Rapid and Accurate Identification by Real-Time PCR of Biotoxin-Producing Dinoflagellates from the Family Gymnodiniaceae

Abstract
:1. Introduction
2. Results and Discussion

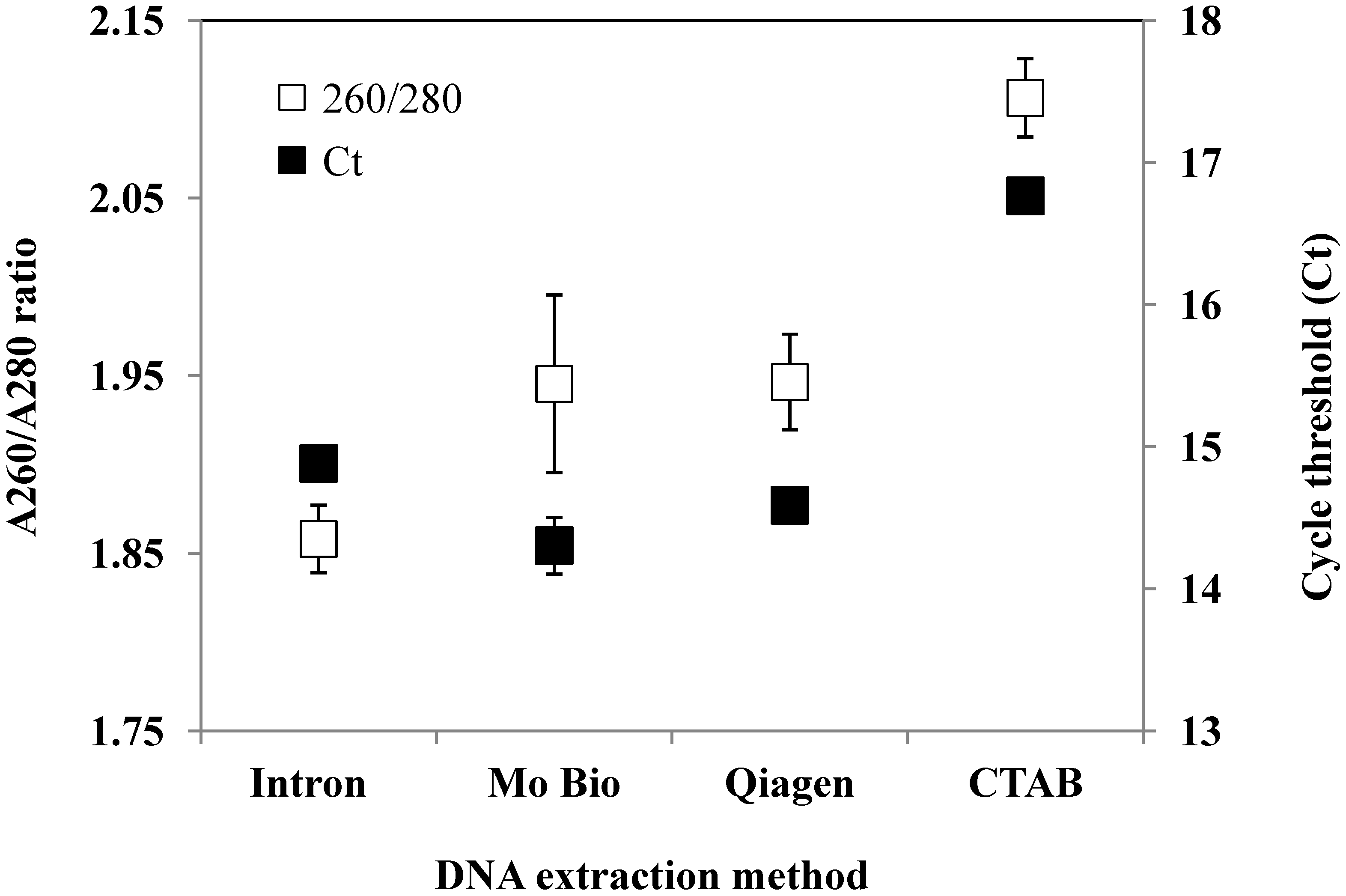
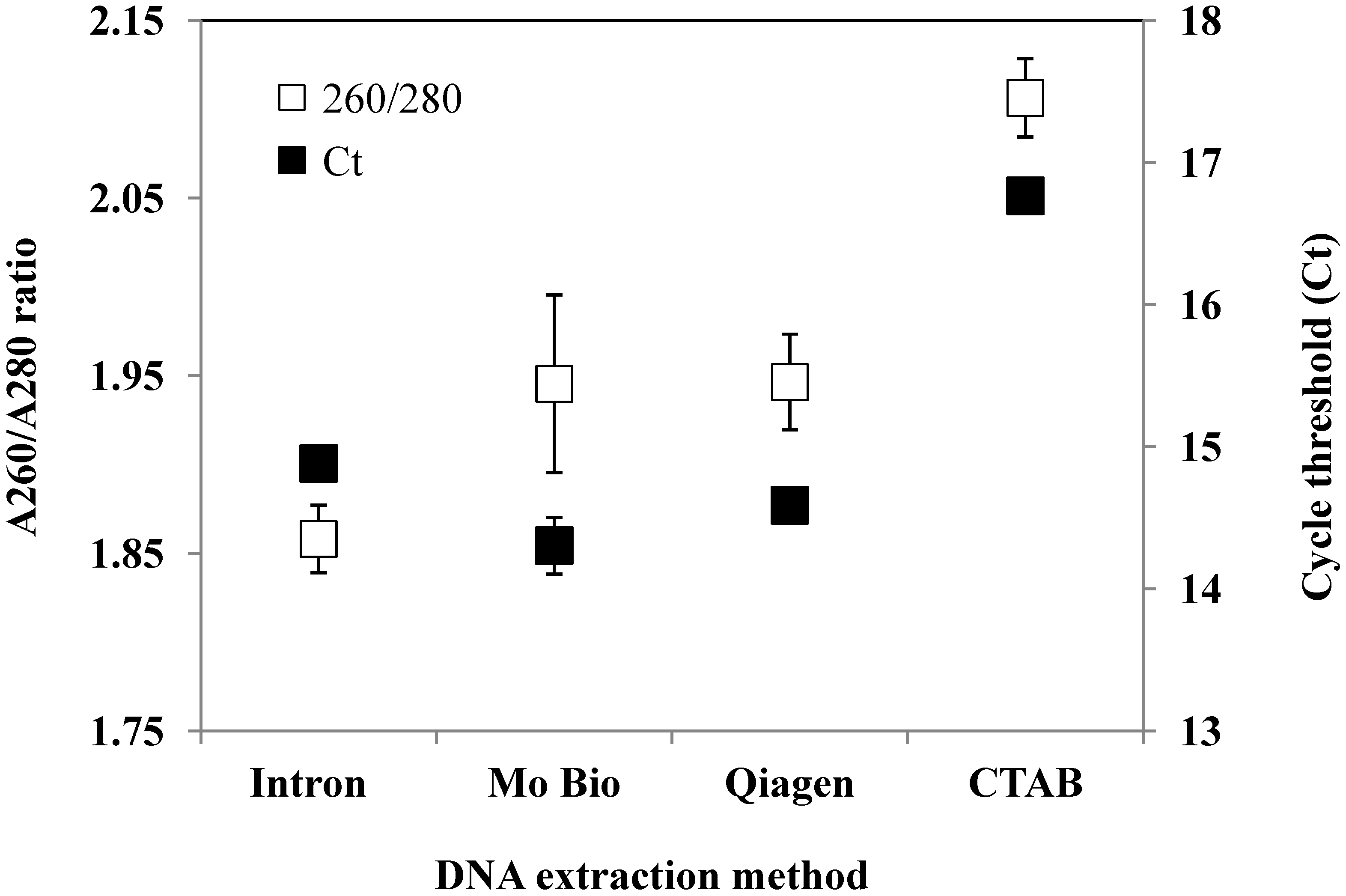{kind=link}
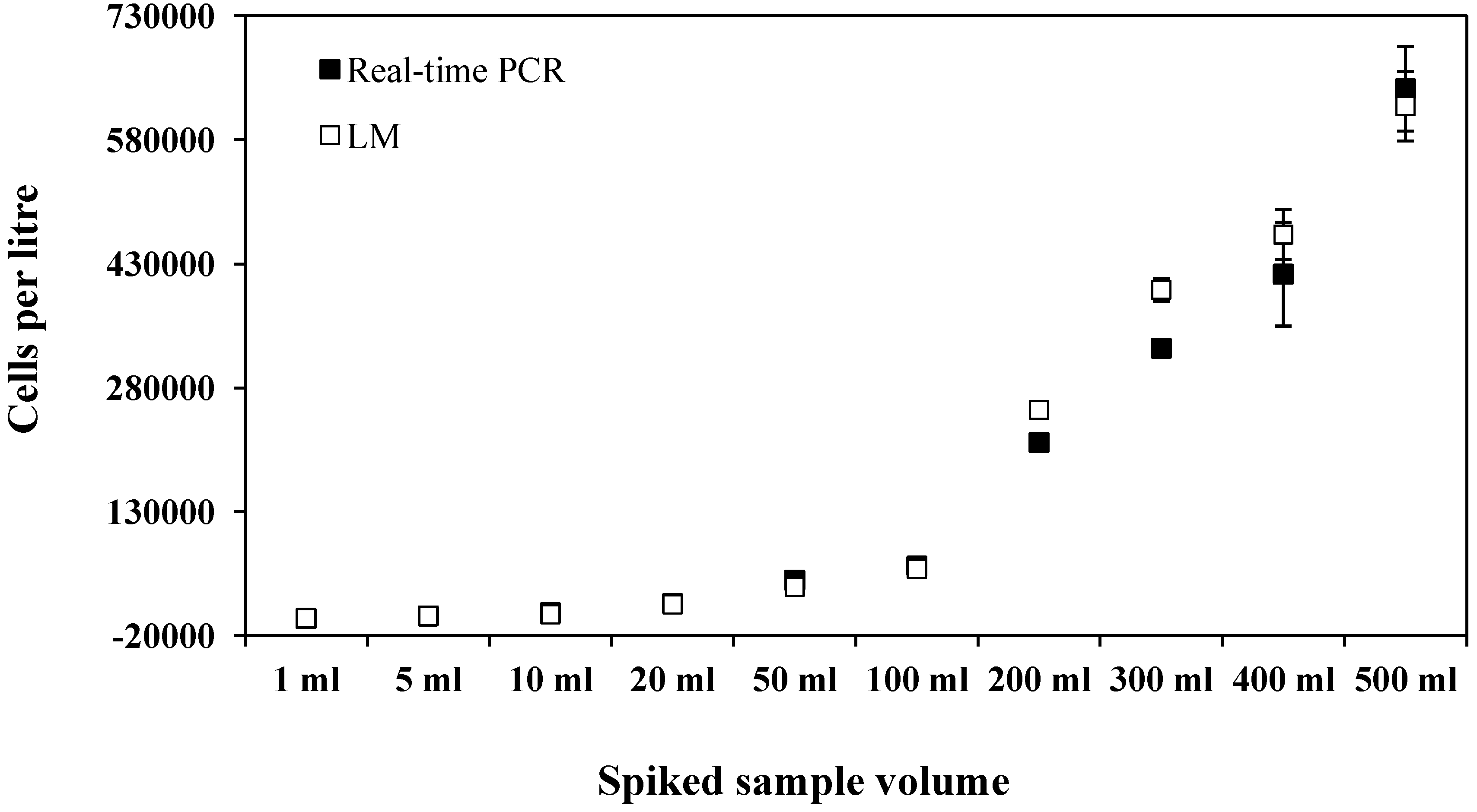
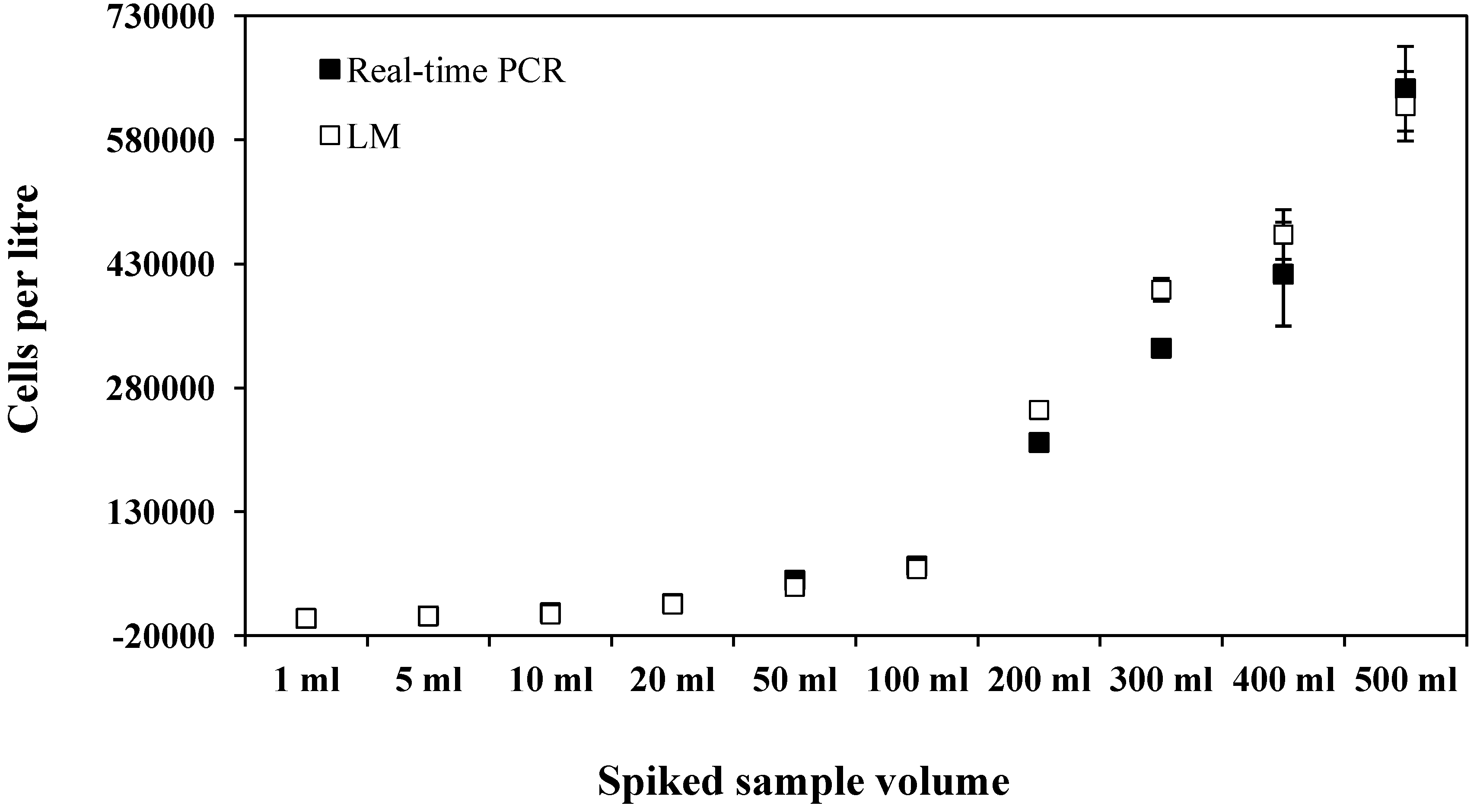{kind=link}
{kind=link}
| Target Species | Primer Name and Sequence | Product Size (bp) | Final Concentration |
|---|---|---|---|
| Gymnodinium aureolum | GA519-F: GGACATGGTAGCCTGCC | 153 | 500 nM |
| GA683-R: GTCAGGAAGGTGCTCAGC | 500 nM | ||
| GA560-P: 6FAM-CAGAACTCACTGTCATATTGCTCCTCC-BHQ-1 | 50 nM | ||
| Gymnodinium catenatum | GC397-F: CTTGGTGAGATTGTCGCAC | 93 | 500 nM |
| GC471-R: GCAAGAAACATCACACCGA | 1000 nM | ||
| GC426-P: 6FAM-TGATCACCTTCTATTCCAGCGAAAGC-BHQ-1 | 80 nM | ||
| Karenia brevisulcata | KBS460-F: GATCTGGATGCGATACTGAAT | 153 | 300 nM |
| KBS585-R: AGCACTGCTACAAGACATATAA | 900 nM | ||
| KBS544-P: 6FAM-TGACTGAATGTCCCTAGTTGAACTC-BHQ-1 | 50 nM | ||
| Karenia mikimotoi | KM541-F: CGAGTGACTGAATGTCCTCA | 112 | 500 nM |
| KM645-R: CCAACAACCTTCATGCAGAG | 250 nM | ||
| KM578-P: 6FAM-CTACCAGACACACAGAGAGCAG-BHQ-1 | 50 nM | ||
| Karenia papilionacea | KP449-F: TCTGGATGCGATACTGGTTG | 232 | 1000 nM |
| KP682-R: TACTTATGTCAAGGATGTGTTC | 750 nM | ||
