Palyosulfonoceramides A and B: Unique Sulfonylated Ceramides from the Brazilian Zoanthids Palythoa caribaeorum and Protopalyhtoa variabilis

Abstract
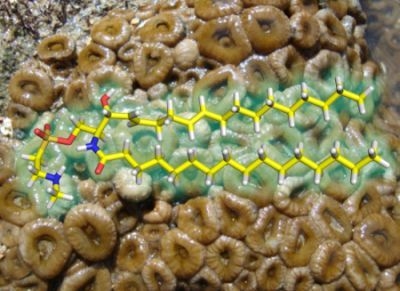
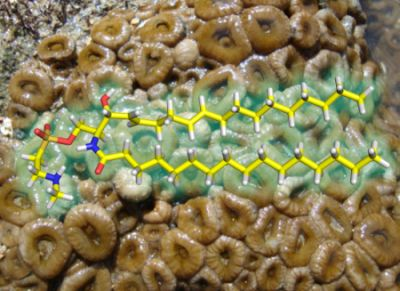
:
1. Introduction

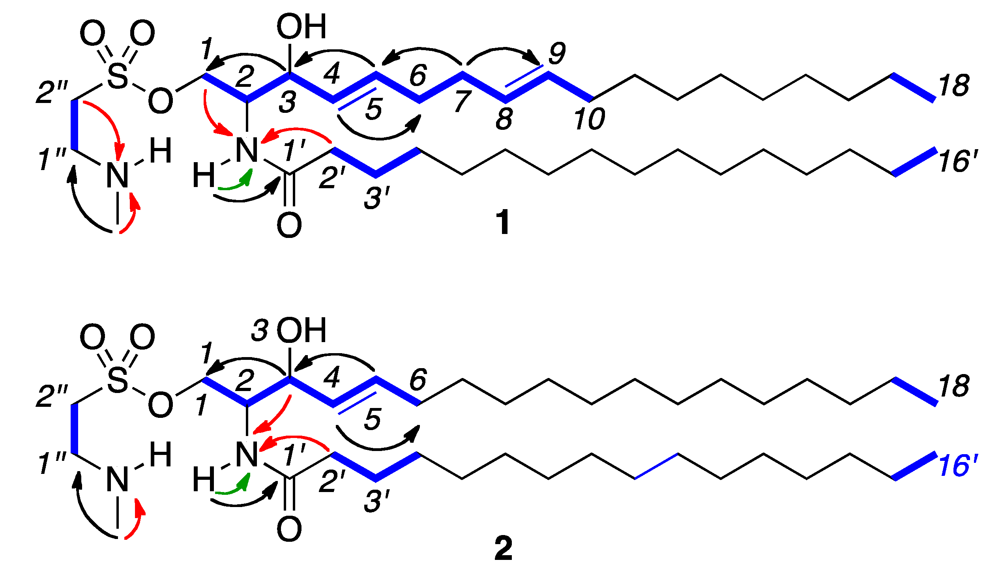
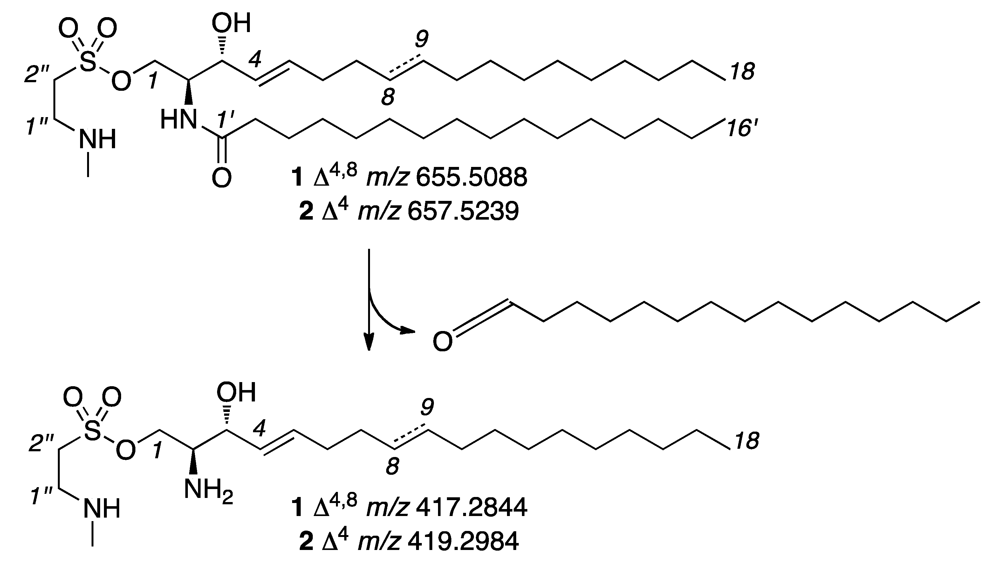
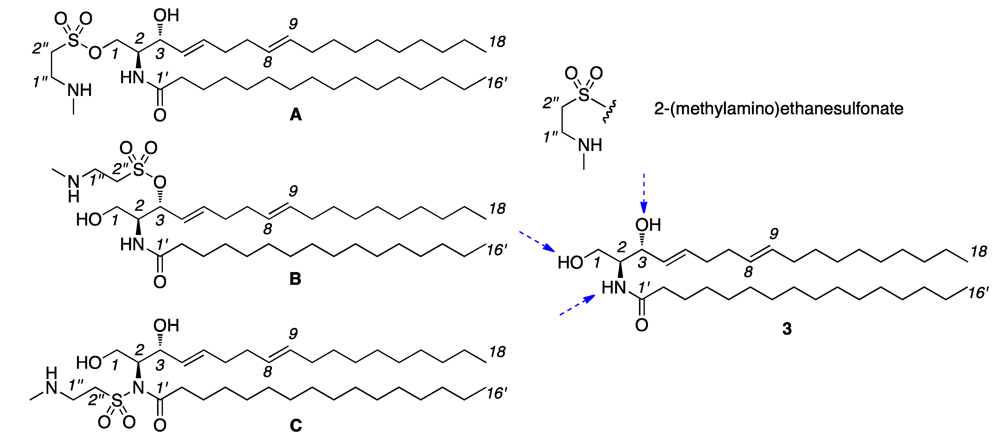
2. Results and Discussion
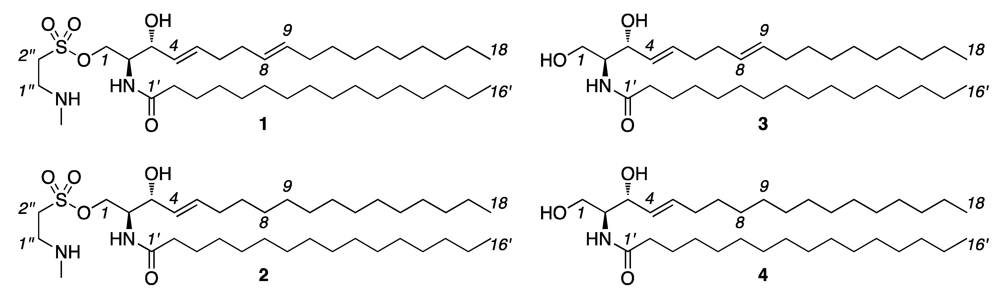

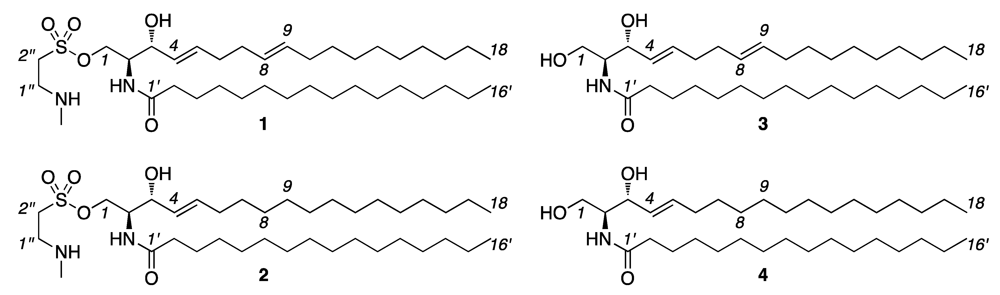
