
Preparation and Characterization of Chitosan Obtained from Shells of Shrimp (Litopenaeus vannamei Boone)

 and
and
Abstract
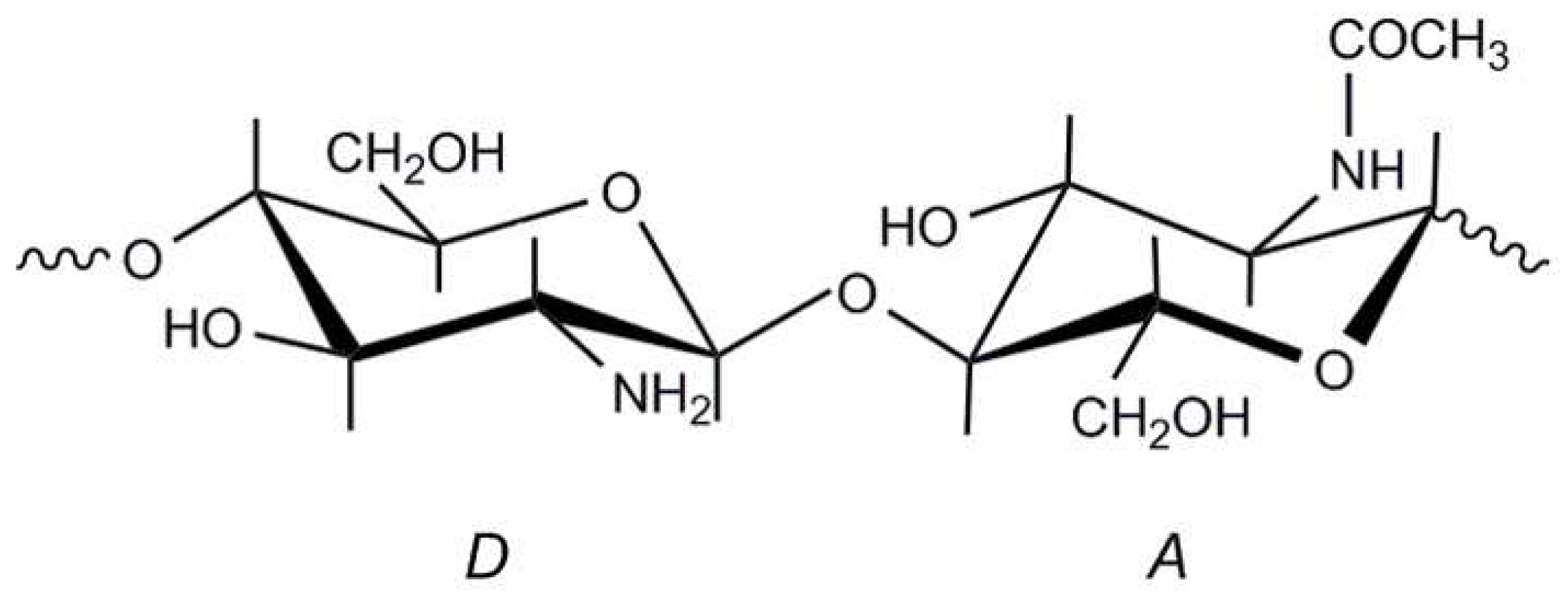
:1. Introduction
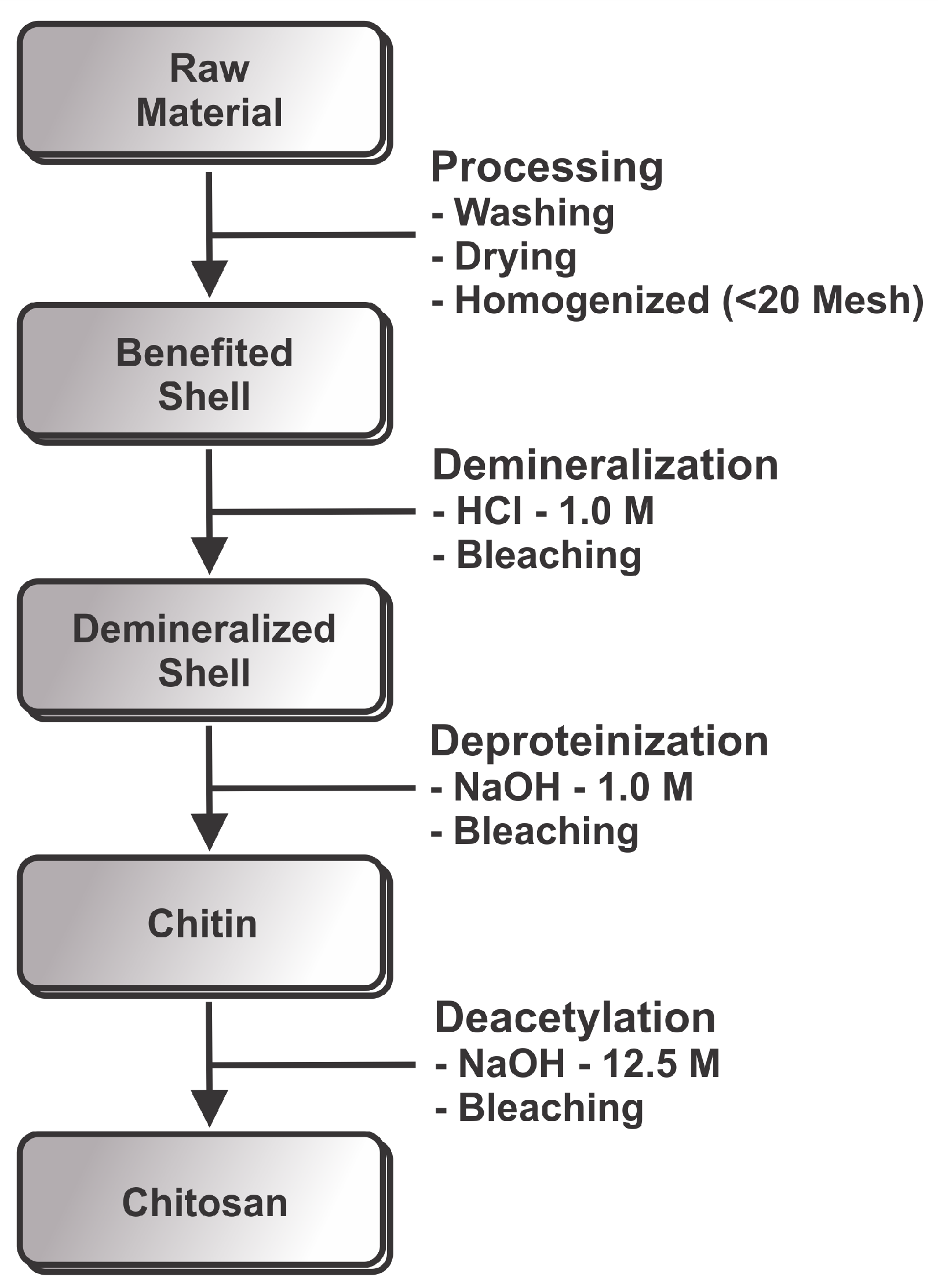
2. Materials and Methods
2.1. Demineralization
2.2. Deproteinization
2.3. Chitosan Production
2.4. Characterization of Samples
2.4.1. Sulfated Ash and Insuluble Content
2.4.2. Scanning Electron Microscopy
2.4.3. Viscosity Average Molecular Weight
2.4.4. Degree of Acetylation
2.4.5. X-ray Diffraction
3. Results and Discussion
3.1. Sulfated Ash and Insoluble Content
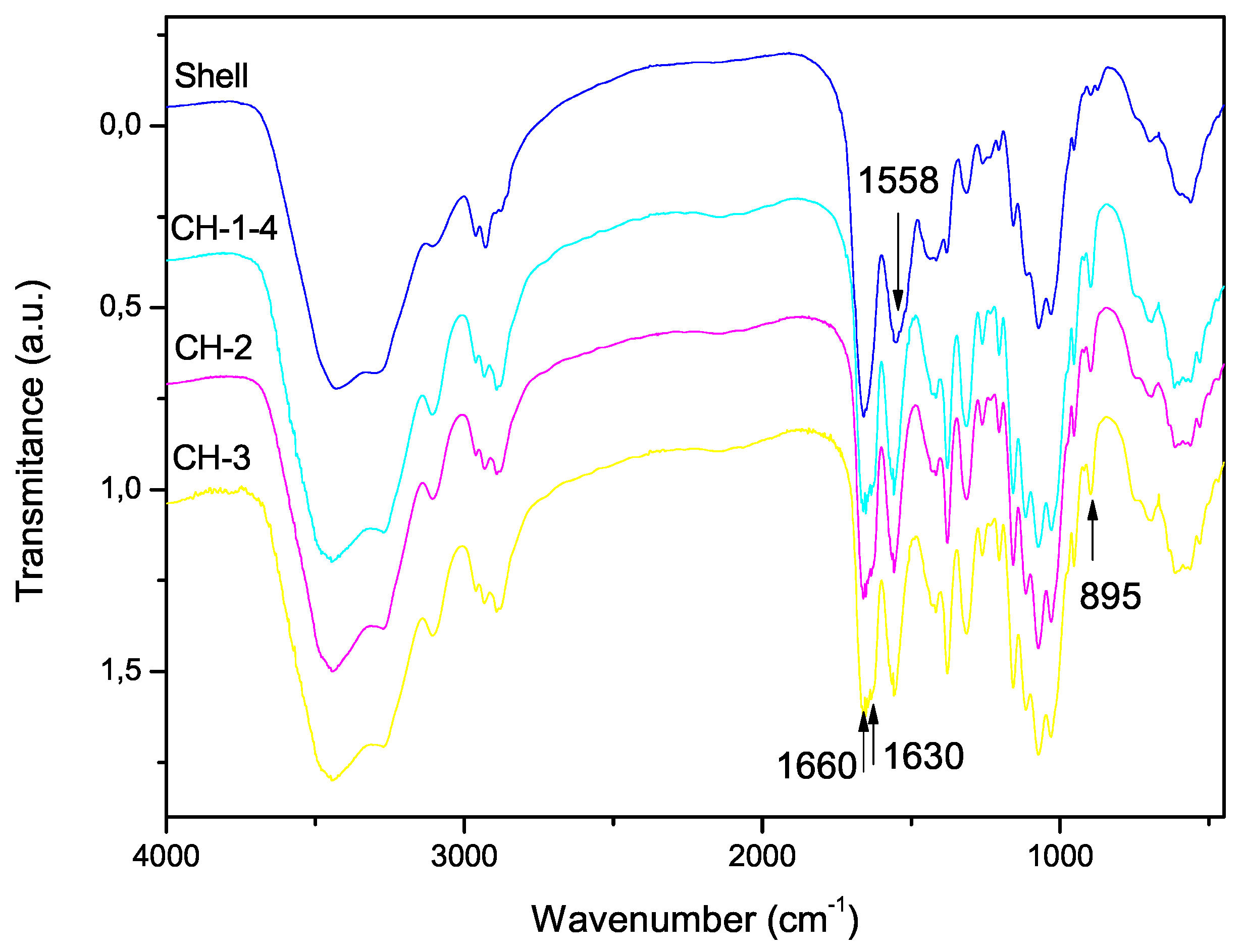
3.2. Scanning Electron Microscopy
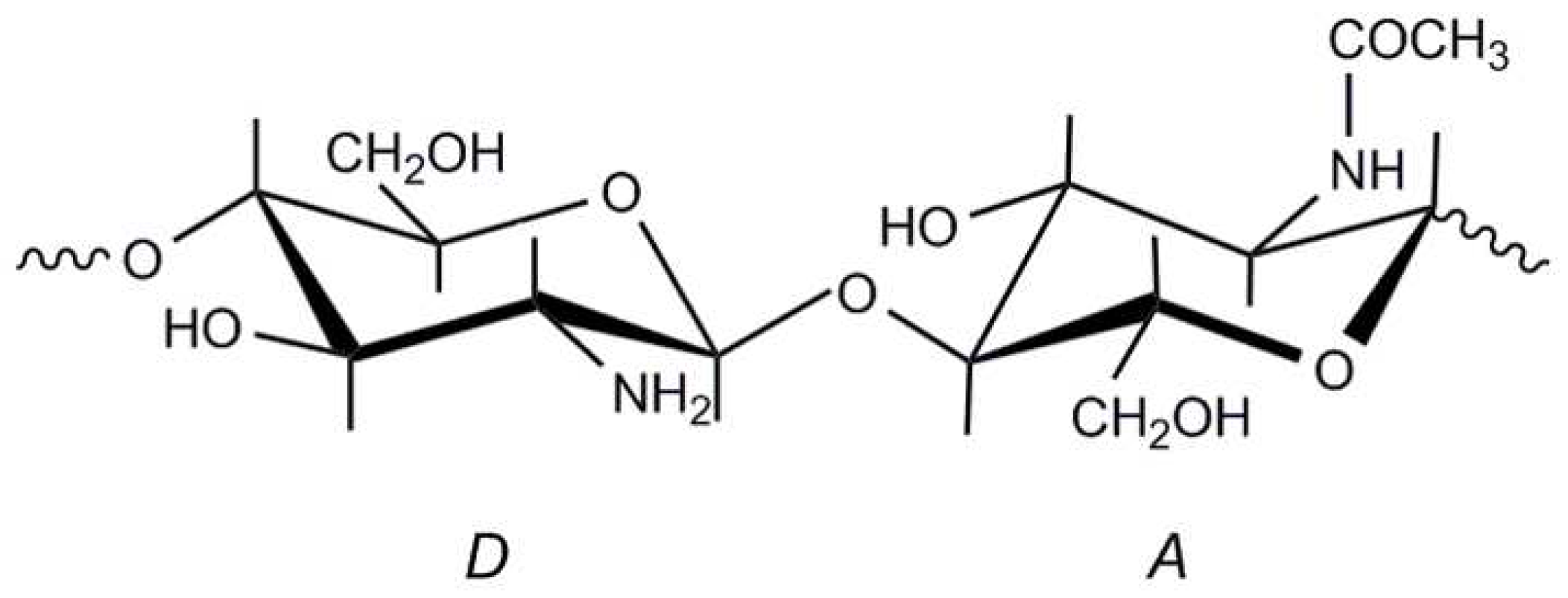
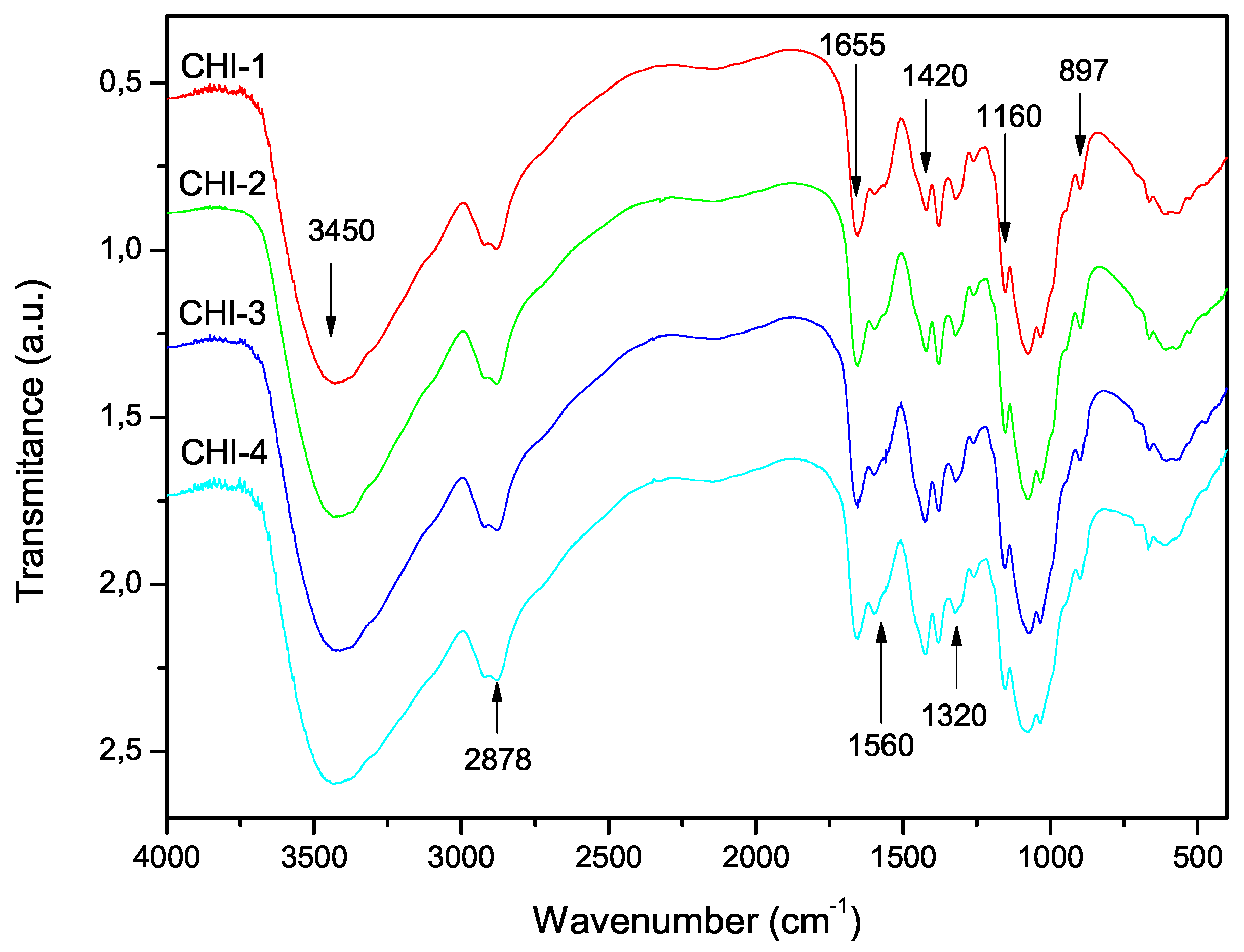
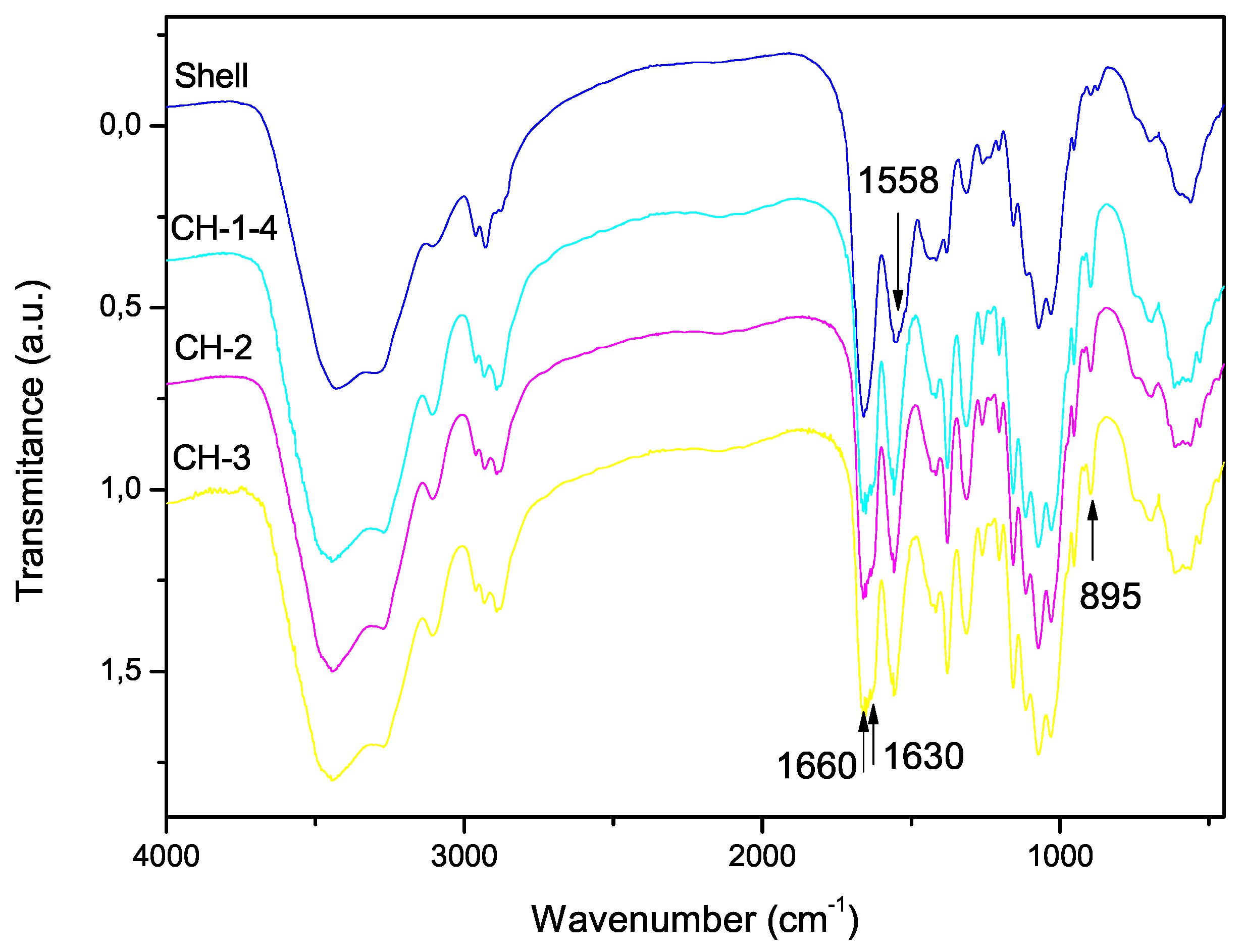
3.3. Degree of Acetylation
3.4. Viscosity Average Molecular Weight
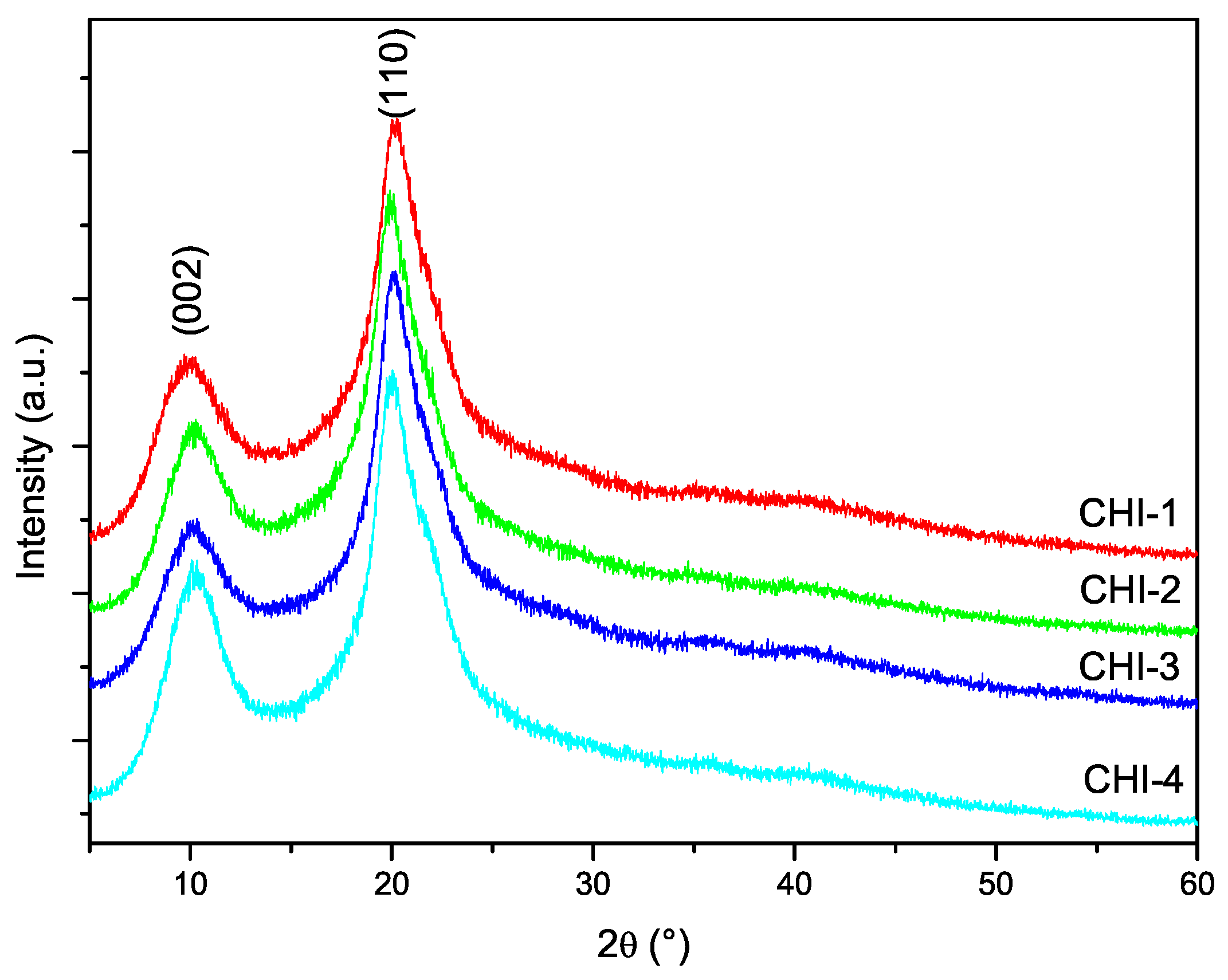
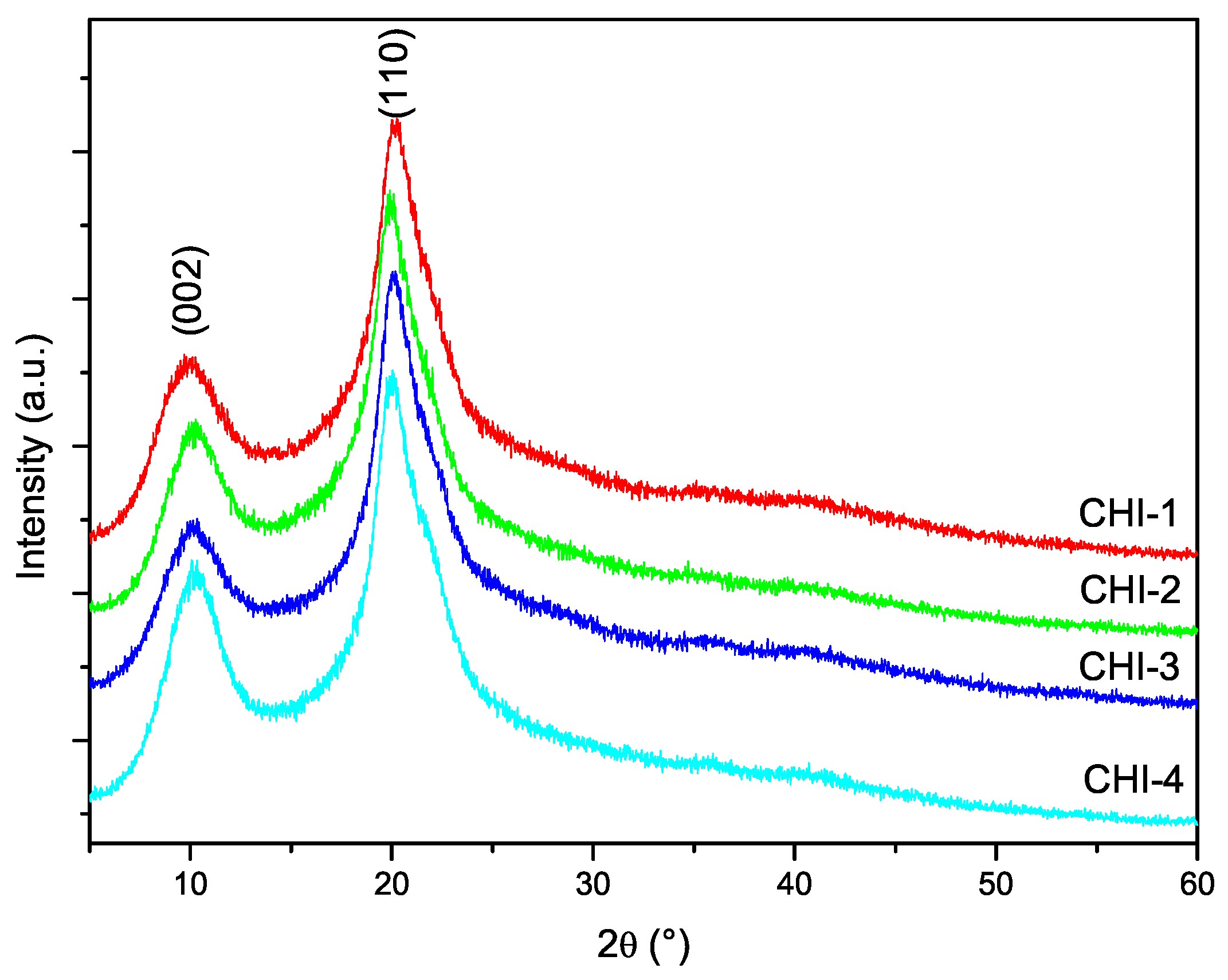
3.5. X-ray Diffraction
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| DOAJ | Directory of Open Access Journals |
| DA | Acetylation Degree |
| EDS | Energy Dispersive Spectroscopy |
| ASTM | American Society for Testing and Materials |
