Asperolide A, a Marine-Derived Tetranorditerpenoid, Induces G2/M Arrest in Human NCI-H460 Lung Carcinoma Cells, Is Mediated by p53-p21 Stabilization and Modulated by Ras/Raf/MEK/ERK Signaling Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
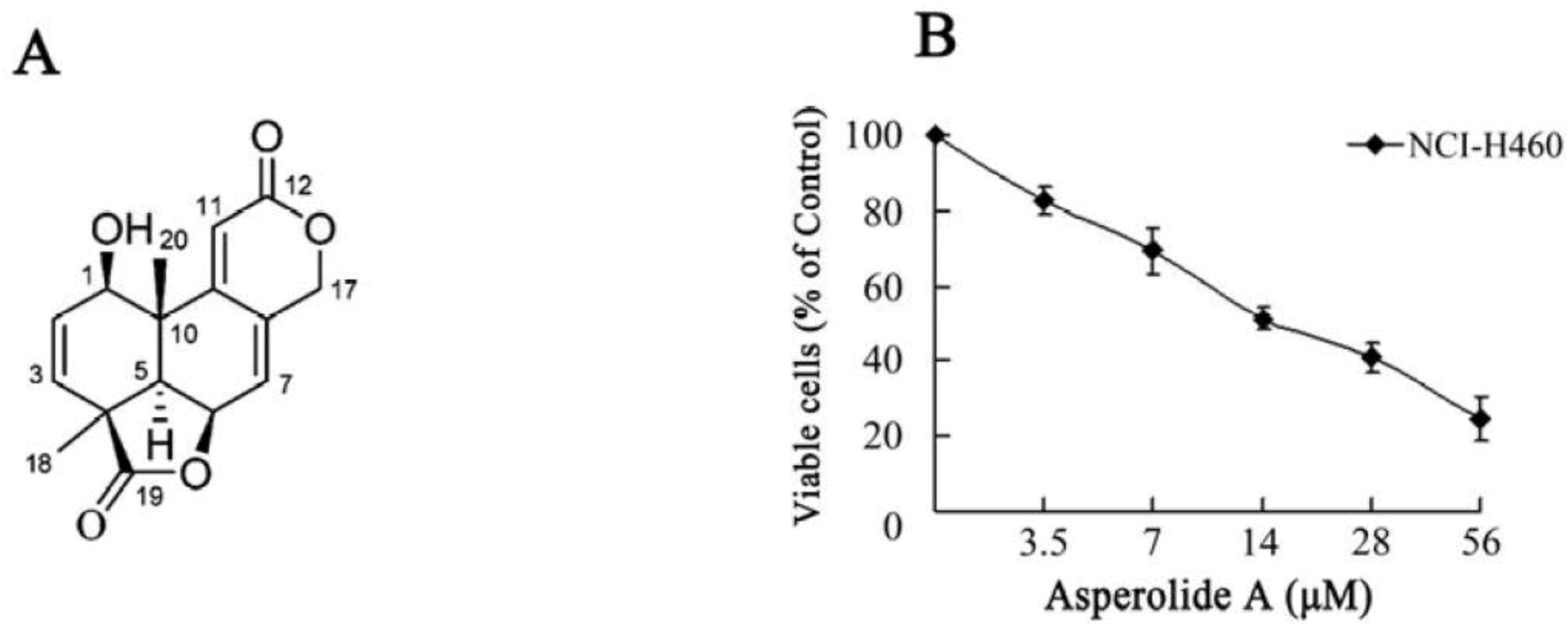
