A New Benzofuran Glycoside and Indole Alkaloids from a Sponge-Associated Rare Actinomycete, Amycolatopsis sp.

 and
and
Abstract
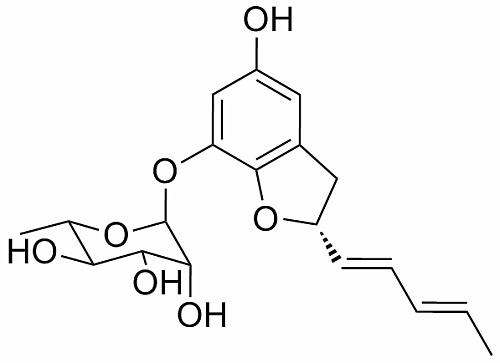
:
1. Introduction
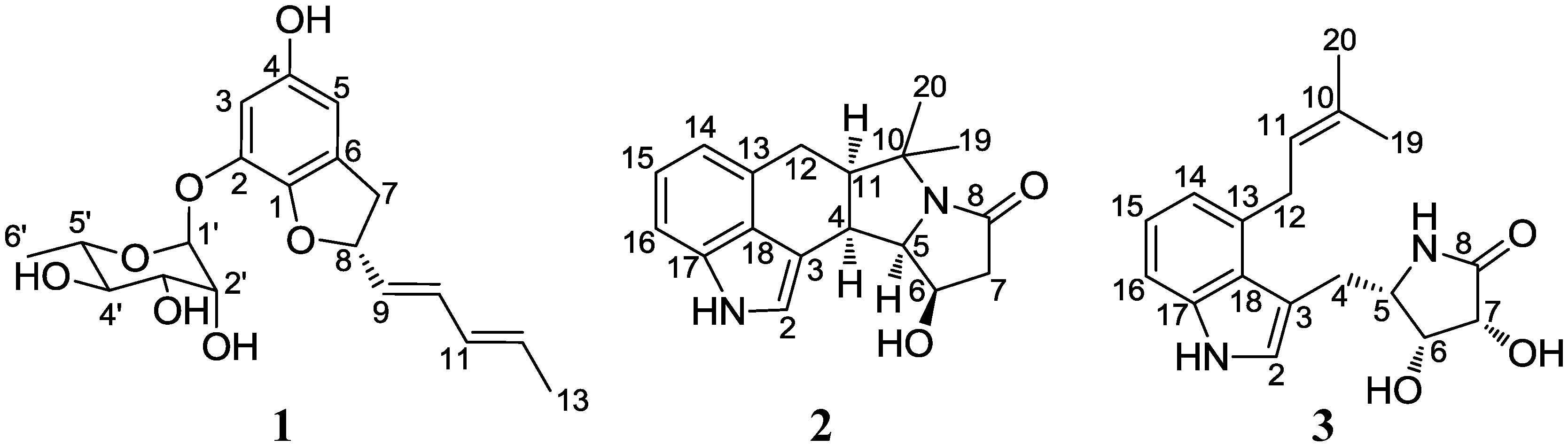
2. Results and Discussion
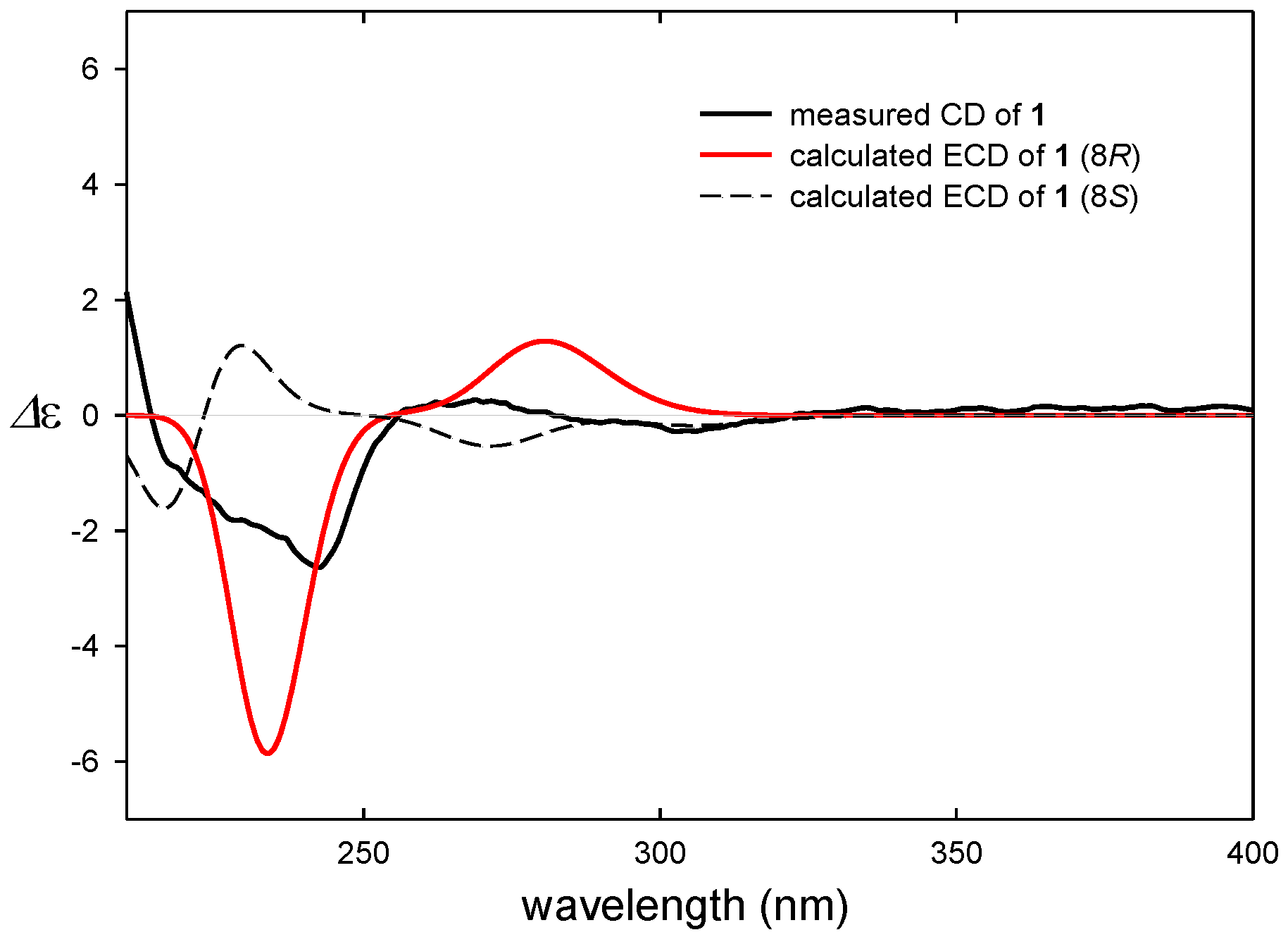
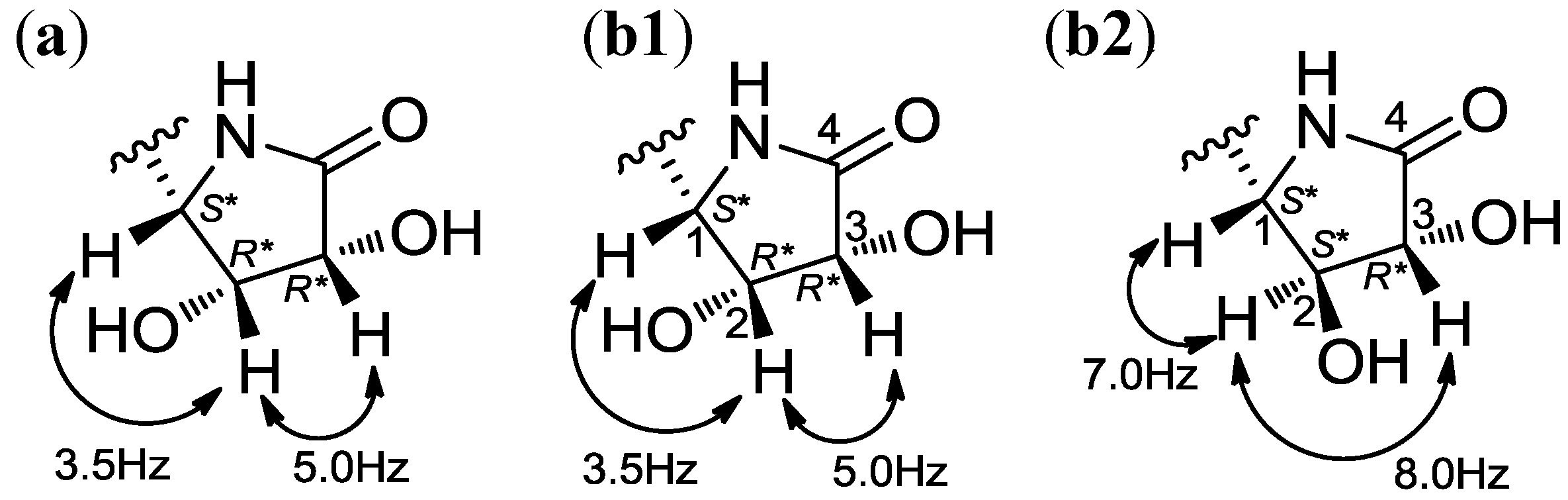
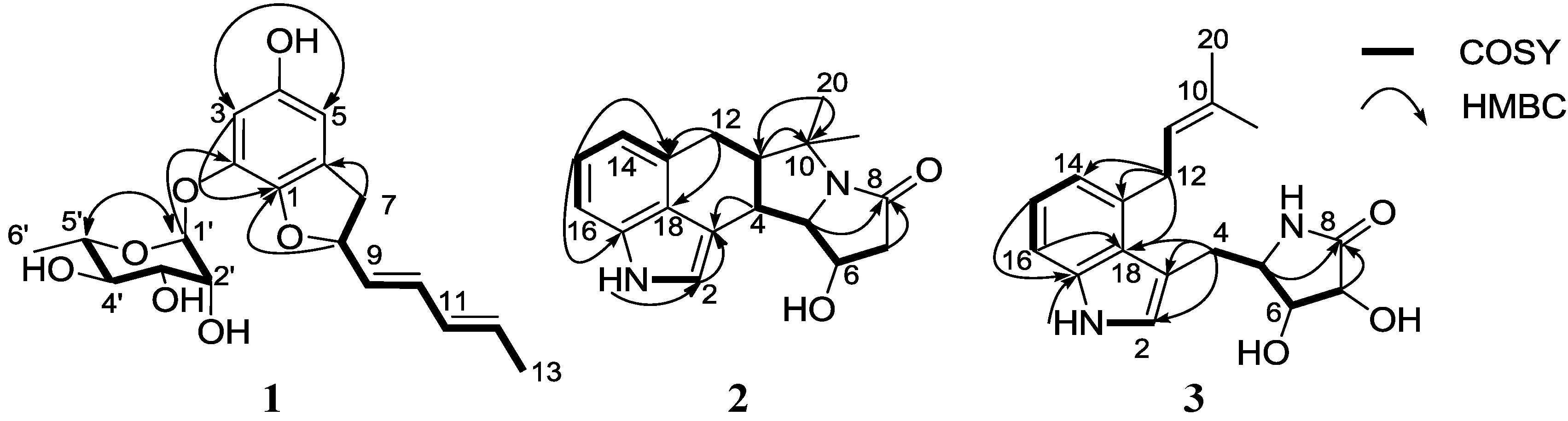
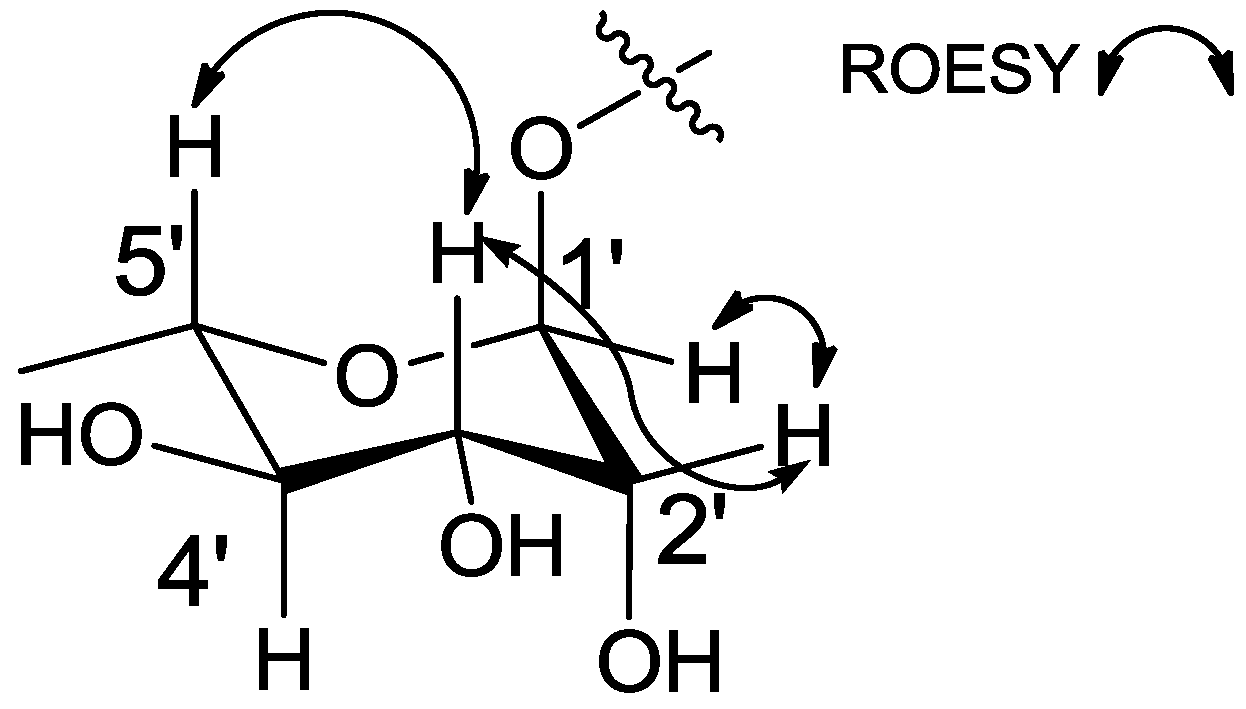
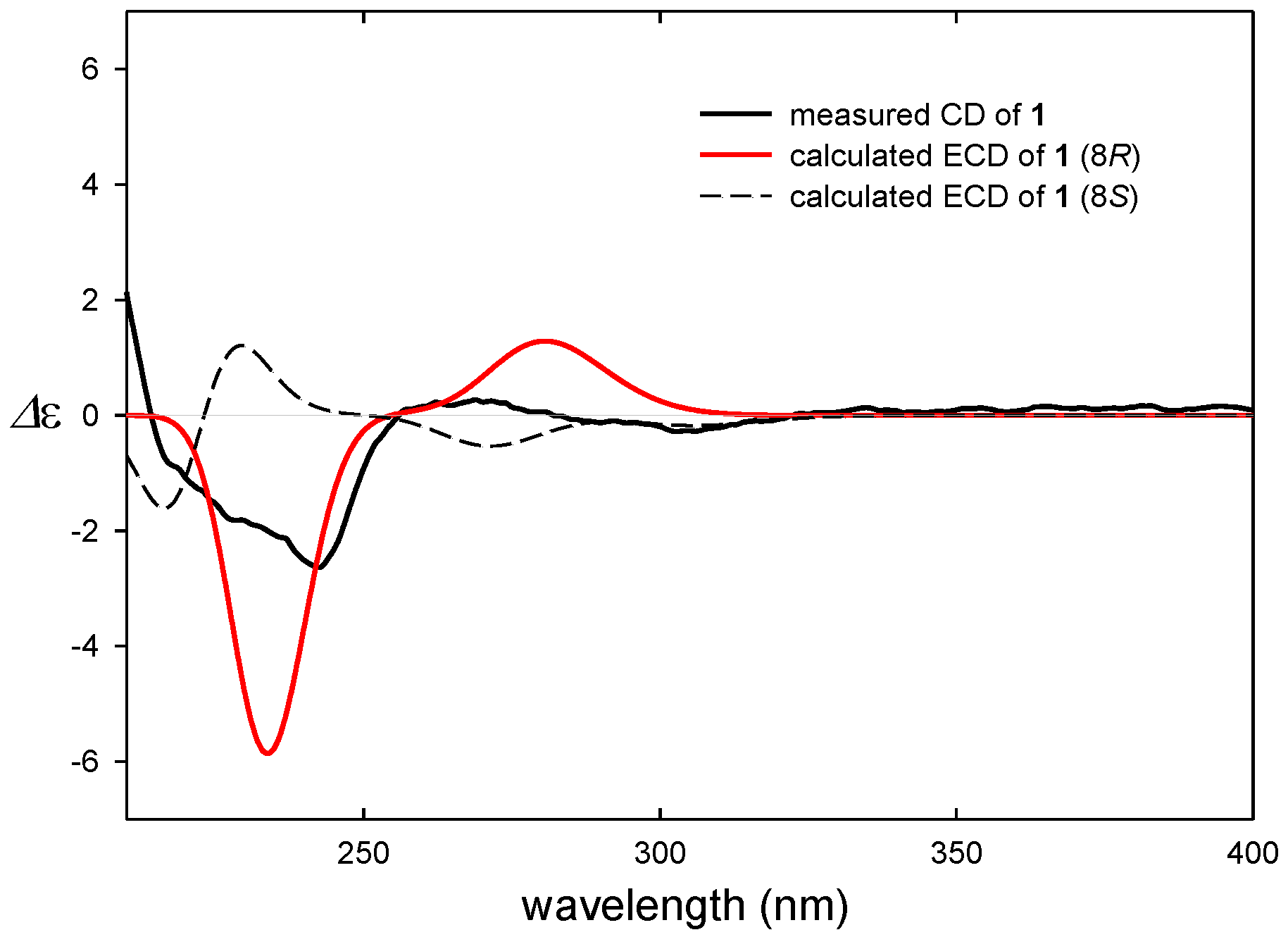
2.1. Structural Elucidation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C/H | δH a | Mult (J in Hz) | δC b | Type |
|---|---|---|---|---|
| 1 | 143.2 | C | ||
| 2 | 141.5 | C | ||
| 3 | 7.25 | d (1.5) | 106.9 | CH |
| 4 | 154.1 | C | ||
| 4-OH | 11.07 | OH | ||
| 5 | 6.83 | d (1.5) | 107.7 | CH |
| 6 | 130.7 | C | ||
| 7a | 3.27 | dd (16.0, 8.0) | 37.8 | CH2 |
| 7b | 2.95 | dd (16.0, 8.0) | ||
| 8 | 5.26 | ddd (8.0, 8.0, 8.0) | 84.5 | CH |
| 9 | 5.78 | dd (15.0, 8.0) | 130.8 | CH |
| 10 | 6.37 | dd (15.0, 10.5) | 133.1 | CH |
| 11 | 6.04 | dd (15.0, 10.5) | 131.7 | CH |
| 12 | 5.67 | dq (15.0, 7.0) | 131.4 | CH |
| 13 | 1.63 | d (7.0) | 18.5 | CH3 |
| 1′ | 6.30 | d (1.0) | 101.5 | CH |
| 2′ | 4.82 | br m | 72.5 | CH |
| 2′-OH | 7.07 | br s | OH | |
| 3′ | 4.75 | br d (9.0) | 72.9 | CH |
| 3′-OH | 6.70 | br s | OH | |