| KP630-P: 6FAM-CTTGTTAGTTACCTGGCATGAGAC-BHQ-1 | 125 nM | ||
| Karenia umbella | KU480-F: ATGTCAACGTCAGTTCACAAT | 161 | 750 nM |
| KU623-R: GCACGAGACGAGGCTTA | 250 nM | ||
| KU542-P: 6FAM-TTCGACTAGGCACATTCAGTCAC-BHQ-1 | 50 nM | ||
| Karlodinium veneficum | KV590-F: TGCCTGGTAGAACTCATGTC | 100 | 1000 nM |
| KV672-R: ACGAGTAACAGAAGCTACAAG | 1000 nM | ||
| KDV640-P: 6FAM-TGTTCTCATTACCTGCGTCTGGG-BHQ-1 | 50 nM | ||
| Takayama tasmanica | TT533-F: ACTTCTGGGTGACTGAACGT | 134 | 100 nM |
| TT665-R: CCACGTCCTGTCCCATGC | 1000 nM | ||
| TT616-P: 6FAM-CTGGGCTTTGTTCACTGCTCTTAA-BHQ-1 | 125 nM |
| Species Name | CICCM Code | Accession Number |
|---|---|---|
| Gymnodinium aureolum | CAWD59, 87 | AY947659 |
| Gymnodinium catenatum | CAWD102, 101, 109, 126 | AY036128 |
| Gymnodinium cf. impudicum | CAWD139 | |
| Gymnodinium cf. microreticulatum | CAWD191 | |
| Gymnodinium chlorophorum | CAWD62 | |
| Gymnodinium impudicum | CAWD03 | |
| Gymnodinium instriatum | CAWD137 | |
| Gymnodinium simplex | CAWD86 | |
| Gymnodinium sp. | CAWD172 | |
| Karenia bidigitata | CAWD80 | |
| Karenia brevis | CAWD08 | |
| Karenia brevisulcata | CAWD82 | AY243032 |
| Karenia mikimotoi | CAWD63, 117, 133, 134, 192 | U92249 |
| Karenia papilionacea | CAWD91 | U92252 |
| Karenia selliformis | CAWD79 | |