2.1. Asperolide A Inhibits the Proliferation of NCI-H460 Lung Cancer Cells
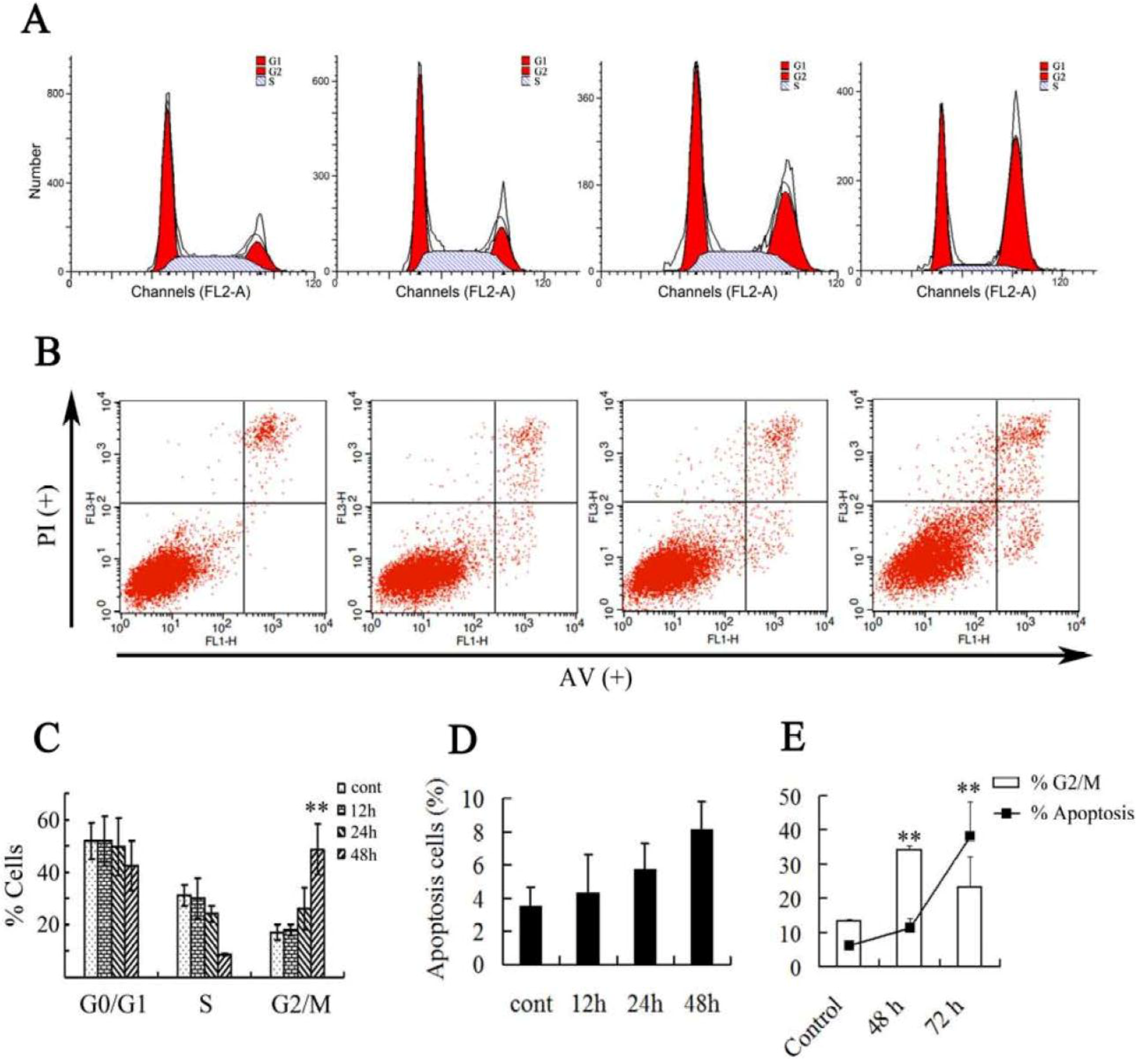
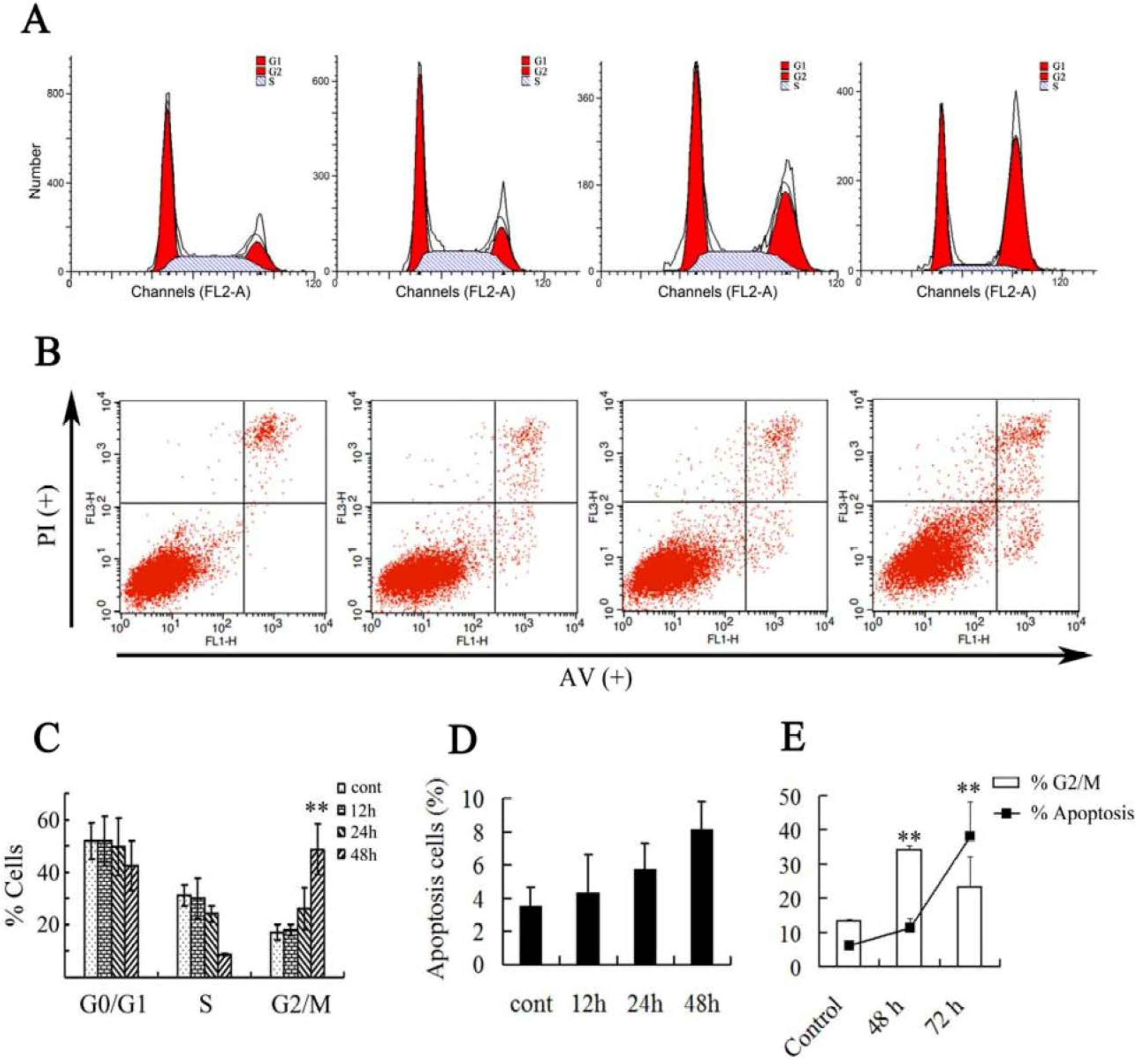
2.2. Effect of Asperolide A on Cell Cycle Progression and Apoptosis
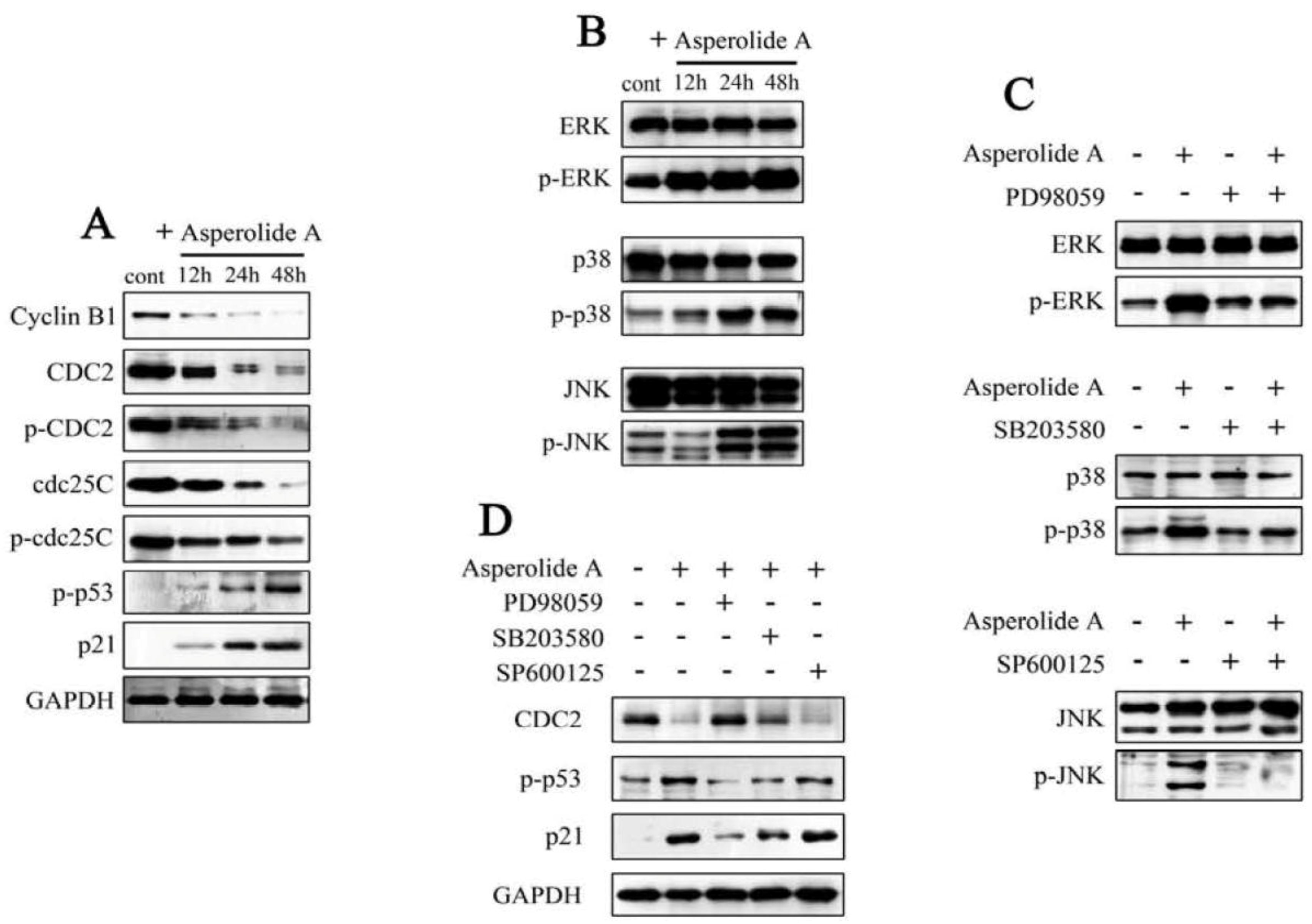
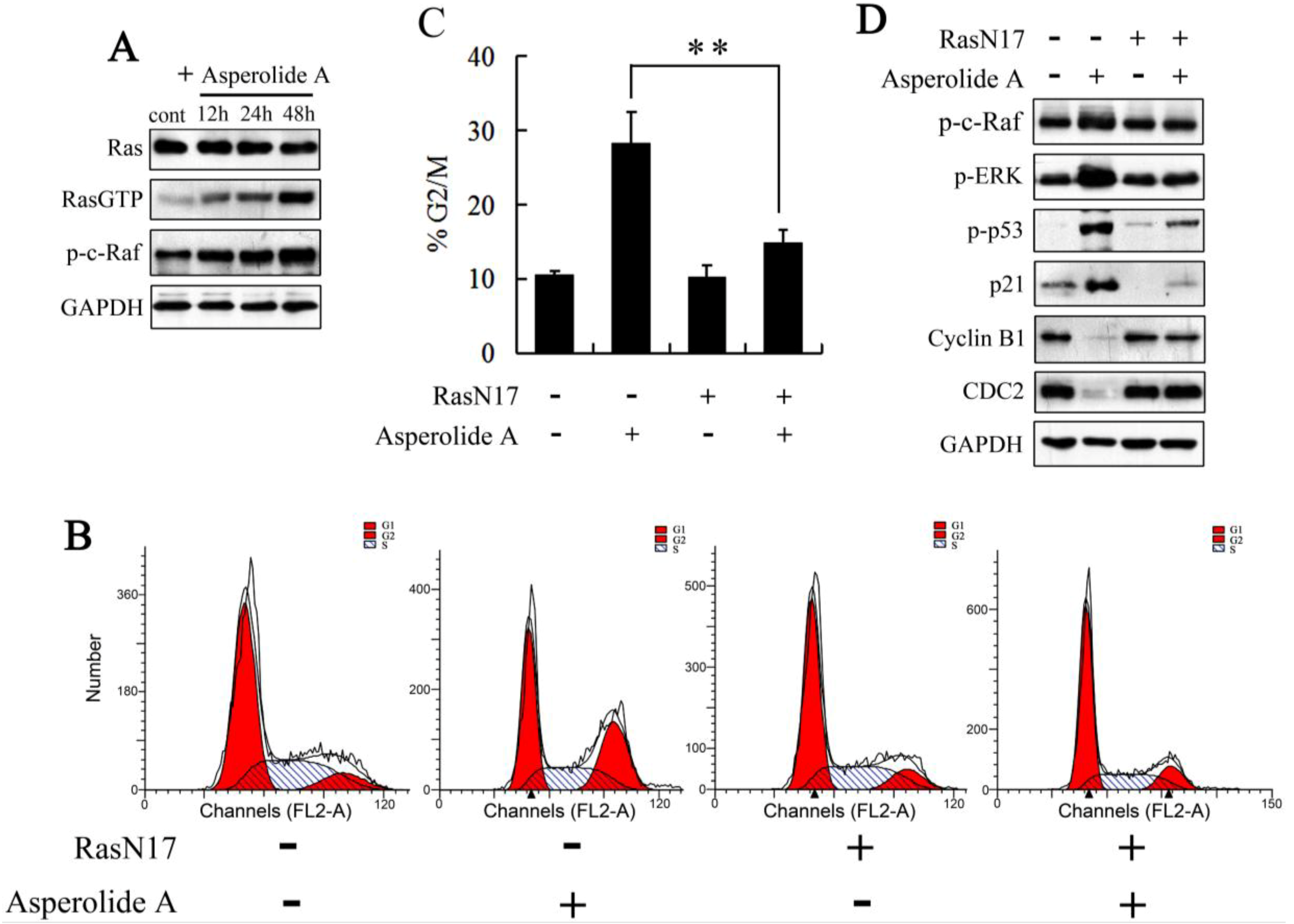
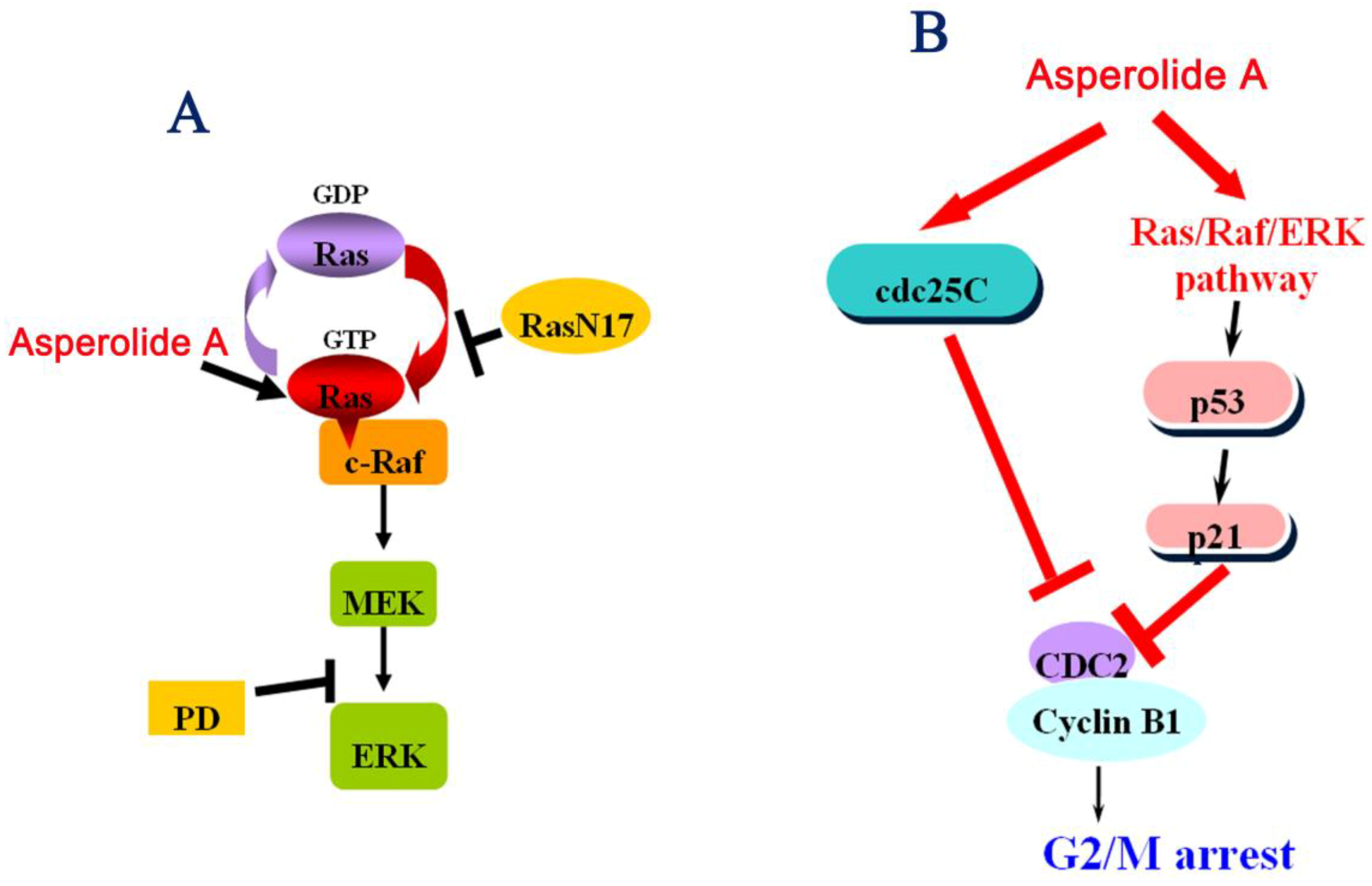