| FTIR | Fourier Transform Infrared Spectroscopy |
| UV | UV-Visible Spectroscopy |
| XRD | X-Ray Diffraction |
| WAXD | Wide Angle X-Ray Diffraction |
| CrI | Crystallinity Index |
| SEM | Scanning Electron Microscope |
| SD | Standard Deviation |
| OPAS | Organization Pan American Health |
| CAPES | Coordination for the Improvement of Higher Education Personnel |
References
- Dash, M.; Chiellini, F.; Ottenbrite, R.M.; Chiellini, E. Chitosan—A versatile semi-synthetic polymer in biomedical applications. Prog. Polym. Sci. 2011, 36, 981–1014. [Google Scholar] [CrossRef]
- Vo, T.-S.; Ng, D.-H.; Kim, S.-K. Cosmeceutical compounds from marine sources. In Kirk-Othmer Chemical Technology of Cosmetics; Seidel, A., Bickford, M., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 483–499. [Google Scholar]
- Khor, E.; Wan, A.C.A. Chitin: Fulfilling a Biomaterials Promise, 2nd ed.; Elsevier Ltd.: Waltham, MA, USA, 2013. [Google Scholar]
- Bouhenna, M.; Salah, R.; Bakour, R.; Drouiche, N.; Abdi, N.; Grib, H.; Lounici, H.; Mameri, N. Effects of chitin and its derivatives on human cancer cells lines. Environ. Sci. Pollut. Res. 2015, 22, 15579–15586. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.A.; Aljaeid, B.M. Preparation, characterization, and potential application of chitosan, chitosan derivatives, and chitosan metal nanoparticles in pharmaceutical drug delivery. Drug Des. Dev. Ther. 2016, 10, 483–507. [Google Scholar] [CrossRef] [PubMed]
- Sharp, R.G. A Review of the Applications of Chitin and Its Derivatives in Agriculture to Modify Plant-Microbial Interactions and Improve Crop Yields. Agronomy 2013, 3, 757–793. [Google Scholar] [CrossRef]
- Bautista-Banos, S.; Romanazzi, G.; Jiménez-Aparicio, A. (Eds.) Chitosan in the Preservation of Agricultural Commodities; Academic Press, Elsevier: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Luo, Y.; Wang, Q. Recent Advances of Chitosan and Its Derivatives for Novel Applications in Food Science. J. Food Process. Beverages 2013, 1, 1–13. [Google Scholar]
- No, L.H.K.; Meyers, S.P. Application of chitosan for treatment of wastewaters. Rev. Environ. Contam. Toxicol. 2000, 1, 1–27. [Google Scholar]
- Peniche, C.; Argüelles-Monal, W. Chitosan Based Polyelectrolyte Complexes. In Natural and Synthetic Polymers: Challenges and Perspectives; Wiley-VCH: Weinheim, Germany, 2001; Volume 168, pp. 103–116. [Google Scholar]