| 4′ | 4.35 | br m | 74.2 | CH |
| 4′-OH | 6.87 | br s | OH | |
| 5′ | 4.52 | m | 71.4 | CH |
| 6′ | 1.56 | d (6.0) | 18.9 | CH3 |
| C/H | δH a | Mult (J in Hz) | δC b | Type |
|---|---|---|---|---|
| 1 | 11.79 | NH | ||
| 2 | 7.46 | d (2.0) | 120.8 | CH |
| 3 | 111.1 | C | ||
| 4 | 3.76 | dd (10.0, 6.0) | 39.7 | CH |
| 5 | 4.18 | dd (10.0, 7.0) | 73.6 | CH |
| 6 | 4.83 | ddd (7.0, 7.0, 6.0) | 73.9 | CH |
| 7 | 2.99 | m | 46.8 | CH2 |
| 8 | 170.4 | C | ||
| 9 | N | |||
| 10 | 60.6 | C | ||
| 11 | 2.47 | m | 53.2 | CH |
| 12a | 3.13 | dd (12.0, 6.0) | 27.0 | CH2 |
| 12b | 3.03 | dd (12.0, 6.0) | ||
| 13 | 129.7 | C | ||
| 14 | 7.08 | d (7.5) | 116.1 | CH |
| 15 | 7.34 | dd (8.0, 7.5) | 122.4 | CH |
| 16 | 7.43 | d (8.0) | 109.0 | CH |
| 17 | 134.5 | C | ||
| 18 | 126.9 | C | ||
| 19 | 1.65 | s | 22.4 | CH3 |
| 20 | 1.74 | s | 26.4 | CH3 |
| C/H | δH a | Mult (J in Hz) | δC b | Type |
|---|---|---|---|---|
| 1 | 12.04 | s | NH | |
| 2 | 7.61 | d (2.0) | 124.9 | CH |
| 3 | 112.6 | C | ||
| 4a | 3.93 | dd(15.0, 7.5) | 27.9 | CH2 |
| 4b | 3.78 | dd(15.0, 7.5) | ||
| 5 | 4.26 | m | 57.1 | CH |
| 6 | 4.73 | dd (5.0, 3.5) | 71.4 | CH |
| 6-OH | 6.78 | OH | ||
| 7 | 4.76 | d (5.0) | 74.2 | CH |
| 7-OH | 8.87 | OH | ||
| 8 | 176.8 | C | ||
| 9 | ND c | NH | ||
| 10 | 131.9 | C | ||
