Antibacterial Polyketides from the Marine Alga-Derived Endophitic Streptomyces sundarbansensis: A Study on Hydroxypyrone Tautomerism

Abstract
:
1. Introduction

2. Results and Discussion
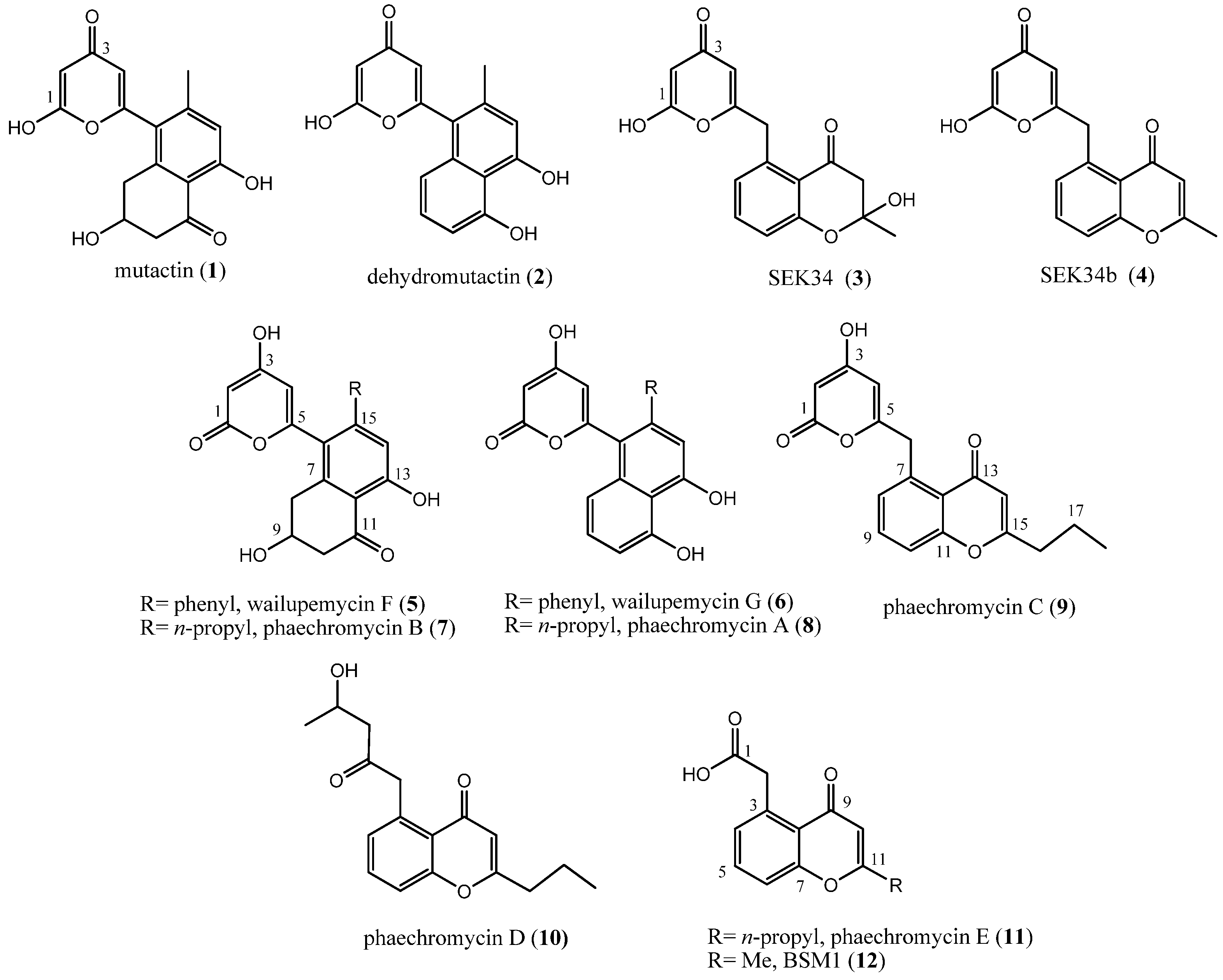
2.1. Production and Structural Characterization of Metabolites


{kind=link}
{kind=link}
{kind=link}
| Position | δH (CD3OD) | δH (CDCl3) | δC, (CDCl3) | HMBC a |
|---|---|---|---|---|
| 1 | – | – | 165.4 | |
| 2 | – | 5.33, s | 89.9 | 3, 4, 5 |
| 3 | 171.3 | |||
| 4 | 5.65, s | 5.75, s | 101.2 | |
| 5 | 166.9 | |||
| 6 | 4.23, dd(16.2) | 4.20, d(16.4) | 38.6 | 1, 4, 8, 12 |
| 4.32, dd(16.2) | 4.28, d(16.4) | |||
| 7 | 133.5 | |||
| 8 | 6.99, d (8.6) | 6.99, s | 125.2 | |
| 9 | 7.48, t (8.1) | 7.39, t (8.2) | 135.4 | 7, 11 |
| 10 | 6.93, d (7.2) | 6.90, d(8.2) | 118.4 | 8, 11, 12 |
| 11 | 158.1 | |||
| 12 | 118.5 | |||
| 13 | 194.0 | |||
| 14 | – | 2.79, d (16.0) | 47.1 | 12,13,15, 16 |
| – | 2.89, d (16.0) | |||
| 15 | 102.3 | |||
| 16 | 1.89, m | 1.88, m | 43.1 | 14, 15, 17, 18 |
| 17 | 1.53, m | 1.57, m | 17.1 | 15, 16, 18 |
| 17 | 1.00, t (7.0) | 0.99 (t, 7.0) | 14.04 | 17, 16 |
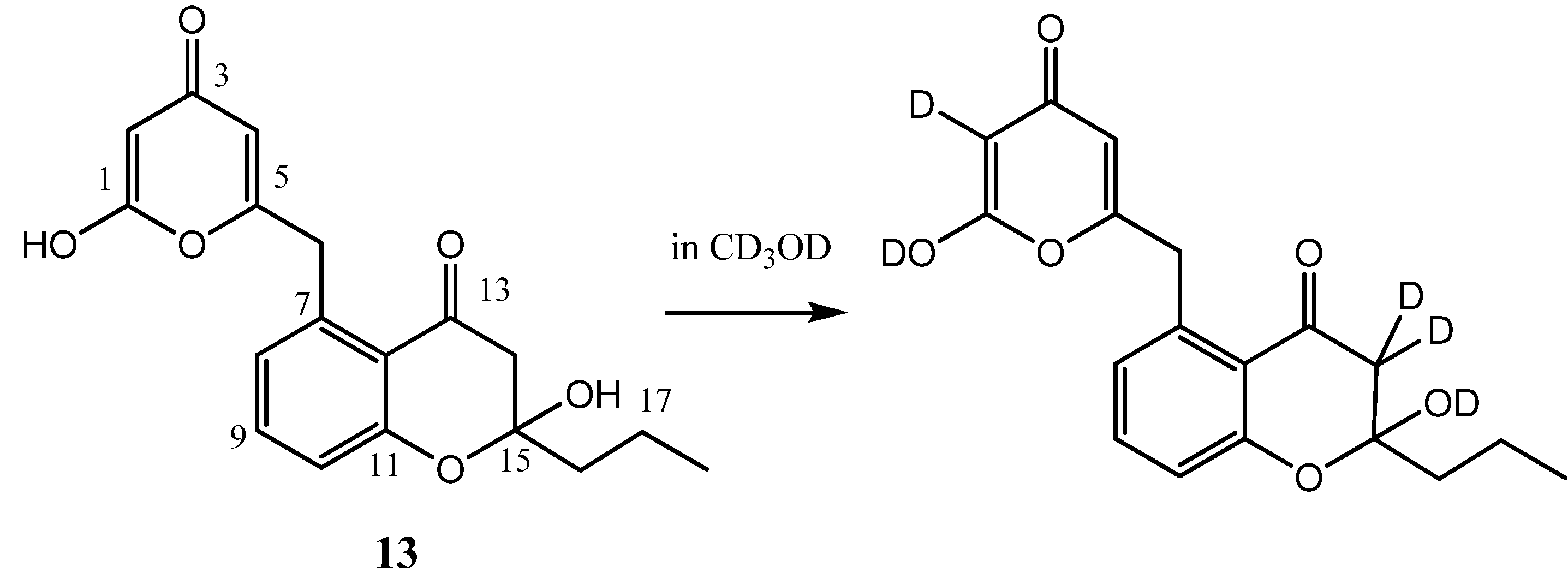
2.2. Hydroxypyrone Tautomerism
 A |  B | ||||
|---|---|---|---|---|---|
| νIRcalcd | |||||
| Compound | νIRexp. | C(1)=O | C(13)=O a | C(3)=O | C(13)=O a |
| 7 | 1637 | 1780 | 1639 | 1686 | 1642 |
| 1620 | |||||
| 1578 | |||||
| 1524 | |||||
| 9 | 1699 | 1780 | 1649 | 1683 | 1662 |
| 1645 | |||||
| 1607 | |||||
| 1598 | |||||
| 13 | 1689 | 1782 | 1678 | 1682 | 1698 |
| 1649 | |||||
| 1603 | |||||
| 1593 | |||||
| R=CH3b | 1738 | 1782 | – | 1692 | – |
| 1645 | |||||
| 1534 | |||||
2.3. Antibacterial Evaluation
| MIC (μg/mL) | |||
|---|---|---|---|
| Compounds | E. coli ATCC 25922 | MRSA ATCC 43300 | P. aeruginosa ATCC 27853 |
| 9 | – | >32 | >32 |
| 11 | >32 | >32 | >32 |
| 13 | 16 | 2 | >32 |
| Vancomycin | – | <2 | – |
| Gentamicin | <2 | <2 | – |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Isolation and Identification of Streptomyces Strain WR1L1S8
