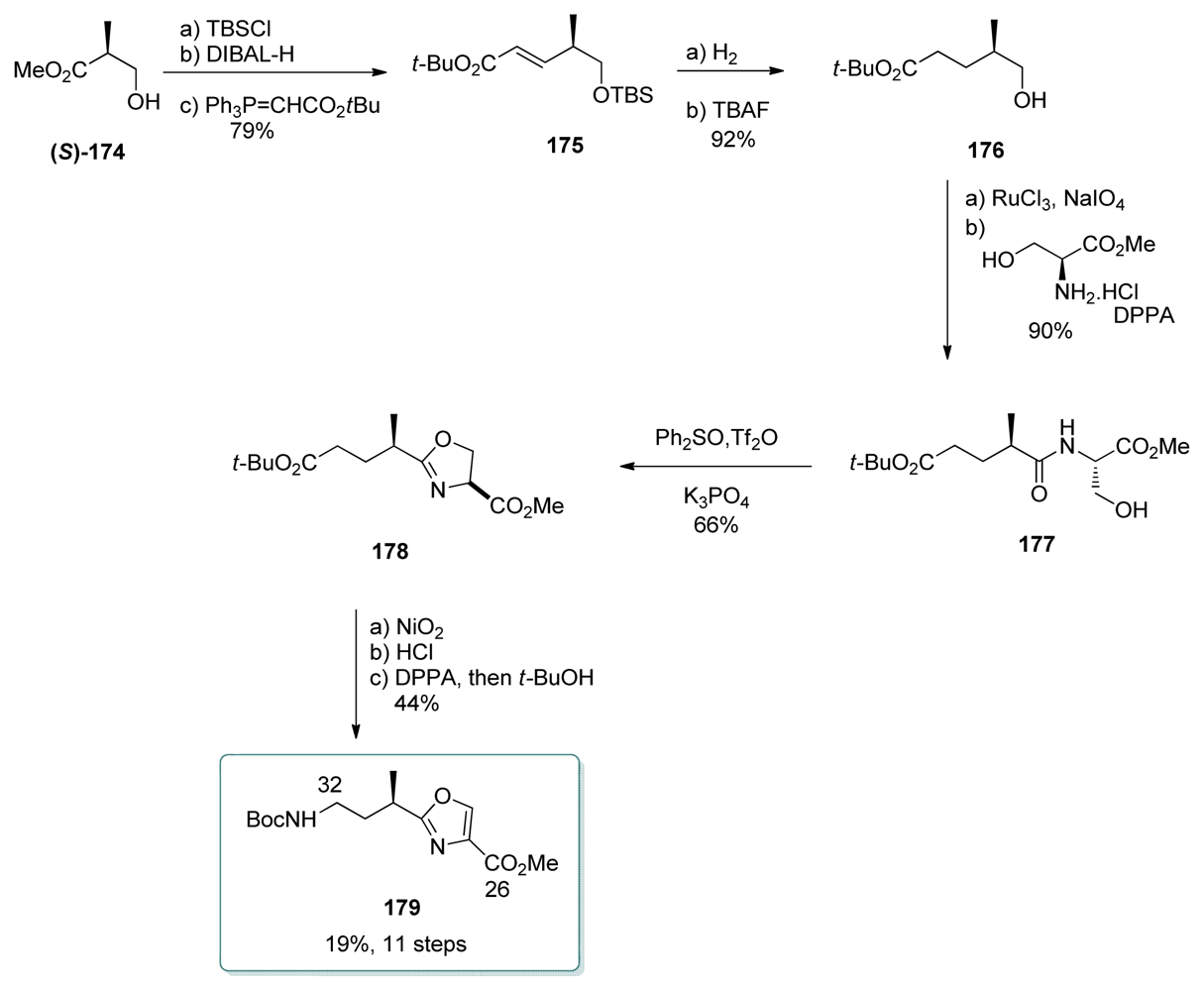
5.4.7. Koskinen [39]
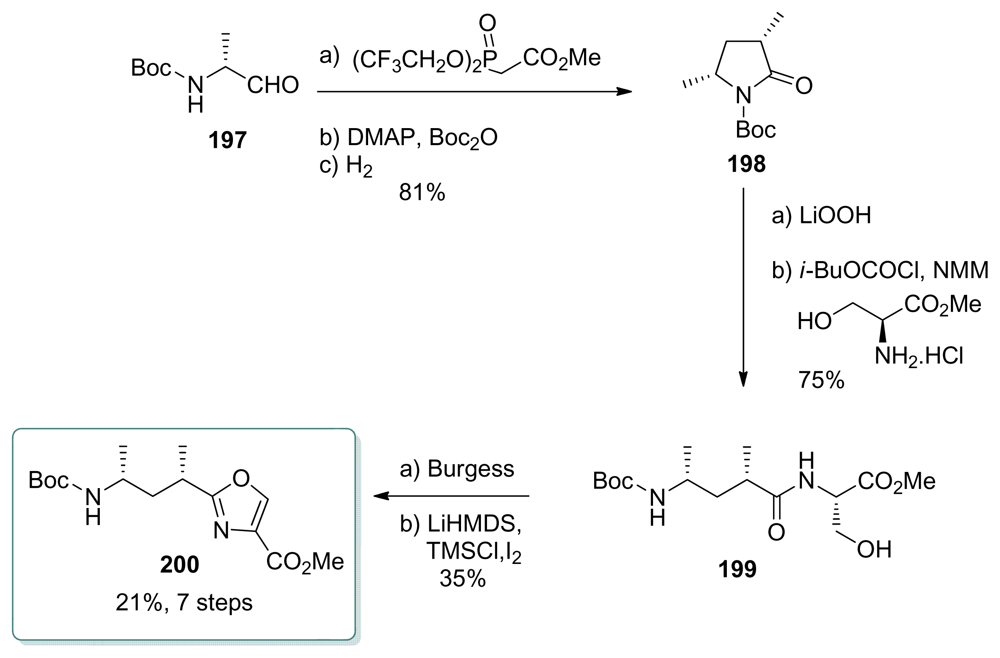
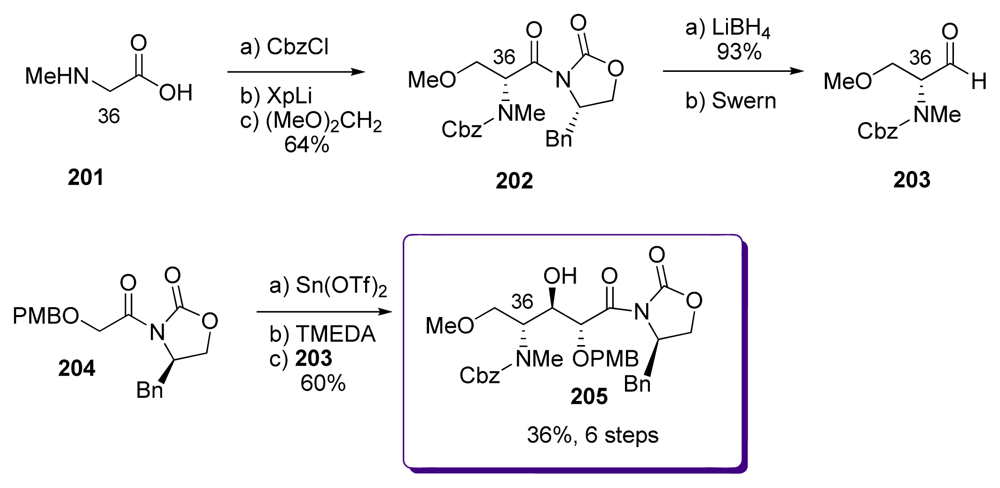
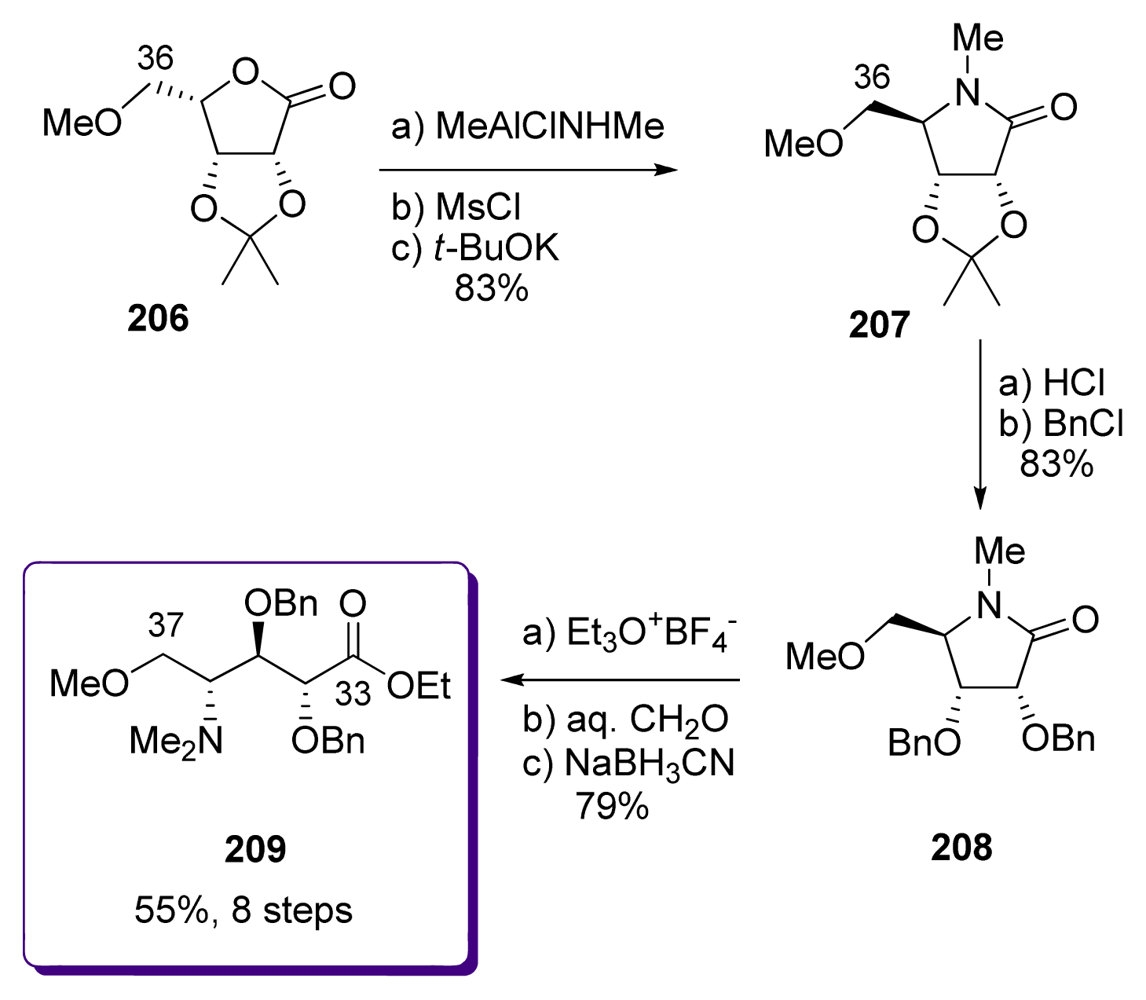
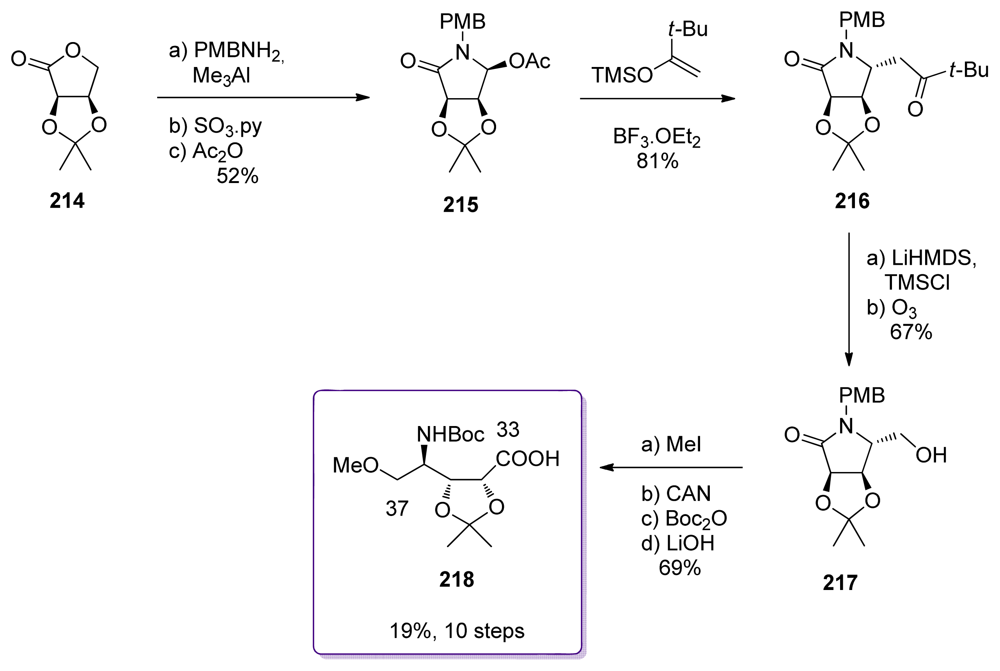
5.5. Syntheses of the C33–C37 amino acid fragment
5.5.1. Evans [18]
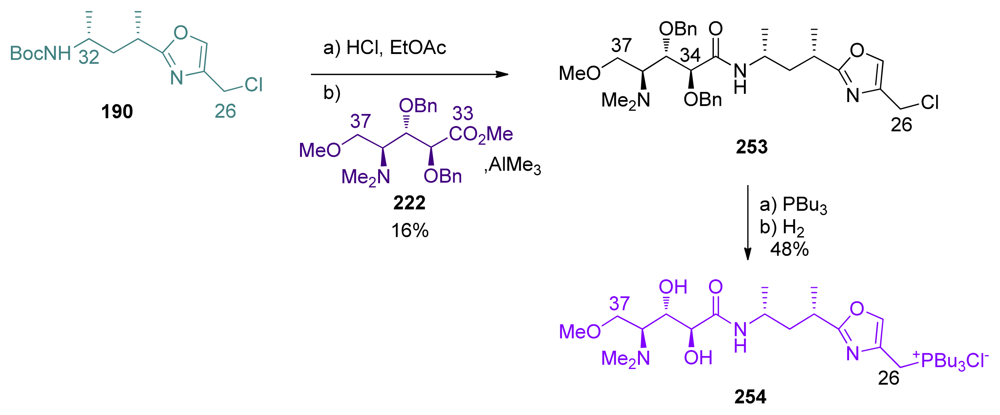
5.5.2. Masamune [19,63]
5.5.3. Shioiri [20,67]
5.5.4. Smith [22,51]
5.5.5. Armstrong [21,65]
5.5.6. Barrett [23,66]
5.5.7. Koskinen [38]
5.6. Finishing the total synthesis: introduction of phosphonate and assembly of fragments
5.6.1. Total synthesis of ent-calyculin A by Evans [18]
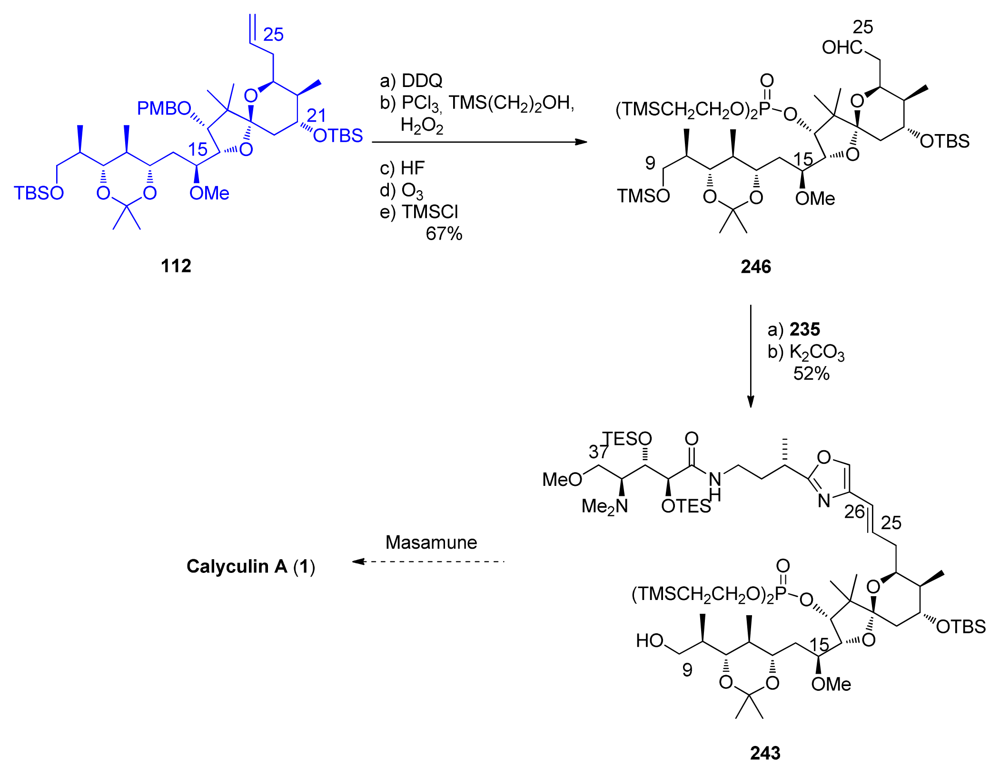
5.6.2. Total synthesis of calyculin A by Masamune [19,63]
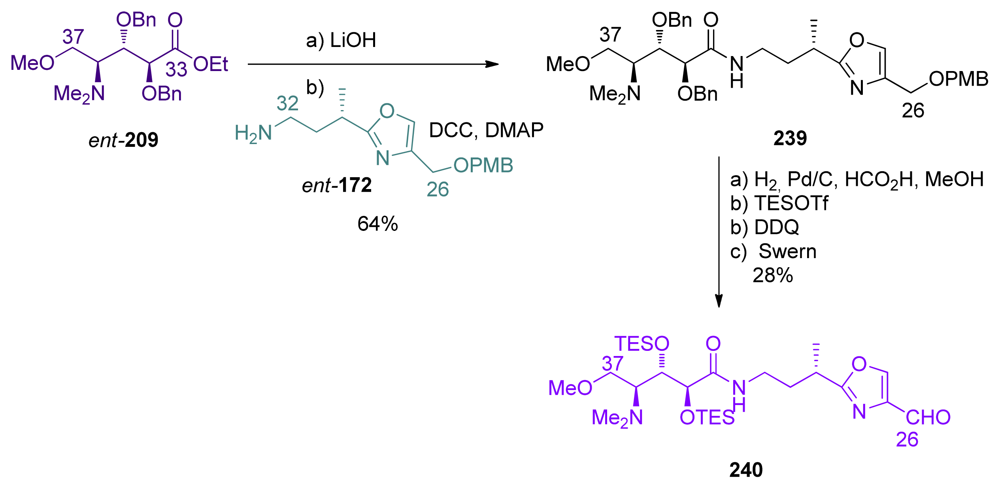
5.6.3. Formal total synthesis of calyculin A by Shioiri [20]
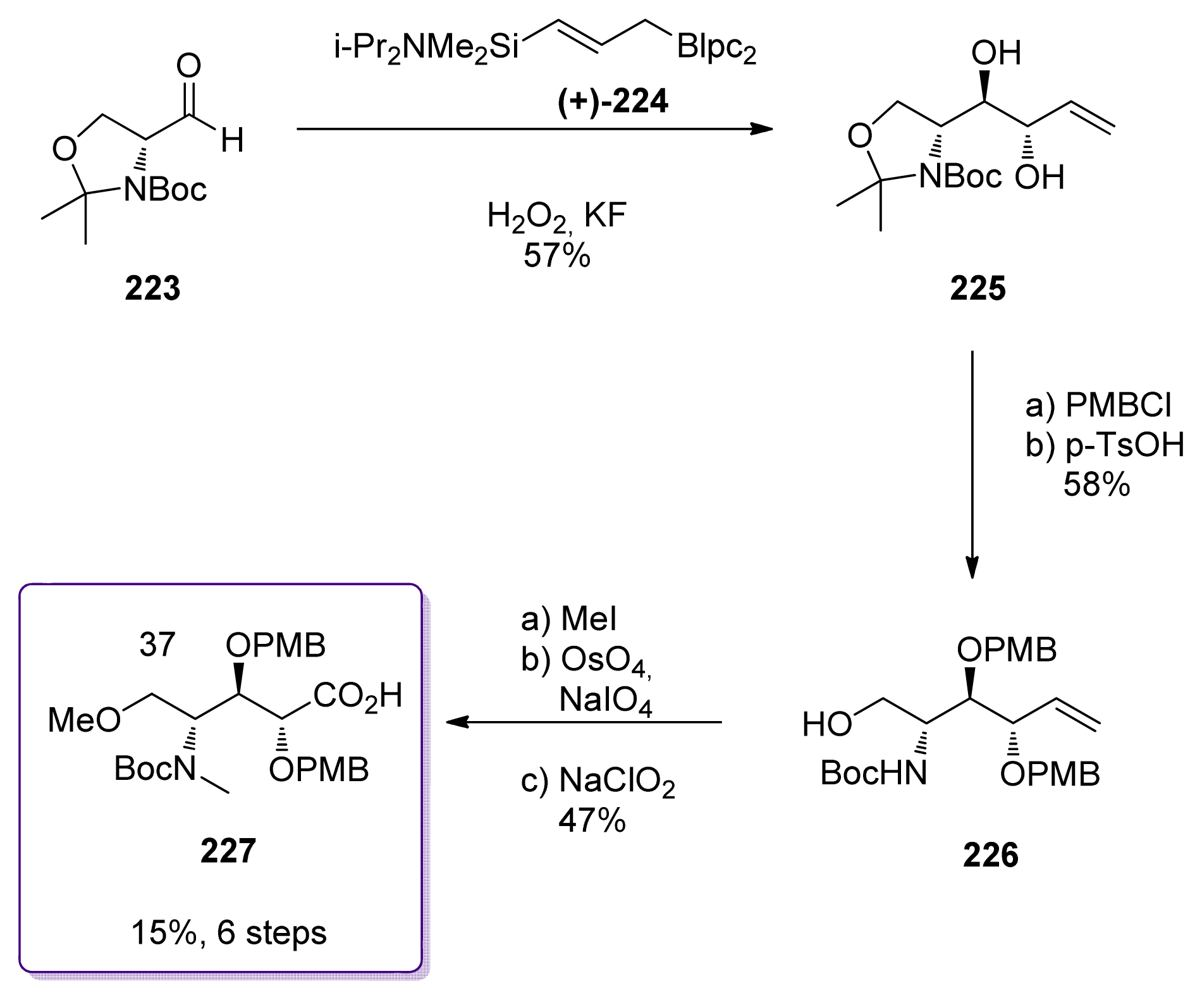
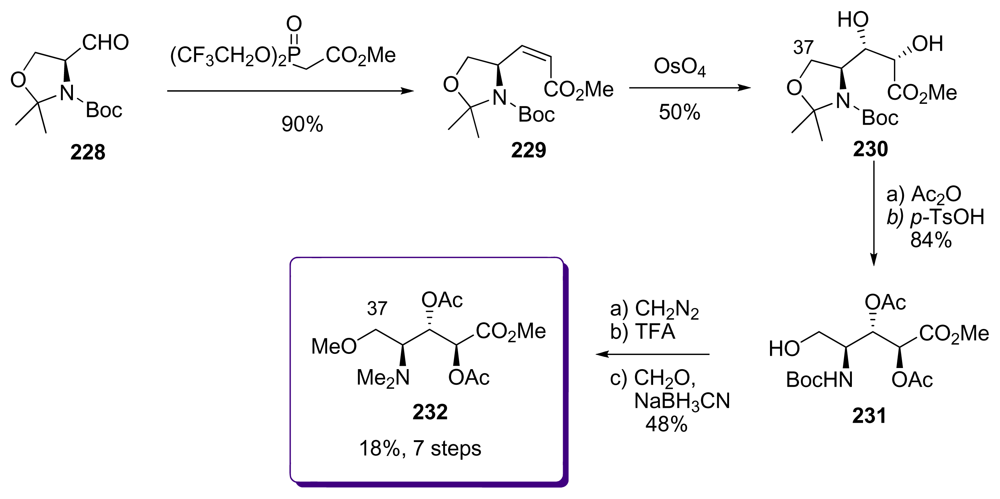
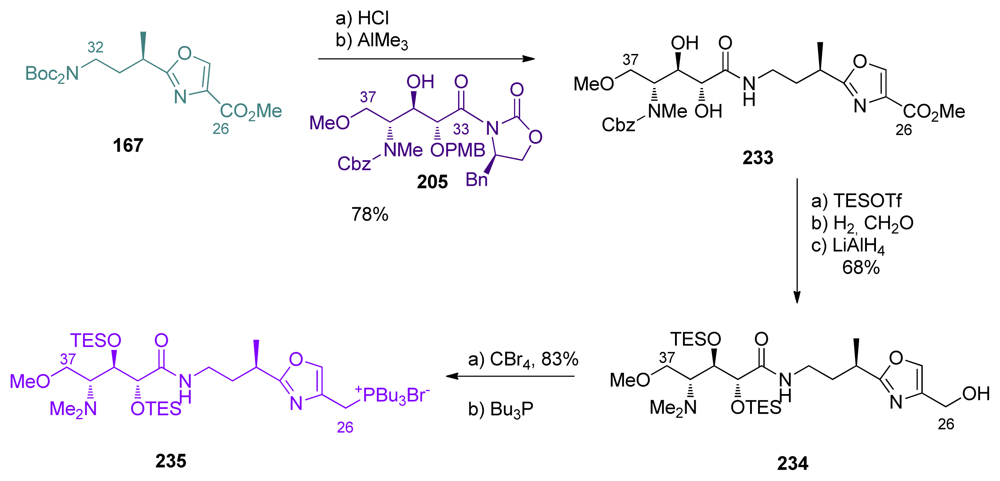
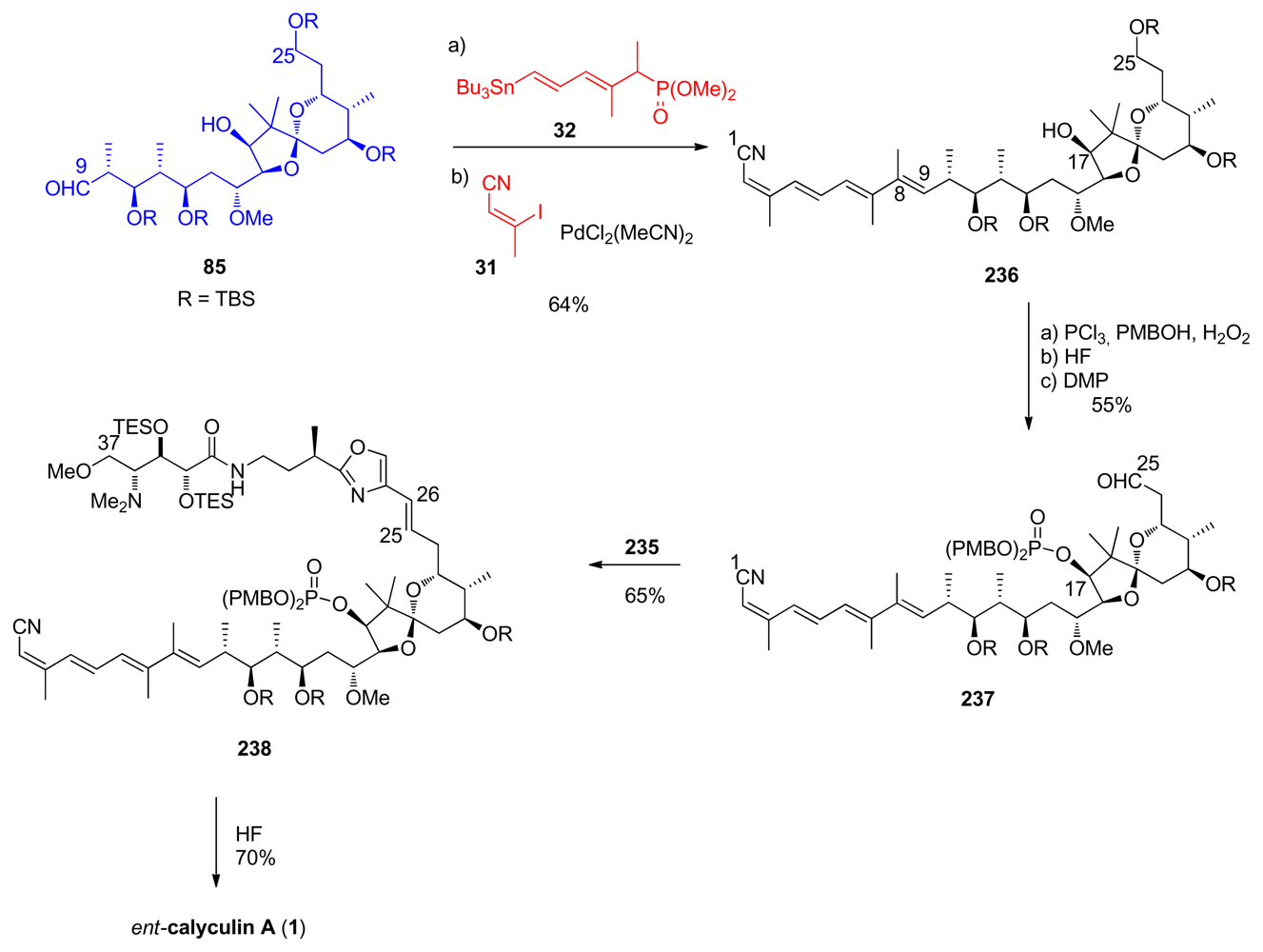
5.6.4. The total synthesis of ent-calyculin A and B by Smith [22,51,69]
5.6.5. Total synthesis of calyculin C by Armstrong [21,65]
5.6.6. Formal total synthesis of ent-calyculin A by Barrett [23]
6. Conclusions
- Masamune and Armstrong have described the total synthesis of natural calyculins A and C, respectively
- Evans and Smith have completed the total synthesis of the enantiomer of naturally occurring calyculin A (and B for Smith)
- Shioiri and Barrett have published highly advanced intermediates, previously prepared by Masamune and Evans respectively and therefore accomplished formal synthesis of natural and non natural calyculin A.
Acknowledgements
- Samples Availability: Available from the authors.
References
- Nakao, Y; Fusetani, N. Enzyme inhibitors from marine invertebrates. J Nat Prod 2007, 70, 689–710. [Google Scholar]
- Barford, D; Das, AK; Egloff, M-P. The strucrure and mechanism of protein phosphatases: Insights into catalysis and regulation. Annu Rev Biophys Biomol Struct 1998, 27, 133–164. [Google Scholar]
- Cohen, PTW. Novel protein serine/threonine phosphatases: Variety is the spice of life. Trends Biochem Sci 1997, 22, 245–251. [Google Scholar]
- McCluskey, A; Sim, ATR; Sakoff, JA. Serine-Threonine protein phosphatase inhibitors: Development of potential therapeutic strategies. J Med Chem 2002, 45, 1151–1175. [Google Scholar]