| 11 | 5.56 | dd (6.0, 6.0) | 125.1 | CH |
| 12 | 4.02 | m | 32.7 | CH2 |
| 13 | 134.8 | C | ||
| 14 | 7.13 | d (7.0) | 120.1 | CH |
| 15 | 7.29 | dd (8.0, 7.0) | 122.4 | CH |
| 16 | 7.53 | d (8.0) | 110.6 | CH |
| 17 | 138.7 | C | ||
| 18 | 126.4 | C | ||
| 19 | 1.67 | s | 25.8 | CH3 |
| 20 | 1.70 | s | 18.0 | CH3 |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Isolation of Bacteria, Cultivation, and Extraction
3.3. Isolation of 1–3
3.3.1. Amycofuran (1)
3.3.2. Amycocyclopiazonic acid (2)
3.3.3. Amycolactam (3)
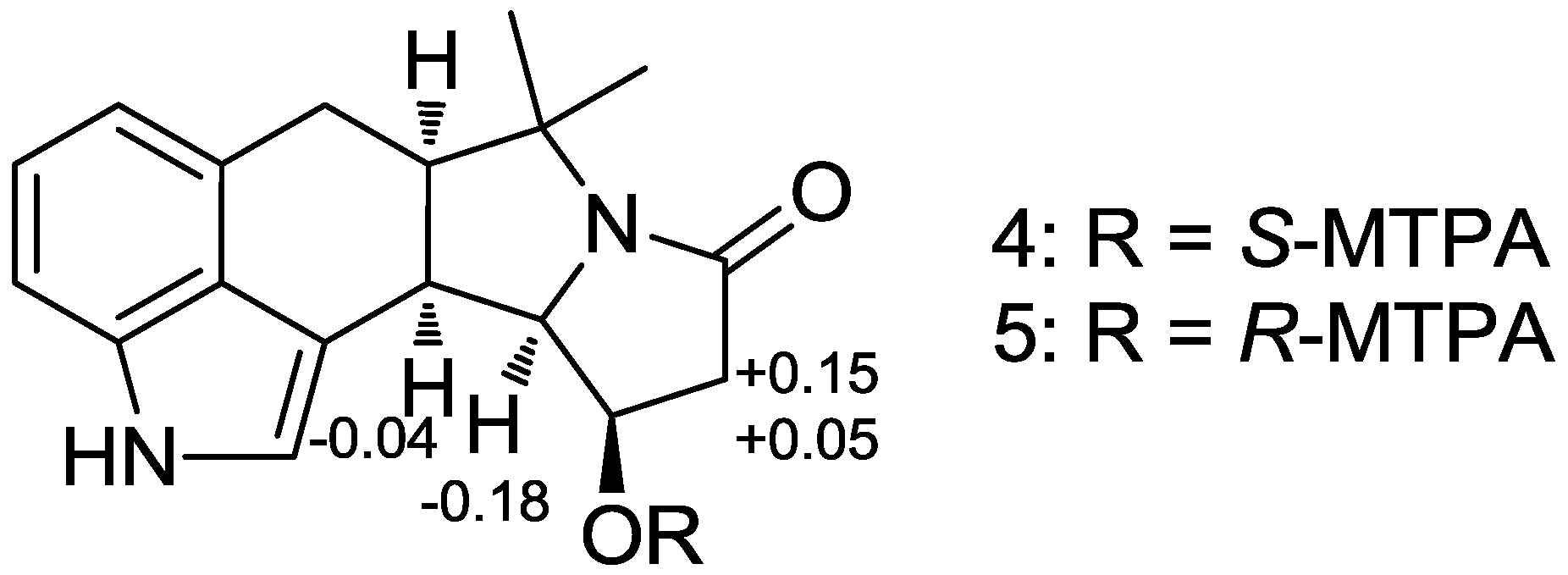
3.4. MTPA Esterification of Amycocyclopiazonic Acid (2)
3.4.1. S-MTPA Ester (4) of Amycocyclopiazonic Acid (2)
3.4.2. R-MTPA Ester (5) of Amycocyclopiazonic Acid (2)
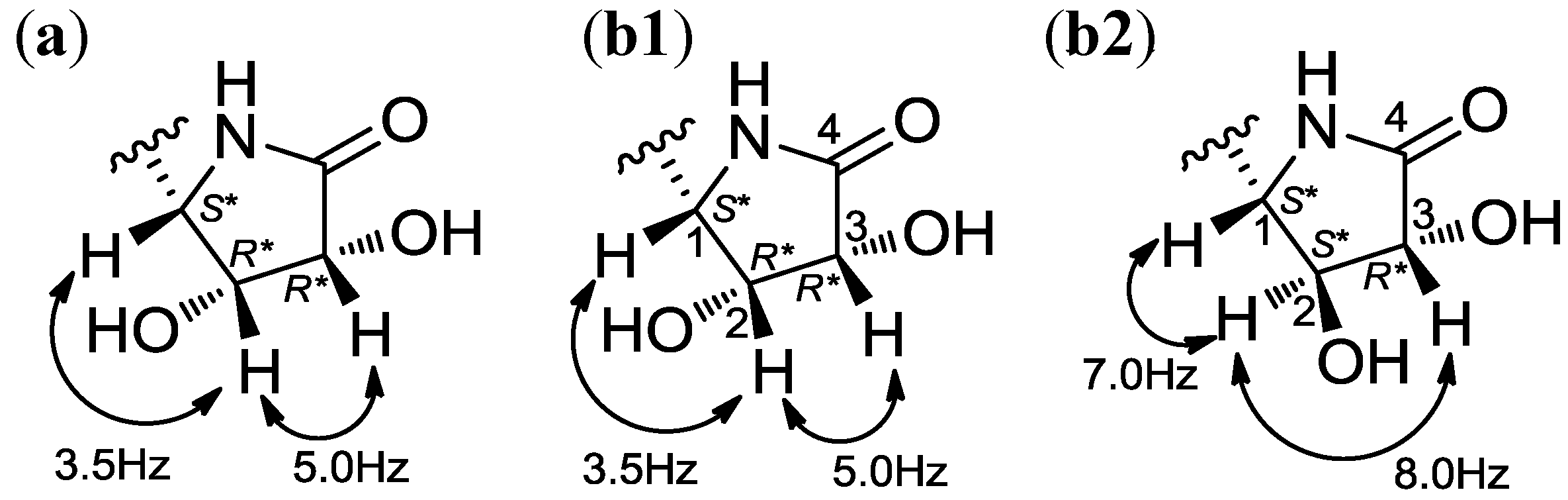
3.5. ECD Computational Calculation
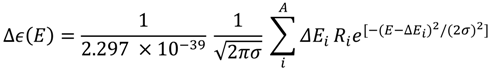
3.6. Evaluation of Anti-Proliferative Activity
4. Conclusions
Acknowledgments
Author Contributions
References
- Monster, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar]
- Liu, Y. Renaissance of marine natural product drug discovery and development. J. Mar. Sci. Res. Dev. 2012, 2, e106. [Google Scholar]
- Hentschel, U.; Piel, J.; Degnan, S.M.; Taylor, M.W. Genomic insights into the marine sponge microbiome. Nat. Rev. Microbiol. 2012, 10, 641–654. [Google Scholar] [CrossRef]