| Karenia umbella | CAWD131, 65 | AY947664 |
| Karlodinium veneficum | CAWD84 | AY947665 |
| Takayama helix | CAWD128 | |
| Takayama tasmanica | CAWD115 | AY947669 |
| Target Species | Lower Limit of Detection (Cells/Reaction, 1 s.f.) | Amplification Efficiency | R2 Value |
|---|---|---|---|
| K. mikimotoi | 0.007 | 101% | 1.00 |
| K. umbella | 0.09 | 105% | 0.99 |
| K. papilionacea | 0.2 | 95% | 0.99 |
| K. brevisulcata | 0.2 | 102% | 0.99 |
| K. veneficum | 0.3 | 93% | 0.98 |
| G. catenatum | 0.006 | 106% | 1.00 |
| G. aureolum | 0.006 | 105% | 0.99 |
| T. tasmanica | 0.09 | 102% | 1.00 |
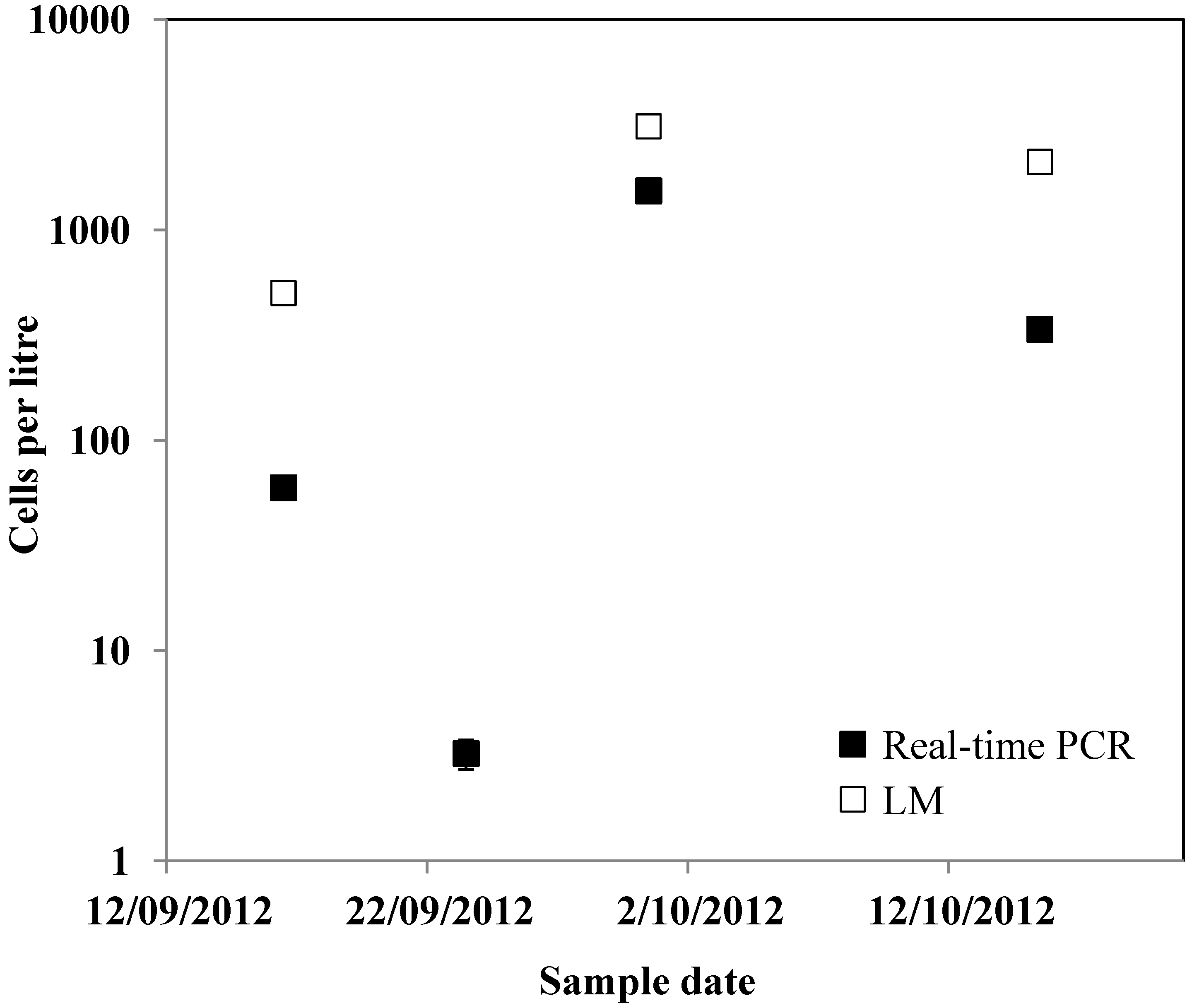
3. Experimental Section
3.1. Culture Maintenance
3.2. DNA Extraction
3.3. Primers and Probe Design for Real-Time PCR Assays
3.4. Real-Time PCR Assay Optimisation, Specificity and Sensitivity
3.5. Determination of Copy Number and Quantification of the Gymnodinium catenatum Assay
3.6. Spiked Environmental and Natural Sample Testing
4. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Daugbjerg, N.; Hansen, G.; Larsen, J.; Moestrup, O. Phylogeny of some of the major genera of dinoflagellates based on ultrastructure and partial LSU rDNA sequence data, including the erection of three new genera of unarmoured dinoflagellates. Phycologia 2000, 39, 302–317. [Google Scholar] [CrossRef]