- Schönthal, AH. Role of serine/threonine protein phosphatase 2A in cancer. Cancer Lett 2001, 170, 1–13. [Google Scholar]
- Sheppeck, JE; Gauss, C-M; Chamberlin, AR. Inhibition of the ser-thr phosphatases PP1 and PP2A by naturally occurring toxins. Bioorg Med Chem 1997, 5, 1739–1750. [Google Scholar]
- Sontag, E. Protein phosphatase 2A: The trojan horse of cellular signaling. Cell Signaling 2001, 13, 7–16. [Google Scholar]
- Kato, Y; Fusetani, N; Matsunaga, S; Hashimoto, K; Koseki, K. Bioactive marine metabolites. 24. Isolation and structure elucidation of calyculins B, C, and D, novel antitumor metabolites, from the marine sponge Discodermia calyx. J Org Chem 1988, 53, 3930–3932. [Google Scholar]
- Bewley, CA; Faulkner, DJ. Lithistid sponges: Star performers or hostes to the stars. Angew Chem Int Ed 1998, 37, 2162–2178. [Google Scholar]
- Dumdei, EJ; Blunt, JW; Munro, MHG; Pannell, LK. Isolation of calyculins, calyculinamides, and swinholide H from the New Zealand deep-water marine sponge lamellomorpha strongylata. J Org Chem 1997, 62, 2636–2639. [Google Scholar]
- Kato, Y; Fusetani, N; Matsunaga, S; Hashimoto, K; Fujita, S; Furuya, T. Bioactive marine metabolites. Part 16. Calyculin A. A novel antitumor metabolite from the marine sponge Discodermia calyx. J Am Chem Soc 1986, 108, 2780–2781. [Google Scholar]
- Kato, Y; Fusetani, N; Matsunaga, S; Hashimoto, K; Koseki, K. Bioactive marine metabolites. 24. Isolation and structure elucidation of calyculins B, C, and D, novel antitumor metabolites, from the marine sponge Discodermia calyx. J Org Chem 2002, 53, 3930–3932. [Google Scholar]
- Matsunaga, S; Fujiki, H; Sakata, D; Fusetani, N. Calyculins E, F, G, and H, additional inhibitors of protein phosphatases 1 and 2A, from the marine sponge discodermia calyx. Tetrahedron 1991, 47, 2999–3006. [Google Scholar]
- Matsunaga, S; Wakimoto, T; Fusetani, N. Isolation of four new calyculins from the marine sponge Discodermia calyx. J Org Chem 1997, 62, 2640–2642. [Google Scholar]
- Matsunaga, S; Wakimoto, T; Fusetani, N; Suganuma, M. Isolation of dephosphonocalyculin A from the marine sponge, Discodermia calyx. Tetrahedron Lett 1997, 38, 3763–3764. [Google Scholar]