- Xu, Y.; Kersten, R.D.; Nam, S.-J.; Lu, L.; Al-Suwailem, A.M.; Zheng, H.; Fenical, W.; Dorrestein, P.C.; Moore, B.S.; Qian, P.-Y. Bacterial biosynthesis and maturation of the didemnin anti-cancer agents. J. Am. Chem. Soc. 2012, 134, 8625–8632. [Google Scholar] [CrossRef]
- Bewley, C.A.; Holland, N.D.; Faulkner, D.J. Two classes of metabolites from Theonella swinhoei are localized in distinct populations of bacterial symbionts. Experientia 1996, 52, 716–722. [Google Scholar] [CrossRef]
- Andrianasolo, E.H.; Groos, H.; Goeger, D.; Musafija-Girt, M.; McPhail, K.; Leal, R.M.; Mooberry, S.L.; Gerwick, W.H. Isolation of swinholide A and related glycosylated derivatives from two field collections of marine cyanobacteria. Org. Lett. 2005, 7, 1375–1378. [Google Scholar] [CrossRef]
- Piel, J.; Hui, D.; Wen, G.; Butzke, D.; Platzer, M.; Fusetani, N.; Matsunaga, S. Antitumor polyketide biosynthesis by an uncultivated bacterial symbiont of the marine sponge Theonella swinhoei. Proc. Natl. Acad. Sci. USA 2004, 101, 16222–16227. [Google Scholar] [CrossRef]
- Fischbach, M.A.; Walsh, C.T. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic machinery, and mechanisms. Chem. Rev. 2006, 106, 3468–3496. [Google Scholar]