2.3. ERK-p53-p21 Activation Contributes to Asperolide A-Induced Cell Cycle Arrest in NCI-H460 Cells

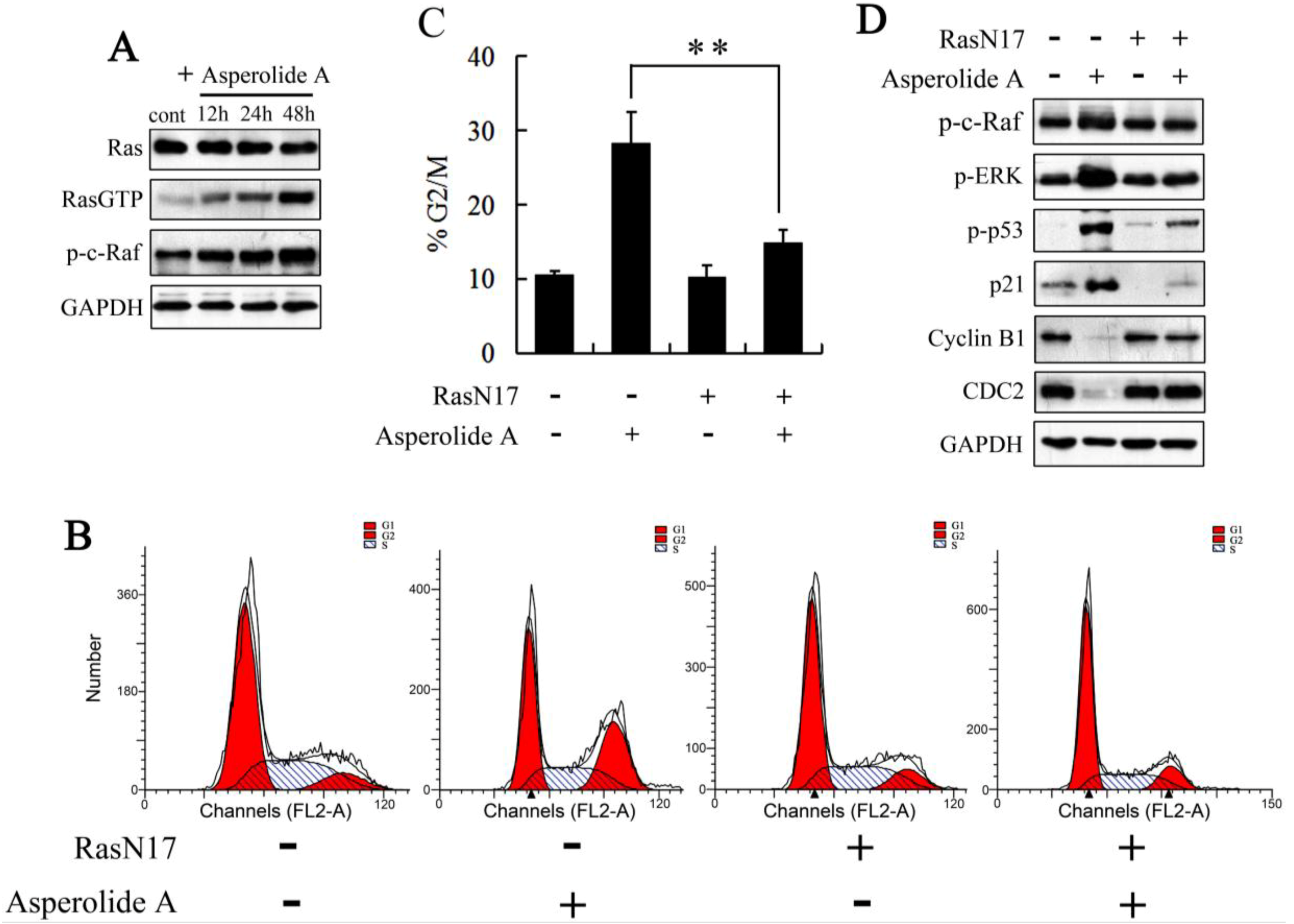
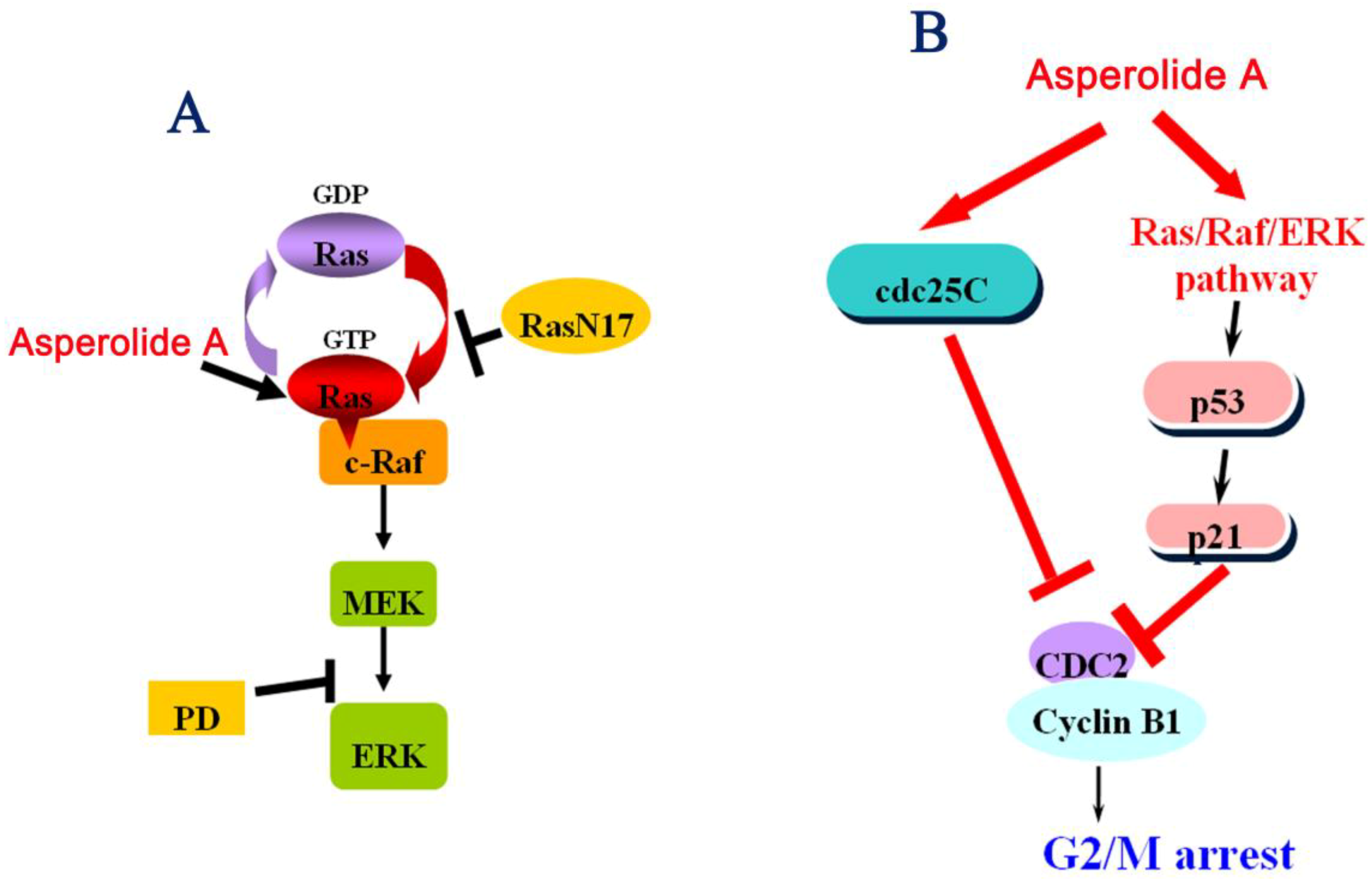
2.4. RasN17 Gene Transfectants Suppress the Asperolide A-Induced G2/M Arrest in NCI-H460 Cells