- Hamada, Y; Tanada, Y; Yokokawa, F; Shioiri, T. Stereoselective synthesis of 2,3-dihydroxy-4-dimethylamino-5-methoxypentanoic acid, a fragment of calyculins -Determination of the absolute configuration of calyculins. Tetrahedron Lett 1991, 32, 5983–5986. [Google Scholar]
- Matsunaga, S; Fusetani, N. Absolute stereochemistry of the calyculins, potent inhibitors of potent phosphatases 1 and 2A. Tetrahedron Lett 1991, 32, 5605–5606. [Google Scholar]
- Evans, DA; Gage, JR; Leighton, JL. Total synthesis of (+)-calyculin A. J Am Chem Soc 1992, 114, 9434–9453. [Google Scholar]
- Tanimoto, N; Gerritz, SW; Sawabe, A; Noda, T; Filla, SA; Masamune, S. The synthesis of naturally occurring (−)-calyculin A. Angew Chem Int Ed 1994, 33, 673–675. [Google Scholar]
- Yokokawa, F; Hamada, Y; Shioiri, T. Total synthesis of calyculin A—Construction of the C(9)–C(37) fragment. Chem Commun 1996, 7, 872. [Google Scholar]
- Ogawa, AK; Armstrong, RW. Total synthesis of calyculin C. J Am Chem Soc 1998, 120, 12435–12442. [Google Scholar]
- Smith, AB; Friestad, GK; Duan, JJW; Barbosa, J; Hull, KG; Iwashima, M; Qiu, Y; Spoors, PG; Bertounesque, E; Salvatore, BA. Total synthesis of (+)-calyculin A and (−)-calyculin B. J Org Chem 1998, 63, 7596–7597. [Google Scholar]
- Anderson, OP; Barrett, AGM; Edmunds, JJ; Hachiya, S-I; Hendrix, JA; Horita, K; Malecha, JW; Parkinson, CJ; VanSickle, A. Applications of crotyldiisopino campheylboranes in synthesis: A formal total synthesis of (+)-calyculin A. Can J Chem 2001, 79, 1562–1592. [Google Scholar]
- Gupta, V; Ogawa, AK; Du, X; Houk, KN; Armstrong, RW. A model for binding of structurally diverse natural product inhibitors of protein phosphatases PP1 and PP2A. J Med Chem 1997, 40, 3199–3206. [Google Scholar]
- Fu, X; Schmitz, FJ; Kelly-Borges, M; McCready, TL; Holmes, CFB. Clavosines A-C from the marine sponge myriastra clavosa: Potent cytotoxins and inhibitors of protein phosphatases 1 and 2A. J Org Chem 1998, 63, 7957–7963. [Google Scholar]
- Kehraus, S; Konig, GM; Wright, AD. A new cytotoxic calyculinamide derivative, geometricin A, from the australian sponge luffariella geometrica. J Nat Prod 2002, 65, 1056–1058. [Google Scholar]
- Edrada, RA; Ebel, R; Supriyono, A; Wray, V; Schupp, P; Steube, K; van Soest, R; Proksch, P. Swinhoeiamide A, a new highly active calyculin derivative from the marine sponge Theonella swinhoei. J Nat Prod 2002, 65, 1168–1172. [Google Scholar]