- Webster, N.S.; Taylor, M.W. Marine sponges and their microbial symbionts: Love and other relationships. Environ. Microbiol. 2012, 14, 335–346. [Google Scholar] [CrossRef]
- Wilson, M.C.; Mori, T.; Ruckert, C.; Uria, A.R.; Helf, M.J.; Takada, K.; Gernert, C.; Steffens, U.A.E.; Heycke, N.; Schmitt, S.; et al. An environmental bacterial taxon with a large and distinct metabolic repertoire. Nature 2014, 506, 58–62. [Google Scholar] [CrossRef]
- Bérdy, J. Thoughts and facts about antibiotics: Where we are now and where we are heading. J. Antibiot. 2012, 65, 385–395. [Google Scholar] [CrossRef]
- Vincente, J.; Stewart, A.; Song, B.; Hill, R.T.; Wright, J.L. Biodiversity of actinomycetes associated with Caribbean sponges and their potential for natural product discovery. Mar. Biotechnol. 2013, 15, 413–424. [Google Scholar] [CrossRef]
- Li, K.; Li, Q.-L.; Ji, N.-Y.; Liu, B.; Zhang, W.; Cao, X.-P. Deoxyuridines from the marine sponge associated actinomycete Streptomyces microflavus. Mar. Drugs 2011, 9, 690–695. [Google Scholar] [CrossRef]
- Wei, R.-B.; Xi, T.; Li, J.; Wang, P.; Li, F.-C.; Lin, Y.-C.; Qin, S. Lobophorin C and D, new kijanimicin derivatives from a marine sponge-associated actinomycetal strain AZS17. Mar. Drugs 2011, 9, 359–368. [Google Scholar] [CrossRef]