- Croisier, F.; Jérôme, C. Chitosan-based biomaterials for tissue engineering. Eur. Polym. J. 2013, 49, 780–792. [Google Scholar]
- Agnihotri, S.A.; Mallikarjuna, N.N.; Aminabhavi, T.M. Recent advances on chitosan-based micro- and nanoparticles in drug delivery. J. Control. Release 2004, 100, 5–28. [Google Scholar] [CrossRef] [PubMed]
- Steinbüchel, A.; De Baets, S.; Vandamme, E.J. Biopolymers, Vol. 6. Polysaccharides II: Polysaccharides from Eukaryotes; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Khoushab, F.; Yamabhai, M. Chitin Research Revisited. Mar. Drugs 2010, 8, 1988–2012. [Google Scholar] [CrossRef] [PubMed]
- Younes, I.; Rinaudo, M. Chitin and chitosan preparation from marine sources. Structure, properties and applications. Mar. Drugs 2015, 13, 1133–1174. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yang, H.; Yan, N. Shell Biorefinery: Dream or Reality? Chem. Eur. J. 2016, 22, 13402–13421. [Google Scholar] [CrossRef] [PubMed]
- Roberts, G.A.F. Chitin Chemistry; The Macmillan Press Ltd.: London, UK, 1992; pp. 54–61. [Google Scholar]