- Wakimoto, T; Matsunaga, S; Takai, A; Fusetani, N. Insight into binding of calyculin A to protein phosphatase 1: Isolation of hemicalyculin A and chemical transformation of calyculin A. Chem Biol 2002, 9, 309–319. [Google Scholar]
- Quinn, RJ; Taylor, C; Suganuma, M; Fujiki, H. The conserved acid binding domain model of inhibitors of protein phosphatases 1 and 2A: Molecular modelling aspects. Bioorg Med Chem Lett 1993, 3, 1029–1034. [Google Scholar]
- Takai, A; Sasaki, K; Nagai, H; Mieskes, G; Isobe, M; Isono, K; Yasumoto, T. Inhibition of spesific binding of okadaic acid to protein phosphatase 2A by microcystin-LR, calyculin-A and tautomycin: Method of analysis of interactions of tight-binding with target protein. Biochem J 1995, 306, 657–665. [Google Scholar]
- Goldberg, J; Huang, H-b; Kwon, Y-g; Greengard, P; Nairn, AC; Kuriyan, J. Three dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1. Nature 1995, 376, 745–753. [Google Scholar]
- Bagu, JR; Sykes, BD; Craig, MM; Holmes, CFB. A molecular basics for different interactions of marine toxins with protein phosphatase-1. J Biol Chem 1997, 272, 5087–5097. [Google Scholar]
- Gauss, C-M; Sheppeck, JE; Nairn, AC; Chamberlin, R. A molecular modeling analysis of the binding interactions between the okadaic acid class of natural product inhibitors and the ser-thr phosphatases, PP1 and PP2A. Bioorg Med Chem 1997, 5, 1751–1773. [Google Scholar]
- Lindvall, MK; Pihko, PM; Koskinen, AMP. The binding model of calyculin A to protein phosphatase.1: A novel spiroketal vector model. J Biol Chem 1997, 272, 23312–23316. [Google Scholar]
- Kita, A; Matsunaga, S; Takai, A; Kataiwa, H; Wakimoto, T; Fusetani, N; Isobe, M; Miki, K. Crystal structure of the complex between calyculin A and the catalytic subunit of protein phosphatase 1. Structure 2002, 10, 715–724. [Google Scholar]
- McCready, TL; Islam, BF; Schmitz, FJ; Luu, HA; Dawson, JF; Holmes, CFB. Inhibition of protein phosphatase-1 by clavosines A and B- novel members of the calyculun family of toxins. J Biol Chem 2002, 9, 4192–4198. [Google Scholar]
- Volter, KE; Embrey, KJ; Pierens, GK; Quinn, RJ. A study of the binding requirements of calyculin A and dephosphonocalyculin A with PP1, development of a molecular recognition model for the binding interactions of the okadaic acid class of compounds with PP1. Eur J Pharm Sci 2001, 12, 181–194. [Google Scholar]