- Pretsch, E.; Bȕhlmann, P.; Affolter, C. Structure Determination of Organic Compounds—Tables of Spectral Data; Springer: New York, NY, USA, 2000; p. 153. [Google Scholar]
- Berova, N.; Di Bari, L.; Pescitelli, G. Application of electronic circular dichroism in configurational and conformational analysis of organic compounds. Chem. Soc. Rev. 2007, 36, 914–931. [Google Scholar] [CrossRef]
- Shi, Y.-M.; Wang, L.-Y.; Zou, X.-S.; Li, X.-N.; Shang, S.-Z.; Gao, Z.-H.; Liang, C.-Q.; Luo, H.-R.; Li, H.-L.; Xiao, W.-L.; et al. Nortriterpenoids from Schisandra chinensis and their absolute configurational assignments by electronic circular dichroism study. Tetrahedron 2014, 70, 859–868. [Google Scholar] [CrossRef]
- Pfefferle, W.; Anke, H.; Bross, M.; Steffan, B.; Vianden, R.; Steglich, W. Asperfuran, a novel antifungal metabolite from Aspergillus oryzae. J. Antibiot. 1990, 43, 648–654. [Google Scholar] [CrossRef]
- Felix, F.; Manuel, S.J.; Emilio, Q.; Riguera, R. Determining the absolute stereochemistry of secondary/secondary diols by 1H NMR: Basis and applications. J. Org. Chem. 2005, 70, 3778–3790. [Google Scholar] [CrossRef]
- Pabba, J.; Rempel, B.P.; Withers, S.G.; Vasella, A. Synthesis of glycaro-1,5-lactams and tetrahydrotetrazolopyridine-5-carboxylates: Inhibitors of β-d-glucuronidase and α-l-iduronidase. Helv. Chim. Acta 2006, 89, 635–666. [Google Scholar]