- de Salas, M.F.; Bolch, C.J.S.; Botes, L.; Nash, G.; Wright, S.W.; Hallegraeff, G.M. Takayama gen. nov (Gymnodiniales, Dinophyceae), a new genus of unarmored dinoflagellates with sigmoid apical grooves, including the description of two new species. J. Phycol. 2003, 39, 1233–1246. [Google Scholar] [CrossRef]
- Brand, L.E.; Campbell, L.; Bresnan, E. Karenia: the biology and ecology of a toxic genus. Harmful Algae 2012, 14, 156–178. [Google Scholar] [CrossRef]
- Taylor, F.J.R.; Fukuyo, Y.; Larsen, J.; Hallegraeff, G.M. Taxonomy of Harmful Dinoflagellates. In Manual on Harmful Marine Microalgae; Hallegraeff, G.M., Anderson, D.M., Cembella, A.D., Eds.; UNESCO Intergovernmental Oceanographic Commission: Pairs, France, 2003; pp. 389–432. [Google Scholar]
- Rhodes, L.L.; Haywood, A.J.; Ballantine, W.J.; MacKenzie, A.L. Algal blooms and climate anomalies in north-east New Zealand, August–December 1992. N. Z. J. Mar. Freshw. Res. 1993, 27, 419–430. [Google Scholar] [CrossRef]
- Wear, R.G.; Gardner, J.P.A. Biological effects of the toxic algal bloom of February and March 1998 on the benthos of Wellington Harbour, New Zealand. Mar. Ecol. Prog. Ser. 2001, 218, 63–76. [Google Scholar] [CrossRef]
- MacKenzie, L.A.; Beauchamp, T. Gymnodinium catenatum in New Zealand: A New Problem for Public Health and the Shellfish Industry; Cawthron Institute: Nelson, New Zealand, 2002. [Google Scholar]