- Koskinen, AMP; Chen, J. Enantiospecific synthesis of polyhydroxy amino acids. Synthesis of the C33-C38 portion of calyculins. Tetrahedron Lett 1991, 32, 6977–6980. [Google Scholar]
- Pihko, PM; Koskinen, AMP. Synthesis of the C26–32 oxazole fragment of calyculin C: A test case for oxazole syntheses. J Org Chem 1998, 63, 92–98. [Google Scholar]
- Pihko, PM; Koskinen, AMP. The triumph of zinc: A short and highly efficient synthesis of the calyculin C1-C9 tetraene building block by Negishi Coupling. Synlett 1999, 12, 1966–1968. [Google Scholar]
- Pihko, PM; Koskinen, AMP; Nissinen, MJ; Rissanen, K. Formation of stable spiro[4.4] ortho ester aminals during the synthesis of the C26-C32 oxazole fragment of calyculin C. J Org Chem 1999, 64, 652–654. [Google Scholar]
- Karisalmi, K; Koskinen, AMP. Studies towards the synthesis of the C(9)-C(20) lactone-dipropionate fragment of calyculin C. Synthesis 2004, 9, 1331–1342. [Google Scholar]
- Karisalmi, K; Koskinen, AMP. Stereoselective synthesis of the C9-C19 lactone-dipropionate fragment of calyculin C. Tetrahedron Lett 2004, 45, 8245–8248. [Google Scholar]
- Rauhala, V; Nevalainen, M; Koskinen, AMP. An expedient synthesis of spiroketals: Model studies for the calyculin C16-C25 fragment. Tetrahedron 2004, 60, 9199–9204. [Google Scholar]
- Rauhala, V; Nättinen, K; Rissanen, K; Koskinen, AMP. The reaction mechanism of spirocylization and stereo selectivity studies for the calyculin C16-C25 fragment. Eur J Org Chem 2005, 4119–4126. [Google Scholar]
- Habrant, D; Stewart, AJW; Koskinen, AMP. Towards the total synthesis of calyculin C: Preparation of the C13-C25 spirocyclic core. Tetrahedron 2009, 65, 7927–7934. [Google Scholar]
- Jacobs, MF; Kitching, W. Spiroacetals of marine origin. Curr Org Chem 1998, 2, 395–436. [Google Scholar]
- Pihko, PM. Chemistry and biology of the calyculins. In Acta Univ Oul, Ser A (Sci Rer Nat); Volume 325, PhD Thesis; Dept Chemistry, University of Oulu: Finland, 1999. [Google Scholar]
- Wang, C; Tobrman, T; Xu, Z; Negishi, E-i. Highly regio- and stereoselective synthesis of (Z)-trisubstituted alkenes via propyne bromoboration and tandem Pd-catalyzed cross-coupling. Org Lett 2009, 11, 4092–4095. [Google Scholar]