- Prasad, K.R.; Pawar, A.B. Enantiospecific total synthesis of (+)-lentiginosine. ARKIVOC 2010, 6, 39–46. [Google Scholar]
- Huang, Y.; Dalton, D.R.; Carroll, P.J. The efficient, enantioselective synthesis of aza sugars from amino acids. 1. The polyhydroxylated pyrrolidines. J. Org. Chem. 1997, 62, 372–376. [Google Scholar] [CrossRef]
- Sonoda, T.; Osada, H.; Uzawa, J.; Isono, K. Actiketal, a new member of the glutarimide antibiotics. J. Antibiot. 1991, 44, 160–163. [Google Scholar] [CrossRef]
- Suzuki, K.; Yahara, S.; Maehata, K.; Uyeda, M. Isoaurostatin, a novel topoisomerase inhibitor produced by Thermomonospora alba. J. Nat. Prod. 2001, 64, 204–207. [Google Scholar] [CrossRef]
- Simonetti, S.O.; Larghi, E.L.; Bracca, A.B.J.; Kaufman, T.S. Angular tricyclic benzofurans and related natural products of fungal origin. Isolation, biological activity and synthesis. Nat. Prod. Rep. 2013, 30, 941–969. [Google Scholar]
- Holzapfel, C.W. Isolation and structure of cyclopiazonic acid, a toxic metabolite of Penicillium cyclopium. Tetrahedron 1968, 24, 2101–2119. [Google Scholar]