3.3. Isolation and Structural Characterization of Compounds 7, 9, 11 and 13
3.4. Computational Details
3.5. Antibacterial Assay
4. Conclusions
Acknowledgments
Supplementary Files
References
- Jensen, P.R.; Mincer, T.J.; Williams, P.G.; Fenical, W. Marine actinomycete diversity and natural product discovery. Antonie van Leeuwenhoek 2005, 87, 43–48. [Google Scholar] [CrossRef]
- Lam, K.S. Discovery of novel metabolites from marine actinomycetes. Curr. Opin. Microbiol. 2006, 9, 245–251. [Google Scholar]
- Berdy, J. Bioactive microbial metabolites. J. Antibiot. 2005, 58, 1–26. [Google Scholar] [CrossRef]
- Pimentel-Elardo, S.M.; Kozytska, S.; Bugni, T.S.; Ireland, C.M.; Moll, H.; Hentschel, U. Anti-Parasitic compounds from Streptomyces sp. strains isolated from Mediterranean sponges. Mar. Drugs 2010, 8, 373–380. [Google Scholar] [CrossRef]
- Bredholt, H.; Fjærvik, E.; Johnsen, G.; Zotchev, S.B. Actinomycetes from sediments in the Trondheim Fjord, Norway: Diversity and biological activity. Mar. Drugs 2008, 6, 12–24. [Google Scholar] [CrossRef]
- Stach, J.E.M.; Maldonado, L.A.; Ward, A.C.; Goodfellow, M.; Bull, A.T. New primers for the class Actinobacteria: Application to marine and terrestrial environments. Environ. Microbiol. 2003, 5, 828–841. [Google Scholar] [CrossRef]
- De Carvalho, C.C.C.R.; Fernandes, P. Production of metabolites as bacterial responses to the marine environment. Mar. Drugs 2010, 8, 705–727. [Google Scholar] [CrossRef]
- Jensen, P.R.; Fenical, W. Marine bacterial diversity as a resource for novel microbial products. J. Ind. Microb. Biotechnol. 1996, 17, 346–351. [Google Scholar] [CrossRef]
- Park, H.B.; Yang, H.O.; Lee, K.R.; Kwon, H.C. Gombapyrones E and F, new α-pyronepolyenes produced by Streptomyces sp. KMC-002. Molecules 2011, 16, 3519–3529. [Google Scholar]
- Zhang, H.I.; He, X.G.; Adefarati, A.; Galluci, J.; Cole, S.P.; Beale, J.M.; Keller, P.J.; Chang, C.J.; Floss, H.G. Mutactin, a novel polyketide from Streptomyces coelicoor. Structure and biosynthetic relationship to Acinorhodin. J. Org. Chem. 1990, 55, 1682–1684. [Google Scholar] [CrossRef]