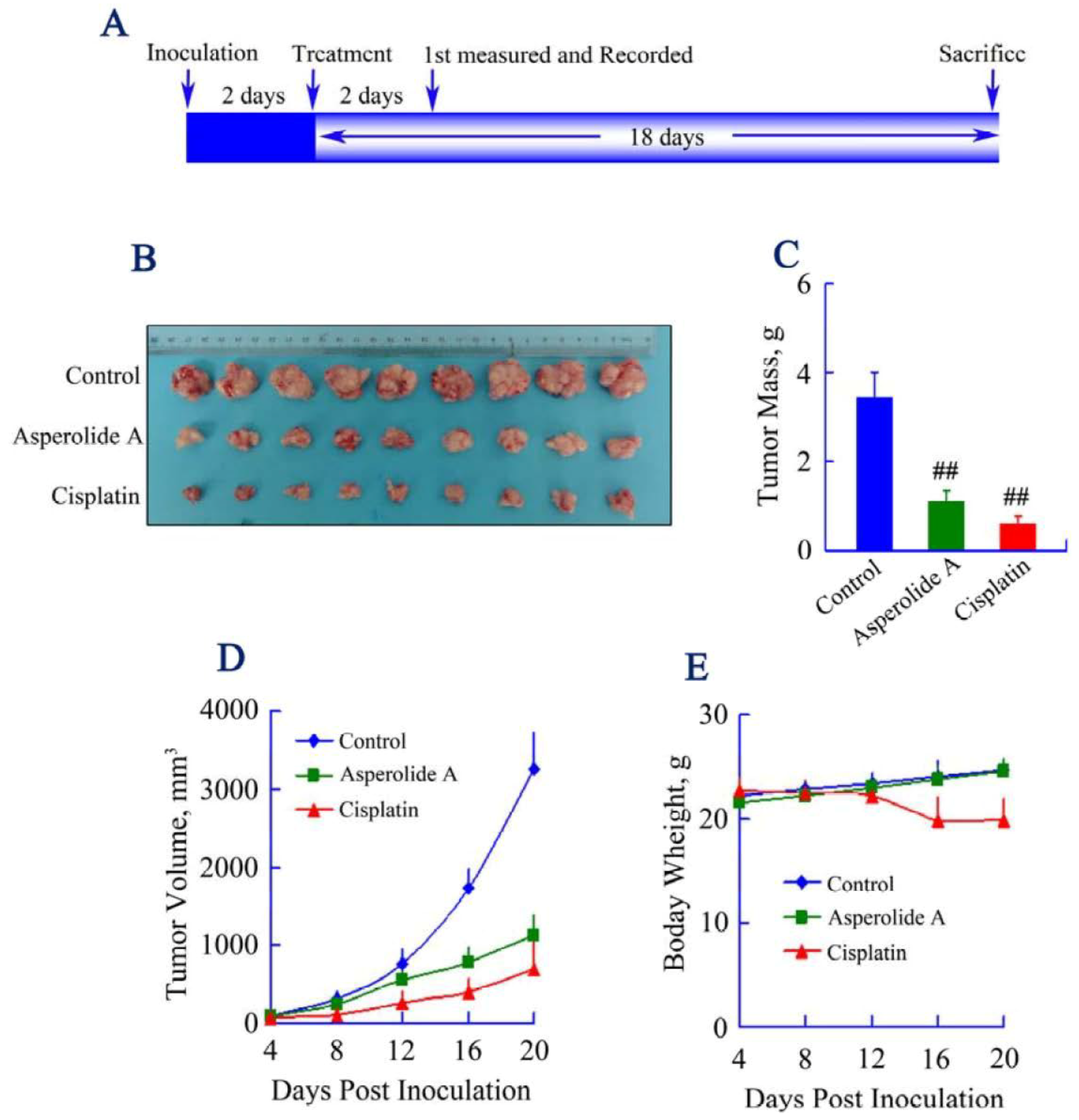
2.5. Asperolide A Inhibits Tumor Xenograft Growth
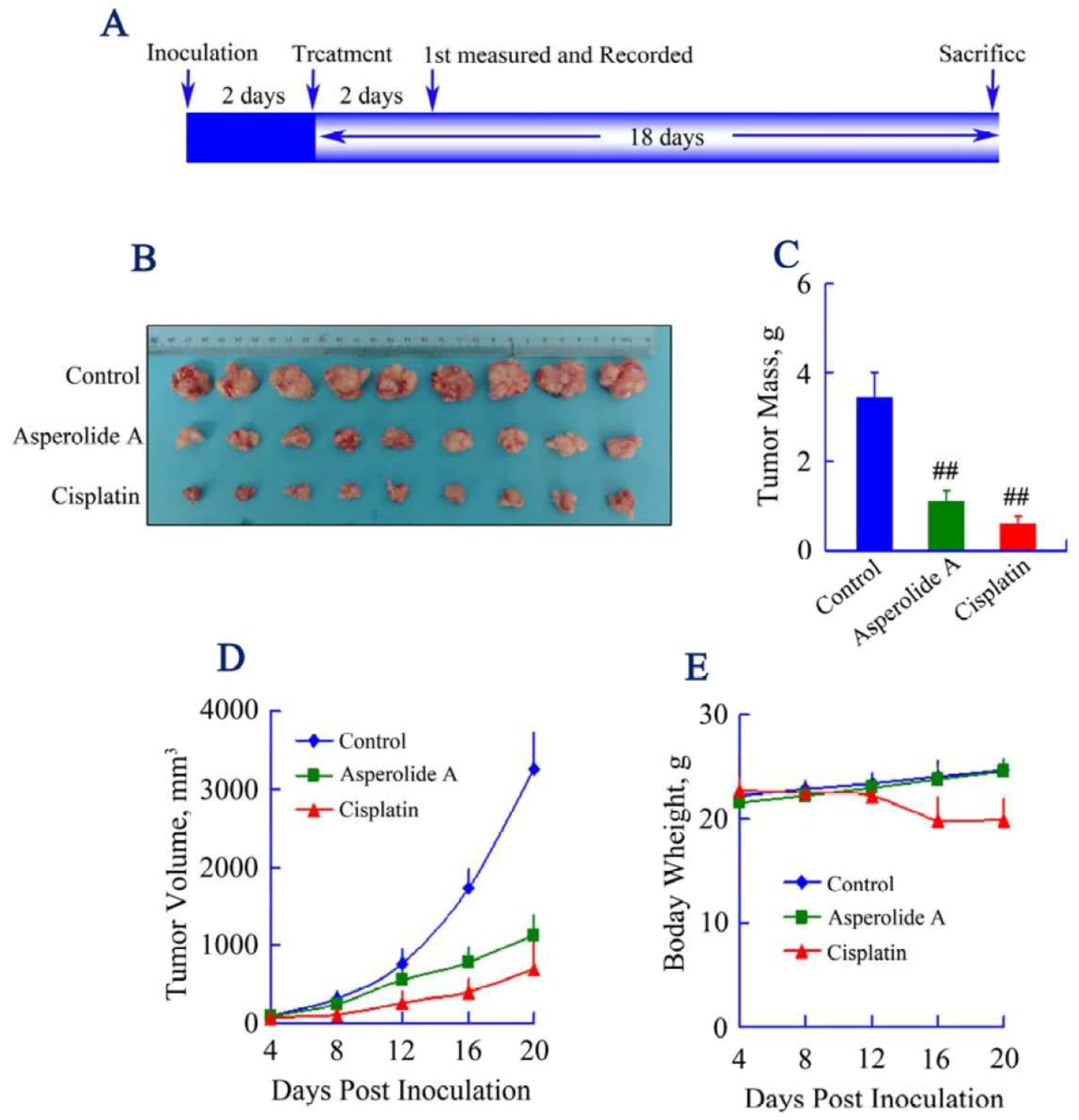
2.6. Discussion
3. Experimental Section
3.1. Materials
3.2. Cell Cultures and Drug Preparation
3.3. Cell Viability Assay
3.4. Cell Apoptosis Assay
3.5. Cell Cycle Analysis
3.6. Western Blot Analysis
3.7. pCMV-RasN17 Vector Transfection
3.8. Mouse Xenograft Model
3.9. Statistical Analysis
4. Conclusions
Acknowledgments
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer. J. Clin. 2011, 6, 69–90. [Google Scholar]
- Navada, S.; Lai, P.; Schwartz, A.G.; Kalemkerian, G.P. Temporal trends in small cell lung cancer: Analysis of the national Surveillance Epidemiology and End-Results (SEER) database. J. Clin. Oncol. 2007, 25, 5570–5577. [Google Scholar] [CrossRef]
- American Cancer Society Surveillance Research, Cancer Facts and Figures 2004; American Cancer Society: Atlanta, GA, USA, 2004; pp. 1–60.