- Irwin, A.; Hallegraeff, G.M.; McMinn, A.; Harrison, J.; Heijnis, H. Cyst and radionuclide evidence demonstrate historic Gymnodinium catenatum dinoflagellate populations in Manukau and Hokianga Harbours, New Zealand. Harmful Algae 2003, 2, 61–74. [Google Scholar] [CrossRef]
- Yang, Z.B.; Takayama, H.; Matsuoka, K.; Hodgkiss, I.J. Karenia digitata sp nov. (Gymnodiniales, Dinophyceae), a new harmful algal bloom species from the coastal waters of west Japan and Hong Kong. Phycologia 2000, 39, 463–470. [Google Scholar] [CrossRef]
- Yang, Z.B.; Hodgkiss, I.J.; Hansen, G. Karenia longicanalis sp nov. (Dinophyceae): A new bloom-forming species isolated from Hong Kong, May 1998. Bot. Mar. 2001, 44, 67–74. [Google Scholar]
- Haywood, A.J.; Steidinger, K.A.; Truby, E.W.; Bergquist, P.R.; Bergquist, P.L.; Adamson, J.; MacKenzie, L. Comparative morphology and molecular phylogenetic analysis of three new species of the genus Karenia (Dinophyceae) from New Zealand. J. Phycol. 2004, 40, 165–179. [Google Scholar] [CrossRef]
- Jasperse, J.A. Marine Toxins and New Zealand Shellfish: Proceedings of a Workshop on Research Issues, 10–11 June 1993; Royal Society of New Zealand: Wellington, New Zealand; p. 1993.