- Brugnerotto, J.; Lizardi, J.; Goycoolea, F.; Argüelles-Monal, W.; Desbrieres, J.; Rinaudo, M. An infrared investigation in relation with chitin and chitosan characterization. Polymer 2001, 42, 3569–3580. [Google Scholar] [CrossRef]
- Qin, Y.; Lu, X.M.; Sun, N.; Rogers, R.D. Dissolution or extraction of crustacean shells using ionic liquids to obtain high molecular weight purified chitin and direct production of chitin films and fibers. Green Chem. 2010, 12, 968–971. [Google Scholar] [CrossRef]
- Setoguchi, T.; Kato, T.; Yamamoto, K.; Kadokawa, J.-I. Facile production of chitin from crab shells using ionic liquid and citric acid. Int. J. Biol. Macromol. 2012, 50, 861–864. [Google Scholar] [CrossRef] [PubMed]
- Beaney, P.; Lizardi-Mendoza, J.; Healy, M. Comparison of chitins produced by chemical and bioprocessing methods. J. Chem. Technol. Biotechnol. 2005, 80, 145–150. [Google Scholar] [CrossRef]
- Arbia, W.; Arbia, L.; Adour, L.; Amrane, A. Chitin extraction from crustacean shells using biological methods—A review. Food Technol. Biotechnol. 2013, 51, 12–25. [Google Scholar]
- Synowiecki, J. The recovery of protein hydrolyzate during enzymatic isolation of chitin from shrimp Crangon processing discard. Food Chem. 2000, 68, 147–152. [Google Scholar] [CrossRef]