- McDaniel, R.; Ebert-Khosla, S.; Hopwood, D.A.; Khosla, C. Engineered biosynthesis of novel polyketides: ActVII and actIV genes encode aromatase and cyclase enzymes, respectively. J. Am. Chem. Soc. 1994, 116, 10855–10859. [Google Scholar]
- McDaniel, R.; Ebert-Khosla, S.; Fu, H.; Hopwood, D.A.; Khosla, C. Engineered biosynthesis of novel polyketides: Influence of a downstream enzyme on the catalytic specificity of a minimal aromatic polyketidesynthase. Proc. Natl. Acad. Sci. USA 1994, 91, 11542–11546. [Google Scholar] [CrossRef]
- Xiang, L.; Kalaitziz, J.A.; Nilsen, G.; Chen, L.; Moore, B.S. Mutational analysis of the enterocinfavorskii biosynthetic rearrangement. Org. Lett. 2002, 4, 957–960. [Google Scholar] [CrossRef]
- Graziani, E.I.; Ritacco, F.V.; Bernan, V.S.; Telliez, J.B. Phaeochromycins A–E, anti-inflammatory polyketides isolated from the soil actinomycete Streptomyces phaeochromogenes LL-P018. J. Nat. Prod. 2005, 68, 1262–1265. [Google Scholar]
- Kalaitziz, J.A.; Moore, B.S. Heterologous biosynthesis of truncated hexaketides derived from the actinorhodin polyketide synthase. J. Nat. Prod. 2004, 67, 1419–1422. [Google Scholar] [CrossRef]
- Arumugam, M.; Mitra, A.; Pramanik, A.; Saha, M.; Gachhui, R.; Mukherjee, J. Streptomyces sundarbansensis sp. nov., an actinomycete that produces 2-allyloxyphenol. Int. J. Syst. Evol. Microbiol. 2011, 61, 2664–2669. [Google Scholar]
- Lord, R.C.; Phillips, W.D. Exchange reactions of γ-pyrone and synthesis of deuterated pyrones. J. Am. Chem. Soc. 1952, 74, 2429–2430. [Google Scholar] [CrossRef]
- Fitzgerald, J.T.; Ridley, C.P.; Khosla, C. Engineered biosynthesis of the antiparasitic agent frenolicin band rationally designed analogs in a heterologous host. J. Antibiot. 2011, 64, 759–762. [Google Scholar] [CrossRef]
- Goel, A.; Ram, V.J. Natural and synthetic 2H-pyran-2-ones and their versatility in organic synthesis. Tetrahedron 2009, 65, 7865–7913. [Google Scholar] [CrossRef]