- Filla, SA. Discodermolide synthesis: Fragments A and BCD; PhD Thesis; Dept Chemistry, Massachusetts Institute of Technology: Cambridge, MA, USA, 1994. [Google Scholar]
- Smith, AB; Friestad, GK; Duan, JJW; Barbosa, J; Hull, KG; Iwashima, M; Qiu, Y; Spoors, PG; Bertounesque, E; Salvatore, BA. Total synthesis of (+)-calyculin A and (−)-calyculin B: Cyanotetraene construction, asymmetric synthesis of the C(26–37) oxazole, fragment assembly, and final elaboration. J Am Chem Soc 1999, 121, 10478–10486. [Google Scholar]
- Scarlato, GR; DeMattei, JA; Chong, LS; Ogawa, AK; Lin, MR; Armstrong, RW. Asymmetric synthesis of calyculin C. 1. Synthesis of the C1–C25 fragment. J Org Chem 1996, 61, 6139–6152. [Google Scholar]
- Barrett, AGM; Edmunds, JJ; Hendrix, JA; Horita, K; Parkinson, CJ. Stereocontrolled synthesis of calyculin A: Construction of the C(1)–C(14) tetraene nitrile unit. Chem Commun 1992, 1238–1240. [Google Scholar]
- Brown, HC; Bhat, KS. Enantiomeric (Z)- and (E)-crotyldiisopinocampheylboranes. Synthesis in high optical purity of all four possible stereoisomers of b-methylhomoalyl alcohols. J Am Chem Soc 1986, 108, 293–294. [Google Scholar]
- Brown, HC; Jadhav, PK; Bhat, KS. Chiral synthesis via organoboranes. 13 A highly diastereoselective and enentioselective addition of [(Z)-g-alkoxyallyl]diisopinocampheylboranes to aldehydes. J Am Chem Soc 1988, 110, 1535–1538. [Google Scholar]
- Trost, BM; Flygare, JA. A synthesis of the spiroketal subunit of (−)-calyculin A. Tetrahedron Lett 1994, 35, 4059–4062. [Google Scholar]
- Yokokawa, F; Hamada, Y; Shioiri, T. A synthesis of the oxazole part of calyculins; Part 2. Synlett 1992, 151–152. [Google Scholar]
- Takebuchi, K; Hamada, Y; Shioiri, T. Synthesis of the C13–C19 unit in the spiroketal fragment of calyculins. Tetrahedron Lett 1994, 35, 5239–5242. [Google Scholar]
- Smith, AB; Duan, JJW; Hull, KG; Salvatore, BA. Calyculin synthetic studies. Stereoselective construction of the C(14)–C(25) spiroketal subunit. Tetrahedron Lett 1991, 32, 4855–4858. [Google Scholar]
- Smith, AB; Friestad, GK; Duan, JJW; Barbosa, J; Hull, KG; Iwashima, M; Qiu, Y; Spoors, PG; Bertounesque, E; Salvatore, BA. Total synthesis of (+)-calyculin A and (−)-calyculin B: Asymmetric synthesis of the C(9–25) spiroketal dipropionate subunit. J Am Chem Soc 1999, 121, 10468–10477. [Google Scholar]