- Molina, J.R.; Yang, P.; Cassivi, S.D. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar]
- Shen, L.; Li, Z.; Shen, S.; Niu, X.; Yu, Y.; Li, Z.; Liao, M.; Chen, Z.; Lu, S. The synergistic effect of EGFR tyrosine kinase inhibitor gefitinib in combination with aromatase inhibitor anastrozole in non-small cell lung cancer cell lines. Lung Cancer 2012, 78, 193–200. [Google Scholar]
- Omenn, G.S.; Goodman, G.E.; Thornquist, M.D.; Balmes, J.; Cullen, M.R.; Glass, A.; Keogh, J.P.; Meyskens, F.L., Jr.; Valanis, B.; Williams, J.H., Jr.; et al. Risk factors for lung cancer and for intervention effects in CARET, the β-Carotene and Retinol Efficacy Trial. J. Natl. Cancer Inst. 1996, 88, 1550–1559. [Google Scholar] [CrossRef]
- Das, A.; Bortner, J.; Desai, D.; Amin, S.; El-Bayoumy, K. The selenium analog of the chemopreventive compound S,S′-(1,4-phenylenebis[1,2-ethanediyl]) bisisothiourea is a remarkable inducer of apoptosis and inhibitor of cell growth in human non-small cell lung cancer. Chem. Biol. Interact. 2009, 180, 158–164. [Google Scholar] [CrossRef]
- Reungwetwattana, T.; Weroha, S.J.; Molina, J.R. Oncogenic pathways, molecularly targeted therapies, and highlighted clinical trials in non-small-cell lung cancer (NSCLC). Clin. Lung Cancer 2012, 13, 252–266. [Google Scholar] [CrossRef]
- Salomon, D.S.; Brandt, R.; Ciardiello, F. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit. Rev. Oncol. Hematol. 1995, 19, 183–232. [Google Scholar] [CrossRef]
- Woessmann, W.; Chen, X.; Borkhardt, A. Ras-mediated activation of ERK by cisplatin induces cell death independently of p53 in osteosarcoma and neuroblastoma cell lines. Cancer Chemother. Pharmacol. 2002, 50, 397–404. [Google Scholar] [CrossRef]
- Bacus, S.S.; Gudkov, A.V.; Lowe, M.; Lyass, L.; Yung, Y.; Komarov, A.P. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene 2001, 20, 147–155. [Google Scholar] [CrossRef]
- Tang, D.; Wu, D.; Hirao, A.; Lahti, J.M.; Liu, L.; Mazza, B. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J. Biol. Chem. 2002, 277, 12710–12717. [Google Scholar]
- Luk, P.P.; Galettis, P.; Links, M. ERK phosphorylation predicts synergism between gemcitabine and the epidermal growth factor receptor inhibitor AG1478. Lung Cancer 2011, 73, 274–282. [Google Scholar] [CrossRef]
- Smalley, K.S. A pivotal role for ERK in the oncogenic behaviour of malignant melanoma? Int. J. Cancer. 2003, 104, 527–532. [Google Scholar] [CrossRef]
- Sun, H.F.; Li, X.M.; Meng, L.; Cui, C.M.; Gao, S.S.; Li, C.S.; Huang, C.G.; Wang, B.G. Asperolides A–C, tetranorlabdane diterpenoids from the marine alga-derived endophytic fungus Aspergillus wentii EN-48. J. Nat. Prod. 2012, 75, 148–152. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]
- Han, Y.H.; Kim, S.Z.; Kim, S.H.; Park, W.H. Arsenic trioxide inhibits the growth of Calu-6 cells via inducing a G2 arrest of the cell cycle and apoptosis accompanied with the depletion of GSH. Cancer Lett. 2008, 270, 40–55. [Google Scholar] [CrossRef]
- Porter, L.A.; Donoghue, D.J. Cyclin B1 and CDK1: Nuclear localization and upstream regulators. Prog. Cell Cycle Res. 2003, 5, 335–347. [Google Scholar]
- Roshak, A.K.; Capper, E.A.; Imburgia, C.; Fornwald, J.; Scott, G.; Marshall, L.A. The human polo-like kinase, PLK, regulates cdc2/cyclin B through phosphorylation and activation of the cdc25C phosphatas. Cell Signal. 2000, 12, 405–411. [Google Scholar]
- Meek, D.W. The role of p53 in the response to mitotic spindle damage. Pathol. Biol. 2000, 48, 246–254. [Google Scholar]
- Yin, X.Y.; Grove, L.; Datta, N.S.; Long, M.W.; Prochownik, E.V. c-myc overexpression and p53 loss cooperate to promote genomic instability. Oncogene 1999, 18, 1177–1184. [Google Scholar] [CrossRef]
- Bates, S.; Vousden, K.H. Mechanisms of p53-mediated apoptosis. Cell. Mol. Life Sci. 1999, 55, 28–37. [Google Scholar] [CrossRef]
- Vogt Sionov, R.; Haupt, Y. The cellular response to p53: The decision between life and death. Oncogene 1999, 18, 6145–6157. [Google Scholar] [CrossRef]
- Vousden, K.H. p53: Death star. Cell 2000, 103, 691–694. [Google Scholar] [CrossRef]
- Chen, X.; Ko, L.J.; Jayaraman, L.; Prives, C. p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response o tumor cells. Genes Dev. 1996, 10, 2438–2451. [Google Scholar] [CrossRef]