- Tsaih, M.L.; Chen, R.H. The effect of reaction time and temperature during heterogenous alkali deacetylation on degree of deacetylation and molecular weight of resulting chitosan. J. Appl. Polym. Sci. 2003, 88, 2917–2923. [Google Scholar] [CrossRef]
- Lertwattanaseri, T.; Ichikawa, N.; Mizoguchi, T.; Tanaka, Y.; Chirachanchai, S. Microwave technique for efficient deacetylation of chitin nanowhiskers to a chitosan nanoscaffold. Carbohydr. Res. 2009, 344, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Hamer, S.N.; Cord-Landwehr, S.; Biarnés, X.; Planas, A.; Waegeman, H.; Moerschbacher, B.M. Enzymatic production of defined chitosan oligomers with a specific pattern of acetylation using a combination of chitin oligosaccharide deacetylases. Sci. Rep. 2015, 5, 8716. [Google Scholar] [CrossRef] [PubMed]
- Allwin, S.I.; Jeyasanta, K.I.; Patterson, J. Extraction of Chitosan from White Shrimp (Litopenaeus vannamei). Processing Waste and Examination of its Bioactive Potentials. Adv. Biol. Res. 2015, 9, 389–396. [Google Scholar]
- Ploydee, E.; Chaiyanan, S. Production of High Viscosity Chitosan from Biologically Purified Chitin Isolated by Microbial Fermentation and Deproteinization. Int. J. Polym. Sci. 2014, 2014, 162173. [Google Scholar] [CrossRef]