- Barrett, AGM; Edmunds, JJ; Horita, K; Parkinson, CJ. Stereocontrolled synthesis of calyculin A: Construction of the C(15)–C(25) spiroketal unit. Chem Commun 1992, 1236–1238. [Google Scholar]
- Chemler, SR; Roush, WR. Stereochemistry of the allylation and crotylation reactions of a-methyl-b-hydroxy aldehydes with allyl- andcrotyltrifluorosilanes. Synthesis of anti-anti-dipropionate stereotriads and stereodivergent pathways for the reactions with 2,3-anti and 2,3-syn-a-methyl-b-hydroxy aldehydes. J Org Chem 2003, 68, 1319–1333. [Google Scholar]
- Vaccaro, HA; Levy, DE; Sawabe, A; Jaetsch, T; Masamune, S. Towards the synthesis of calyculin: A synthetic intermediate corresponding to the C(26)–C(37) fragment. Tetrahedron Lett 1992, 33, 1937–1940. [Google Scholar]
- Yokokawa, F; Hamada, Y; Shioiri, T. A synthesis of the oxazole part of calyculins; Part 1. Synlett 1992, 149–150. [Google Scholar]
- Ogawa, AK; DeMattei, JA; Scarlato, GR; Tellew, JE; Chong, LS; Armstrong, RW. Asymmetric synthesis of calyculin C. 2. synthesis of the C26–C37 fragment and model Wittig couplings. J Org Chem 1996, 61, 6153–6161. [Google Scholar]
- Barrett, AGM; Edmunds, JJ; Malecha, JW; Parkinson, CJ. Stereocontrolled synthesis of calyculin A: Construction of the C(26)–C(37) amide-oxazole unit. Chem Commun 1992, 1240–1242. [Google Scholar]
- Yokokawa, F; Hamada, Y; Shioiri, T. Synthesis of the C26–C37 fragment of calyculin A having natural configuration. Synlett 1992, 703–705. [Google Scholar]
- Sawabe, A; Filla, SA; Masamune, S. Use of 2-trimethylsilylethyl as a protecting group in phosphate monoester synthesis. Tetrahedron Lett 1992, 33, 7685–7686. [Google Scholar]
- Smith, AB; Salvatore, BA; Hull, KG; Duan, JJW. Calyculin synthetic studies. 2. Stereocontrolled assembly of the C(9)-C(13) dithiane and C(26)-C(37) oxazole intermediates. Tetrahedron Lett 1991, 32, 4859–4862. [Google Scholar]






























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of the inhibitor | Isolation origin | Structural Scaffold | IC50 nM a | Properties | Ref. | |
|---|---|---|---|---|---|---|
| PP1 | PP2A | |||||
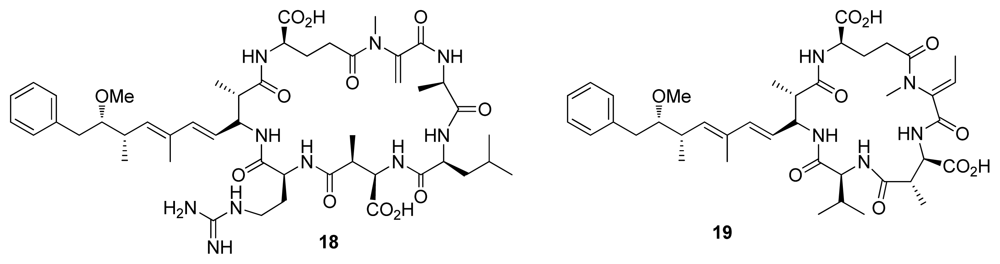
| Microcystin-LR (18) | Blue green algae | Cyclic peptide | 0.3–0.6 | 0.04–2.0 | Liver toxin | [24] |
| Nodularin-V (19) | Blue green algae | Cyclic peptide | 0.5–3 | 0.03–1.0 | Liver toxin | [24] |