- Seixas de Melo, J.; Quinteiro, G.; Pina, J.; Breda, S.; Fausto, R. Spectroscopic characterization of α- and γ-pyrones and their substituted 4-hydrxy and 4-methoxy derivative: An integrated infrared, photophysical and theoretical study. J. Mol. Struct. 2001, 565–566, 59–67. [Google Scholar]
- Tenover, F.C. Mechanisms of antimicrobial resistance in bacteria. Am. J. Med. 2006, 119, 3–10. [Google Scholar] [CrossRef]
- Gibbons, S. Anti-Staphylococcal plant natural products. Nat. Prod. Rep. 2004, 21, 263–277. [Google Scholar] [CrossRef]
- Gilbert, K.E.; Midland, M.M. PCMODEL for Windows, version 7.00; Serena Software: Bloomington, IN, USA, 1999. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T., Jr.; Kudin, K.N.; Burant, J.C.; et al. Gaussian Revision E. 01; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Merrick, J.P.; Moran, D.; Radom, L. An evaluation of harmonic vibrational frequency scale factors. J. Phys. Chem. A 2007, 111, 11683–11700. [Google Scholar]
- Samples Availability: Available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Djinni, I.; Defant, A.; Kecha, M.; Mancini, I. Antibacterial Polyketides from the Marine Alga-Derived Endophitic Streptomyces sundarbansensis: A Study on Hydroxypyrone Tautomerism. Mar. Drugs 2013, 11, 124-135. https://doi.org/10.3390/md11010124
Djinni I, Defant A, Kecha M, Mancini I. Antibacterial Polyketides from the Marine Alga-Derived Endophitic Streptomyces sundarbansensis: A Study on Hydroxypyrone Tautomerism. Marine Drugs. 2013; 11(1):124-135. https://doi.org/10.3390/md11010124
Chicago/Turabian StyleDjinni, Ibtissem, Andrea Defant, Mouloud Kecha, and Ines Mancini. 2013. "Antibacterial Polyketides from the Marine Alga-Derived Endophitic Streptomyces sundarbansensis: A Study on Hydroxypyrone Tautomerism" Marine Drugs 11, no. 1: 124-135. https://doi.org/10.3390/md11010124
APA StyleDjinni, I., Defant, A., Kecha, M., & Mancini, I. (2013). Antibacterial Polyketides from the Marine Alga-Derived Endophitic Streptomyces sundarbansensis: A Study on Hydroxypyrone Tautomerism. Marine Drugs, 11(1), 124-135. https://doi.org/10.3390/md11010124