- Hajji, S.; Younes, I.; Ghorbel-Bellaaj, O.; Hajji, R.; Rinaudo, M.; Nasri, M.; Jellouli, K. Structural differences between chitin and chitosan extracted from three different marine sources. Int. J. Biol. Macromol. 2014, 65, 298–306. [Google Scholar] [CrossRef] [PubMed]
- De Andrade, S.M.B.; Ladchumananandasivam, R.; da Rocha, B.G.; Belarmino, D.D.; Galvão, A.O. The Use of Exoskeletons of Shrimp Litopenaeus vanammei) and Crab (Ucides cordatus) for the extraction of Chitosan and Production of nanomembrane. Mater. Sci. Appl. 2012, 3, 495–508. [Google Scholar]
- Moura, C.; Muszinski, P.; Schmidt, C.; Almeida, J.; Pinto, L. Quitina e quitosana produzidas a partir de resíduos de camarão e siri: avaliação do processo em escala piloto. Vetor Rio Grande 2006, 16, 37–45. [Google Scholar]
- Lamarque, G.; Cretenet, M.; Viton, C.; Domard, A. New Route of Deacetylation of α- and β-Chitins by Means of Freeze-Pump Out-Thaw Cycles. Biomacromolecules 2005, 6, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Agência Nacional de Vigilância Sanitária Farmacopéia Brasileira; Anvisa: Brasilia, DF, Brazil, 2010.