- Todd, K. A Review of NSP Monitoring in New Zealand in Support of a New Programme; Cawthron Report No. 660; Marine Biotoxin Technical Committee: Nelson, New Zealand, 2003. [Google Scholar]
- MacKenzie, L.A.; Haywood, A.J.; Adamson, J.; Truman, P.; Till, D.; Seki, T.; Satake, M.; Yasumoto, T. Gymnodimine Contamination of Shellfish in New Zealand. In Harmful and Toxic Algal Blooms; Yasumoto, T., Oshima, Y., Fukuyo, Y., Eds.; Intergovernmental Oceanographic Commission of UNESCO: Paris, France, 1996; pp. 97–100. [Google Scholar]
- Holland, P.T.; Shi, F.; Satake, M.; Hamamoto, Y.; Ito, E.; Beuzenberg, V.; McNabb, P.; Munday, R.; Briggs, L.; Truman, P.; et al. Novel toxins produced by the dinoflagellate Karenia brevisulcata. Harmful Algae 2012, 13, 47–57. [Google Scholar] [CrossRef]
- Satake, M.; Shoji, M.; Oshima, Y.; Naoki, H.; Fujita, T.; Yasumoto, T. Gymnocin-A, a cytotoxic polyether from the notorious red tide dinoflagellate, Gymnodinium mikimotoi. Tetrahedron Lett. 2002, 43, 5829–5832. [Google Scholar] [CrossRef]
- Satake, M.; Tanaka, Y.; Ishikura, Y.; Oshima, Y.; Naoki, H.; Yasumoto, T. Gymnocin-B with the largest contiguous polyether rings from the red tide dinoflagellate, Karenia (formerly Gymnodinium) mikimotoi. Tetrahedron Lett. 2005, 46, 3537–3540. [Google Scholar] [CrossRef]
- de Salas, M.F.; Bolch, C.J.S.; Hallegraeff, G.M. Karlodinium australe sp. nov. (Gymnodiniales, Dinophyceae), a new potentially ichthyotoxic unarmoured dinoflagellate from lagoonal habitats of south-eastern Australia. Phycologia 2005, 44, 640–650. [Google Scholar] [CrossRef]
- García Camacho, F.; Gallardo Rodríguez, J.; Sánchez Mirón, A.; Cerón García, M.C.; Belarbi, E.H.; Chisti, Y.; Molina Grima, E. Biotechnological significance of toxic marine dinoflagellates. Biotechnol. Adv. 2007, 25, 176–194. [Google Scholar] [CrossRef]
- Waters, A.L.; Hill, R.T.; Place, A.R.; Hamann, M.T. The expanding role of marine microbes in pharmaceutical development. Curr. Opin. Biotechnol. 2010, 21, 780–786. [Google Scholar] [CrossRef]
- Beuzenberg, V.; Mountfort, D.; Holland, P.; Shi, F.; MacKenzie, L. Optimization of growth and production of toxins by three dinoflagellates in photobioreactor cultures. J. Appl. Phycol. 2012, 24, 1023–1033. [Google Scholar] [CrossRef]
- Selwood, A.I.; van Ginkel, R.; Wilkins, A.L.; Munday, R.; Ramsdell, J.S.; Jensen, D.J.; Cooney, J.M.; Miles, C.O. Semisynthesis of S-Desoxybrevetoxin-B2 and Brevetoxin-B2, and assessment of their acute toxicities. Chem. Res. Toxicol. 2008, 21, 944–950. [Google Scholar] [CrossRef]
- Rhodes, L.L.; Scholin, C.; Tyrrell, J.; Adamson, J.; Todd, K. The integration of DNA probes into New Zealand’s routine phytoplankton monitoring programmes. In Harmful Algal Blooms; Hallegraeff, G.M., Blackburn, S.I., Bolch, C.J., Lewis, R.J., Eds.; Intergovernmental Oceanographic Commission of UNESCO: Paris, France, 2001; pp. 429–432. [Google Scholar]
- Rhodes, L.L.; Smith, K.F.; Moisan, C. Shifts and stasis in marine HAB monitoring in New Zealand. Environ. Sci. Pollut. Res. 2013, 20, 6872–6877. [Google Scholar] [CrossRef]
- NZFSA. Non-Commercial Marine Biotoxin Monitoring Programme: NZFSA VA Operating and Response Manual; New Zealand Food Safety Authority (NZFSA): Wellington, New Zealand, 2010; p. 39. [Google Scholar]
- Rhodes, L.L.; Smith, K.F.; de Salas, M. DNA probes, targeting large sub-unit rRNA, for the rapid identification of the paralytic shellfish poison producing dinoflagellate, Gymnodinium catenatum. N. Z. J. Mar. Freshw. Res. 2007, 41, 385–390. [Google Scholar] [CrossRef]
- Burkholder, J.M. Implications of harmful microalgae and heterotrophic dinoflagellates in management of sustainable marine fisheries. Ecol. Appl. 1998, 8, S37–S62. [Google Scholar]
- Van Dolah, F.M. Marine algal toxins: origins, health effects, and their increased occurrence. Environ. Health Perspect. 2000, 108, 133–141. [Google Scholar] [CrossRef]
- Landsberg, J.H. The effects of harmful algal blooms on aquatic organisms. Rev. Fish. Sci. 2002, 10, 113–390. [Google Scholar] [CrossRef]
- Hallegraeff, G.M. Ocean climate change, phytoplankton community responses, and harmful algal blooms: a formidable predictive challenge. J. Phycol. 2010, 46, 220–235. [Google Scholar] [CrossRef]