- Liu, X.; Walsh, C.T. Characterization of cyclo-acetoacetyl-l-tryptophan dimethylallytransferase in cyclopiazonic acid biosynthesis: Substrate promiscuity and site directed mutagenesis studies. Biochemistry 2009, 48, 11032–11044. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kwon, Y.; Kim, S.-H.; Shin, Y.; Bae, M.; Kim, B.-Y.; Lee, S.K.; Oh, K.-B.; Shin, J.; Oh, D.-C. A New Benzofuran Glycoside and Indole Alkaloids from a Sponge-Associated Rare Actinomycete, Amycolatopsis sp. Mar. Drugs 2014, 12, 2326-2340. https://doi.org/10.3390/md12042326
Kwon Y, Kim S-H, Shin Y, Bae M, Kim B-Y, Lee SK, Oh K-B, Shin J, Oh D-C. A New Benzofuran Glycoside and Indole Alkaloids from a Sponge-Associated Rare Actinomycete, Amycolatopsis sp. Marine Drugs. 2014; 12(4):2326-2340. https://doi.org/10.3390/md12042326
Chicago/Turabian StyleKwon, Yun, Seong-Hwan Kim, Yoonho Shin, Munhyung Bae, Byung-Yong Kim, Sang Kook Lee, Ki-Bong Oh, Jongheon Shin, and Dong-Chan Oh. 2014. "A New Benzofuran Glycoside and Indole Alkaloids from a Sponge-Associated Rare Actinomycete, Amycolatopsis sp." Marine Drugs 12, no. 4: 2326-2340. https://doi.org/10.3390/md12042326
APA StyleKwon, Y., Kim, S.-H., Shin, Y., Bae, M., Kim, B.-Y., Lee, S. K., Oh, K.-B., Shin, J., & Oh, D.-C. (2014). A New Benzofuran Glycoside and Indole Alkaloids from a Sponge-Associated Rare Actinomycete, Amycolatopsis sp. Marine Drugs, 12(4), 2326-2340. https://doi.org/10.3390/md12042326