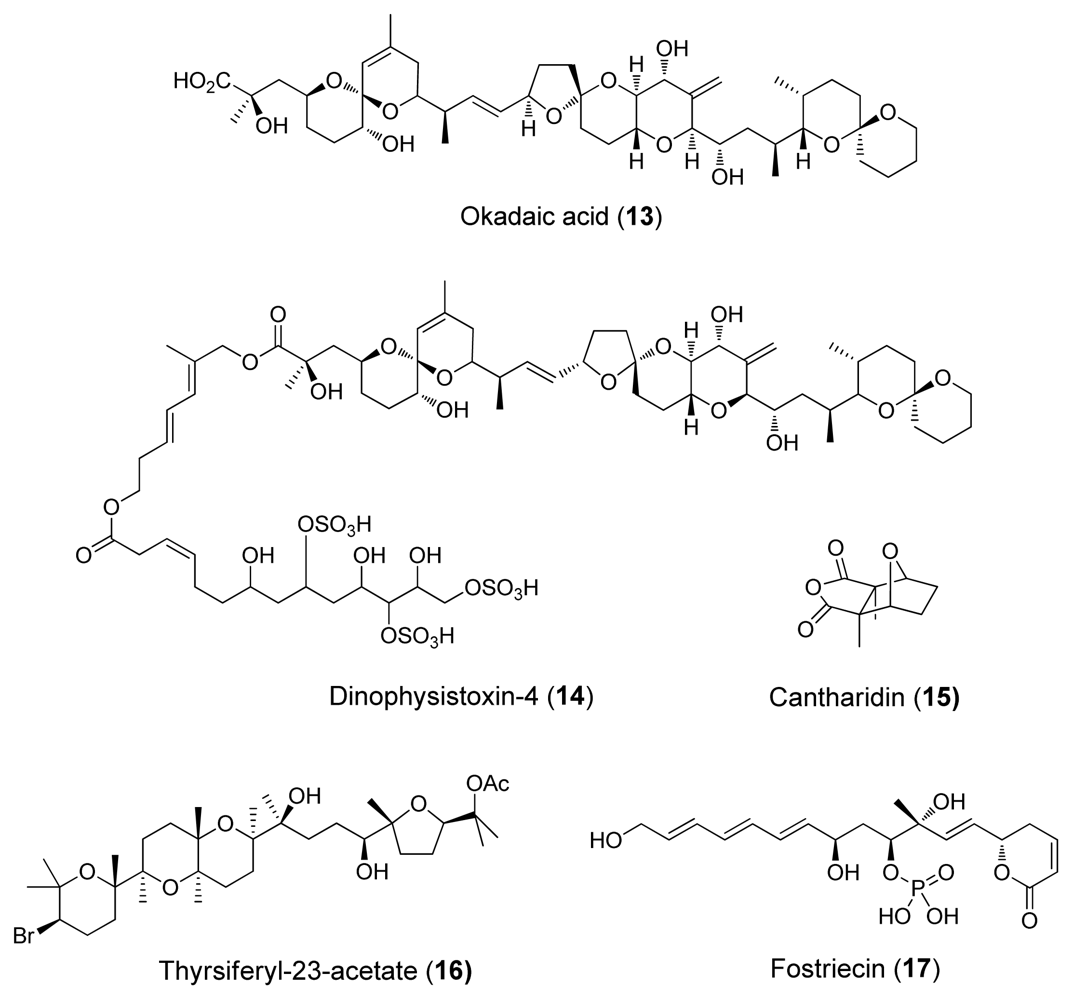
| Cantharidin (15) | Blister beetles | Terpenoid | 0.5–2.0 | 0.2 | Natural defensive toxicant | [6] |
| Thyrsiferyl- 23-acetate (16) | Red algae, L. Obtusa | Terpenoid | >1 | (4–16)·10−3 | [6] | |
| Okadaic acid (13) | Dinoflagellates | Polyketide | 10–1300 | 0.02–1.0 | Tumour promoter | [24] |
| Dinophysistoxin-4 (14) | Dinoflagellates | Polyketide | ~200 | ~2 | [4] | |
| Calyculin A (1) | Marine sponge D. calyx. | Polyketide | 0.4–2.0 | 0.25–3 | Tumour promoter | [24] |
| Calyculin C (3) | Marine sponge D. calyx. | Polyketide | 0.6 | 2.8 | Tumour promoter | [24] |
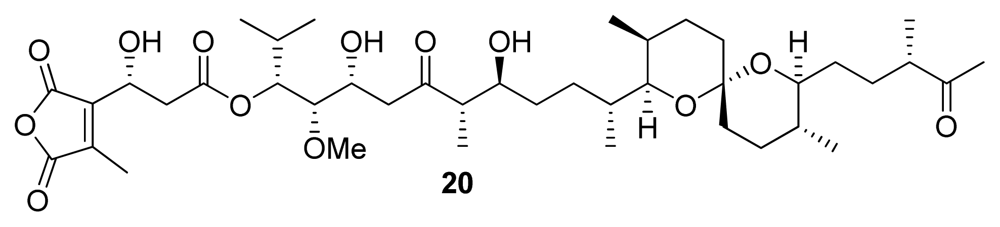
| Tautomycin (20) | Bacterium, Streptomyces verticillatus | Polyketide | 1.1–7.51 | 10–23.1 | Antibiotic | [24] |
| Fostriecins (17) | Bacterium, Streptomyces pulveraceus | Polyketide | 0.131 | 3.4·10−6 | Antitumoric activity | [4] |
| Group | Target | Pub. year | Number of steps a | Overall yield (%) a | Number of steps b | Overall yield (%) b |
|---|---|---|---|---|---|---|
| Evans | ent-Calyculin A | 1992 | 33 | 0.54 | 36 | - |
| Masamune | Calyculin A | 1994 | 43 | 0.31 | 45 | 0.18 |
| Shioiri | Calyculin A | 1996 | 32 | 0.092 | 32 | 0.092 |
| Smith | ent-Calyculin A | 1998 | 35 | 0.89 | 37 | 0.79 |
| Armstrong | Calyculin C | 1998 | 30 | 0.018 | 30 | 0.018 |
| Barrett | ent-Calyculin A | 2001 | 34 | 0.9 | 34 | 0.9 |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fagerholm, A.E.; Habrant, D.; Koskinen, A.M.P. Calyculins and Related Marine Natural Products as Serine- Threonine Protein Phosphatase PP1 and PP2A Inhibitors and Total Syntheses of Calyculin A, B, and C. Mar. Drugs 2010, 8, 122-172. https://doi.org/10.3390/md80100122
Fagerholm AE, Habrant D, Koskinen AMP. Calyculins and Related Marine Natural Products as Serine- Threonine Protein Phosphatase PP1 and PP2A Inhibitors and Total Syntheses of Calyculin A, B, and C. Marine Drugs. 2010; 8(1):122-172. https://doi.org/10.3390/md80100122
Chicago/Turabian StyleFagerholm, Annika E., Damien Habrant, and Ari M. P. Koskinen. 2010. "Calyculins and Related Marine Natural Products as Serine- Threonine Protein Phosphatase PP1 and PP2A Inhibitors and Total Syntheses of Calyculin A, B, and C" Marine Drugs 8, no. 1: 122-172. https://doi.org/10.3390/md80100122