- Godhe, A.; Cusack, C.; Pedersen, J.; Anderson, P.; Anderson, D.M.; Breasnan, E.; Cembella, A.; Dahl, E.; Diercks, S.; Elbrachter, M.; et al. Intercalibration of classical and molecular techniques for identification of Alexandrium fundyense (Dinophyceae) and estimation of cell densities. Harmful Algae 2007, 6, 56–72. [Google Scholar] [CrossRef] [Green Version]
- John, U.; Medlin, L.K.; Groben, R. Development of specific rRNA probes to distinguish between geographic clades of the Alexandrium tamarense species complex. J. Plankton Res. 2005, 27, 199–204. [Google Scholar] [CrossRef]
- Penna, A.; Bertozzini, E.; Battocchi, C.; Galluzzi, L.; Giacobbe, M.G.; Vila, M.; Garces, E.; Luglie, A.; Magnani, M. Monitoring of HAB species in the Mediterranean Sea through molecular methods. J. Plankton Res. 2007, 29, 19–38. [Google Scholar]
- Wood, S.A.; Smith, K.F.; Banks, J.C.; Tremblay, L.A.; Rhodes, L.L.; Mountfort, D.; Cary, S.C.; Pochon, X. Molecular genetic tools for environmental monitoring of New Zealand’s aquatic habitats, past, present and the future. N. Z. J. Mar. Freshw. Res. 2013, 47, 90–119. [Google Scholar] [CrossRef]
- Miller, P.E.; Scholin, C.A. Identification and enumeration of cultured and wild Pseudo-nitzschia (Bacillariophyceae) using species-specific LSU rRNA-targeted fluorescent probes and filter-based whole cell hybridization. J. Phycol. 1998, 34, 371–382. [Google Scholar] [CrossRef]
- Rhodes, L.L.; Scholin, C.; Garthwaite, I. Pseudo-nitzschia in New Zealand and the role of DNA probes and immunoassays in refining marine biotoxin monitoring programmes. Nat. Toxins 1998, 6, 105–111. [Google Scholar] [CrossRef]
- Ayers, K.; Rhodes, L.L.; Tyrrell, J.V.; Gladstone, M.; Scholin, C.A. International accreditation of sandwich hybridisation assay format DNA probes for micro-algae. N. Z. J. Mar. Freshw. Res. 2005, 39, 1225–1231. [Google Scholar] [CrossRef]
- Haywood, A.J.; Scholin, C.A.; Marin, R., III; Steidinger, K.A.; Heil, C.; Ray, J. Molecular detection of the brevetoxin-producing dinoflagellate Karenia brevis and closely related species using rRNA-targeted probes and a semiautomated sandwich hybridization assay. J. Phycol. 2007, 43, 1271–1286. [Google Scholar] [CrossRef]
- Bowers, H.A.; Tengs, T.; Goto, S.; Tomas, C.; Ono, C.; Yoshimatsu, S.; Oldach, D.; Steidinger, K.A.; Landsberg, J.H.; Tomas, C.R.; et al. Development of real-time PCR assays for the detection of Chattonella species in culture and environmental samples. In Harmful Algae 2002; Steidinger, K.A., Landsberg, J.H., Tomas, C.R., Vargo, G.A., Eds.; Florida Institute of Oceanography, and Intergovernmental Oceanographic Commission of UNESCO: Pairs, France, 2004; pp. 231–233. [Google Scholar]
- Coyne, K.J.; Handy, S.M.; Demir, E.; Whereat, E.B.; Hutchins, D.A.; Portune, K.J.; Doblin, M.A.; Cary, S.C. Improved quantitative real-time PCR assays for enumeration of harmful algal species in field samples using an exogenous DNA reference standard. Limnol. Oceanogr. 2005, 3, 381–391. [Google Scholar] [CrossRef]
- Patil, J.; Gunasekera, R.; Deagle, B.; Bax, N.; Blackburn, S. Development and evaluation of a PCR based assay for detection of the toxic dinoflagellate, Gymnodinium catenatum (Graham) in ballast water and environmental samples. Biol. Invasions 2005, 7, 983–994. [Google Scholar] [CrossRef]
- Dyhrman, S.T.; Erdner, D.L.; La Du, J.; Galac, M.; Anderson, D.M. Molecular quantification of toxic Alexandrium fundyense in the Gulf of Maine using real-time PCR. Harmful Algae 2006, 5, 242–250. [Google Scholar] [CrossRef]
- Murray, S.A.; Wiese, M.; Stüken, A.; Brett, S.; Kellmann, R.; Hallegraeff, G.; Neilan, B.A. sxtA-based quantitative molecular assay to identify saxitoxin-producing harmful algal blooms in marine waters. Appl. Environ. Microb. 2011, 77, 7050–7057. [Google Scholar] [CrossRef]
- Perini, F.; Casabianca, A.; Battocchi, C.; Accoroni, S.; Totti, C.; Penna, A. New approach using the real-time PCR method for estimation of the toxic marine dinoflagellate Ostreopsis cf. ovata in marine environment. PLoS One 2011, 6, e17699. [Google Scholar]
- Penna, A.; Galluzzi, L. The quantitative real-time PCR applications in the monitoring of marine harmful algal bloom (HAB) species. Environ. Sci. Pollut. Res. 2013, 20, 6851–6862. [Google Scholar] [CrossRef]