- Duliæ, V.; Stein, G.H.; Far, D.F.; Reed, S.I. Nuclear accumulation of p21Cip1 at the onset of mitosis: A role at the G2/M-phase transition. Mol. Cell. Biol. 1998, 18, 546–557. [Google Scholar]
- Niculescu, A.B., III; Chen, X.; Smeets, M.; Hengst, L.; Prives, C.; Reed, S.I. Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol. Cell. Biol. 1998, 18, 629–643. [Google Scholar]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. p53 in health and disease. Nat. Rev. Mol. Cell. Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Darbinyan, A.; Darbinian, N.; Safak, M.; Radhakrishnan, S.; Giordano, A.; Khalili, K. Evidence for dysregulation of cell cycle by human polyomavirus, JCV, late auxiliary protein. Oncogene 2002, 21, 5574–5581. [Google Scholar] [CrossRef]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef]
- Drukteinis, J.S.; Medrano, T.; Ablordeppey, E.A.; Kitzman, J.M.; Shiverick, K.T. Benzo[a]pyrene, but not 2,3,7,8-TCDD, induces G2/M cell cycle arrest, p21CIP1 and p53 phosphorylation in human choriocarcinoma JEG-3 cells: A distinct signaling pathway. Placenta 2005, 26, S87–S95. [Google Scholar] [CrossRef]
- Moodie, S.A.; Willumsen, B.M.; Weber, M.J.; Wolfman, A. Complexes of Ras-GTP with Raf-1 and mitogen-activated protein kinase kinase. Science 1993, 260, 1658–1661. [Google Scholar]
- Khosravi-Far, R.; Solski, P.A.; Clark, G.J.; Kinch, M.S.; Der, C.J. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol. Cell. Biol. 1995, 15, 6443–6453. [Google Scholar]
- Waskiewicz, A.J.; Cooper, J.A. Mitogen and stress response pathways: MAP kinase cascades and phosphatase regulation in mammals and yeast. Curr. Opin. Cell Biol. 1995, 7, 798–805. [Google Scholar] [CrossRef]
- Campbell, S.L.; Khosravi-Far, R.; Rossman, K.L.; Clark, G.J.; Der, C.J. Increasing complexity of Ras signaling. Oncogene 1998, 17, 1395–1413. [Google Scholar]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar]
- Rush, V.; Klimstra, D.; Venkatraman, E.; Oliver, J.; Martini, N.; Gralla, R.; Kris, M.; Dmitrovsky, E. Aberrant p53 expression predicts clinical resistance to cisplatinum-based chemotherapy in locally advanced non-small cell lung cancer. Cancer Res. 1995, 55, 5038–5042. [Google Scholar]
- Fujiwara, T.; Grimm, E.A.; Mukhopadhyay, T.; Zhang, W.-W.; Owen-Schaub, L.B.; Roth, J.A. Induction of chemosensitivity in human lung cancer cells in vivo by adenovirus-mediated transfer of the wild-type p53 gene. Cancer Res. 1994, 54, 2287–2291. [Google Scholar]
- Fan, S.; El-Deiry, W.S.; Bae, I.; Freeman, J.; Jondle, D.; Bhatia, K.; Fornace, A.J., Jr.; Magrath, I.; Kohn, K.W.; O’Connor, P.M. The p53 gene mutations are associated with decreased sensitivity of human lymphoma cells to DNA-damaging agents. Cancer Res. 1994, 54, 5824–5830. [Google Scholar]
- Giaccone, G. Clinical perspectives on platinum resistance. Drugs 2000, 59, 37–38. [Google Scholar]
- Stordal, B.; Davey, M. Understanding cisplatin resistance using cellular models. IUBMB Life 2007, 59, 696–699. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Wang, W.; Liu, T.; Wu, Y.; Guo, H.; Wang, P. Doxorubicin-loaded glycyrrhetinic acid-modified alginate nanoparticles for liver tumor chemo-therapy. Biomaterials 2012, 33, 2187–2196. [Google Scholar]
- Samples Availability: Available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lv, C.; Sun, W.; Sun, H.; Wei, S.; Chen, R.; Wang, B.; Huang, C. Asperolide A, a Marine-Derived Tetranorditerpenoid, Induces G2/M Arrest in Human NCI-H460 Lung Carcinoma Cells, Is Mediated by p53-p21 Stabilization and Modulated by Ras/Raf/MEK/ERK Signaling Pathway. Mar. Drugs 2013, 11, 316-331. https://doi.org/10.3390/md11020316
Lv C, Sun W, Sun H, Wei S, Chen R, Wang B, Huang C. Asperolide A, a Marine-Derived Tetranorditerpenoid, Induces G2/M Arrest in Human NCI-H460 Lung Carcinoma Cells, Is Mediated by p53-p21 Stabilization and Modulated by Ras/Raf/MEK/ERK Signaling Pathway. Marine Drugs. 2013; 11(2):316-331. https://doi.org/10.3390/md11020316
Chicago/Turabian StyleLv, Cuiting, Wenxia Sun, Haofen Sun, Shanjian Wei, Ruohua Chen, Bingui Wang, and Caiguo Huang. 2013. "Asperolide A, a Marine-Derived Tetranorditerpenoid, Induces G2/M Arrest in Human NCI-H460 Lung Carcinoma Cells, Is Mediated by p53-p21 Stabilization and Modulated by Ras/Raf/MEK/ERK Signaling Pathway" Marine Drugs 11, no. 2: 316-331. https://doi.org/10.3390/md11020316