- ASTM International. F2103-11 Standard Guide for Characterization and Testing of Chitosan Salts as Starting Materials Intended for Use in Biomedical and Tissue-Engineered Medical Product Applications; ASTM International: West Conshohocken, PA, USA, 2011. [Google Scholar]
- Rinaudo, M.; Milas, M.; Le Dung, P. Characterization of chitosan. Influence of ionic strength and degree of acetylation on chain expansion. Int. J. Biol. Macromol. 1993, 15, 281–285. [Google Scholar] [CrossRef]
- Domszy, J.G.; Roberts, G.A. Evaluation of infrared spectroscopic techniques for analysing chitosan. Macromol. Chem. Phys. 1985, 186, 1671–1677. [Google Scholar] [CrossRef]
- Muzzarelli, R.A.; Rocchetti, R. Determination of the degree of acetylation of chitosans by first derivative ultraviolet spectrophotometry. Carbohydr. Polym. 1985, 5, 461–472. [Google Scholar] [CrossRef]
- Osorio-Madrazo, A.; David, L.; Trombotto, S.; Lucas, J.-M.; Peniche-Covas, C.; Domard, A. Kinetics study of the solid-state acid hydrolysis of chitosan: Evolution of the crystallinity and macromolecular structure. Biomacromolecules 2010, 11, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- The European Pharmacopeia; C.o. Europe: Strasburg, France, 2007; pp. 1490–1491.
- The United States Pharmacopeia; The United States Pharmacopeial Convention: Rockville, MD, USA, 2011; pp. 5361–5365.
- Povea, M.B.; Monal, W.A.; Cauich-Rodríguez, J.V.; Pat, A.M.; Rivero, N.B.; Covas, C.P. Interpenetrated chitosan-poly(acrylic acid-co-acrylamide) hydrogels. Synthesis, characterization and sustained protein release studies. Mater. Sci. Appl. 2011, 2, 509–520. [Google Scholar] [CrossRef]
- Kumirska, J.; Czerwicka, M.; Kaczynski, Z.; Bychowska, A.; Brzozowski, K.; Thoming, J.; Stepnowski, P. Application of spectroscopic methods for structural analysis of chitin and chitosan. Mar. Drugs 2010, 8, 1567–1636. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Demineralization Time (h) | Deacetylation Time (h) | Sulfated Ash (%) ± SD | Insoluble Content (%) ± SD |
|---|---|---|---|---|
| CHI-1 | 6 | 4 | ||
| CHI-2 | 2 | 6 | ||
| CHI-3 | 0.5 | 6 | ||
| CHI-4 | 6 | 6 |
| Sample | FTIR | UV-First Derivate ± SD | ||
|---|---|---|---|---|
| Equation (3) ± SD | Equation (4) ± SD | Equation (5) ± SD | ||
| CHI-1 | ||||
| CHI-2 | ||||
| CHI-3 | ||||
| CHI-4 | ||||
| Sample | [] (100 mL/g) ± SD | (g/mol) | CrI (%) |
|---|---|---|---|
| CHI-1 | 45 | ||
| CHI-2 | 40 | ||
| CHI-3 | 40 | ||
| CHI-4 | 46 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Queiroz Antonino, R.S.C.M.; Lia Fook, B.R.P.; De Oliveira Lima, V.A.; De Farias Rached, R.Í.; Lima, E.P.N.; Da Silva Lima, R.J.; Peniche Covas, C.A.; Lia Fook, M.V. Preparation and Characterization of Chitosan Obtained from Shells of Shrimp (Litopenaeus vannamei Boone). Mar. Drugs 2017, 15, 141. https://doi.org/10.3390/md15050141
De Queiroz Antonino RSCM, Lia Fook BRP, De Oliveira Lima VA, De Farias Rached RÍ, Lima EPN, Da Silva Lima RJ, Peniche Covas CA, Lia Fook MV. Preparation and Characterization of Chitosan Obtained from Shells of Shrimp (Litopenaeus vannamei Boone). Marine Drugs. 2017; 15(5):141. https://doi.org/10.3390/md15050141
Chicago/Turabian StyleDe Queiroz Antonino, Rayane Santa Cruz Martins, Bianca Rosa Paschoal Lia Fook, Vítor Alexandre De Oliveira Lima, Raid Ícaro De Farias Rached, Eunice Paloma Nascimento Lima, Rodrigo José Da Silva Lima, Carlos Andrés Peniche Covas, and Marcus Vinícius Lia Fook. 2017. "Preparation and Characterization of Chitosan Obtained from Shells of Shrimp (Litopenaeus vannamei Boone)" Marine Drugs 15, no. 5: 141. https://doi.org/10.3390/md15050141
APA StyleDe Queiroz Antonino, R. S. C. M., Lia Fook, B. R. P., De Oliveira Lima, V. A., De Farias Rached, R. Í., Lima, E. P. N., Da Silva Lima, R. J., Peniche Covas, C. A., & Lia Fook, M. V. (2017). Preparation and Characterization of Chitosan Obtained from Shells of Shrimp (Litopenaeus vannamei Boone). Marine Drugs, 15(5), 141. https://doi.org/10.3390/md15050141