- Vandersea, M.W.; Kibler, S.R.; Holland, W.C.; Tester, P.A.; Schultz, T.F.; Faust, M.A.; Holmes, M.J.; Chinain, M.; Litaker, R.W. Development of semi-quantitative PCR assays for the detection and enumeration of Gambierdiscus species (Gonyaulacales, Dinophyceae). J. Phycol. 2012, 48, 902–915. [Google Scholar] [CrossRef]
- Hariganeya, N.; Tanimoto, Y.; Yamaguchi, H.; Nishimura, T.; Tawong, W.; Sakanari, H.; Yoshimatsu, T.; Sato, S.; Preston, C.M.; Adachi, M. Quantitative PCR method for enumeration of cells of cryptic species of the toxic marine dinoflagellate Ostreopsis spp. in coastal waters of Japan. PLoS One 2013, 8, e57627. [Google Scholar] [CrossRef]
- Gescher, C.; Metfies, K.; Medlin, L.K. The ALEX CHIP—Development of a DNA chip for identification and monitoring of Alexandrium. Harmful Algae 2008, 7, 485–494. [Google Scholar] [CrossRef]
- Galluzzi, L.; Cegna, A.; Casabianca, S.; Penna, A.; Saunders, N.; Magnani, M. Development of an oligonucleotide microarray for the detection and monitoring of marine dinoflagellates. J. Microbiol. Methods 2011, 84, 234–242. [Google Scholar] [CrossRef]
- Zamor, R.M.; Glenn, K.L.; Hambright, K.D. Incorporating molecular tools into routine HAB monitoring programs: using qPCR to track invasive Prymnesium. Harmful Algae 2012, 15, 1–7. [Google Scholar] [CrossRef]
- Yuan, J.; Mi, T.; Zhen, Y.; Yu, Z. Development of a rapid detection and quantification method of Karenia mikimotoi by real-time quantitative PCR. Harmful Algae 2012, 17, 83–91. [Google Scholar] [CrossRef]
- Chang, F.H. Gymnodinium brevisulcatum sp nov (Gymnodiniales, Dinophyceae), a new species isolated from the 1998 summer toxic bloom in Wellington Harbour, New Zealand. Phycologia 1999, 38, 377–384. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bertozzini, E.; Penna, A.; Perini, F.; Garces, E.; Magnani, M. Analysis of rRNA gene content in the Mediterranean dinoflagellate Alexandrium catenella and Alexandrium taylori: Implications for the quantitative real-time PCR-based monitoring methods. J. Appl. Phycol. 2010, 22, 1–9. [Google Scholar] [CrossRef]
- Godhe, A.; Asplund, M.E.; Härnström, K.; Saravanan, V.; Tyagi, A.; Karunasagar, I. Quantification of diatom and dinoflagellate biomasses in coastal marine seawater samples by real-time PCR. Appl. Environ. Microb. 2008, 74, 7174–7182. [Google Scholar] [CrossRef]
- New Zealand Food Safety Authority, Ministry for Primary Industries, Wellington, New Zealand. Unpublished data. 2013.
- Loeblich, A.R.; Smith, V.E. Chloroplast pigments of the marine dinoflagellate Gyrodinium resplendens. Lipids 1968, 3, 5–13. [Google Scholar] [CrossRef]
- Keller, M.D.; Selvin, R.C.; Claus, W.; Guillard, R.R.L. Media for the culture of oceanic ultraphytoplankton. J. Phycol. 1987, 23, 633–638. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Throndsen, J. Preservation and storage. In Phytoplankton Manual; Sournia, A., Ed.; United Nations Educational, Scientific and Cultural Organization (UNESCO): Paris, France, 1978; pp. 69–74. [Google Scholar]
- Utermöhl, H. Zur vervollkommung der quantitativen phytoplankton methodik (Towards a perfection of quantitative phytoplankton methodology). Mitt. Int. Ver. Theor. Angew. Limnol. 1958, 9, 1–38. [Google Scholar]
- Ebenezer, V.; Medlin, L.K.; Ki, J.S. Molecular detection, quantification, and diversity evaluation of microalgae. Mar. Biotechnol. 2012, 14, 129–142. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Smith, K.F.; De Salas, M.; Adamson, J.; Rhodes, L.L. Rapid and Accurate Identification by Real-Time PCR of Biotoxin-Producing Dinoflagellates from the Family Gymnodiniaceae. Mar. Drugs 2014, 12, 1361-1376. https://doi.org/10.3390/md12031361
Smith KF, De Salas M, Adamson J, Rhodes LL. Rapid and Accurate Identification by Real-Time PCR of Biotoxin-Producing Dinoflagellates from the Family Gymnodiniaceae. Marine Drugs. 2014; 12(3):1361-1376. https://doi.org/10.3390/md12031361
Chicago/Turabian StyleSmith, Kirsty F., Miguel De Salas, Janet Adamson, and Lesley L. Rhodes. 2014. "Rapid and Accurate Identification by Real-Time PCR of Biotoxin-Producing Dinoflagellates from the Family Gymnodiniaceae" Marine Drugs 12, no. 3: 1361-1376. https://doi.org/10.3390/md12031361