{kind=link}
{kind=link}
{kind=link}
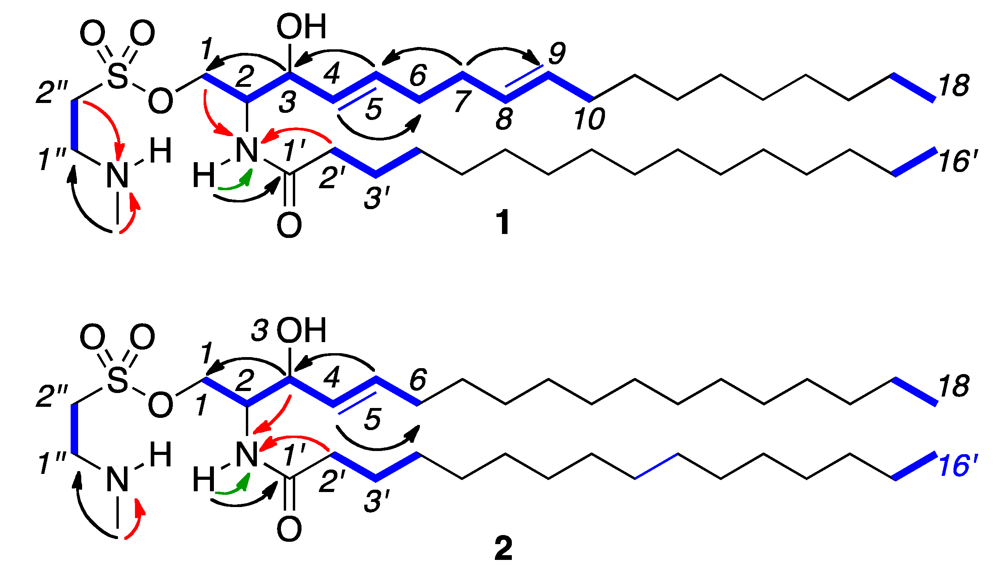{kind=link}
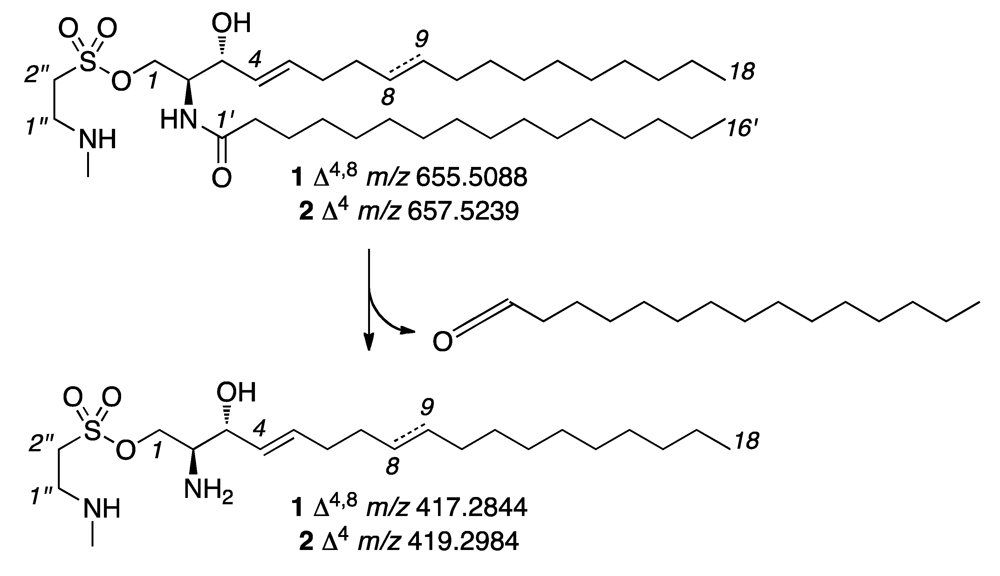{kind=link}
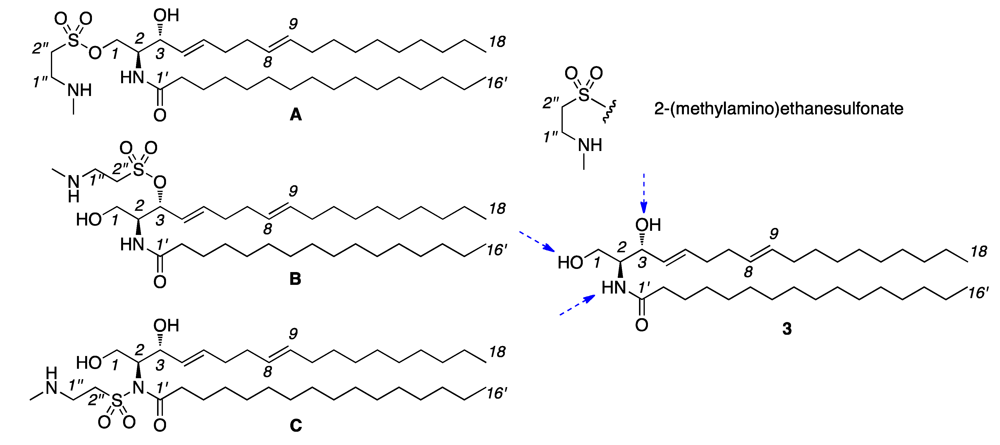{kind=link}
| Palyosulfonoceramide A (1) | Palyosulfonoceramide B (2) | |||
|---|---|---|---|---|
| 1HδH (mult, J in Hz) | 13CδC (type) | 1HδH (mult, J in Hz) | 13CδC (type) | |
| 1a | 4.09 (m) | 64.0 (CH2) | 3.97 (m) | 63.4 (CH2) |
| 1b | 3.91 (m) | 3.74 (m) | ||
| 2 | 3.91 (m) | 54.0 (CH) | 3.74 (m) | 53.9 (CH) |
| 3 | 4.02 (t, 7.3) | 71.4 (CH) | 3.88 (t, 7.2) | 71.1 (CH) |
| 4 | 5.43 (dd, 15.3, 7.1) | 129.4 (CH) | 5.26 (dd, 15.4, 7.1) | 128.9 (CH) |
| 5 | 5.68 (dt, 15.3, 7.1) | 133.8 (CH) | 5.54 (dt, 14.2, 6.7) | 134.3 (CH) |
| 6 | 2.01 (m) | 32.6 (CH2) | 1.83 (dd, 14.2, 7.0) | 32.3 (CH2) |
| 7 | 2.01 (m) | 32.4 (CH2) | 1.08 (m) | 31.8 (CH2) |
| 8 | 5.36 (dd, 5.7, 15.9) a | 131.1 (CH) | 1.08 (m) | 29.5–29.1 (CH2) |
| 9 | 5.34 (dd, 5.0, 15.9) a | 129.2 (CH) | 1.08 (m) | 29.5–29.1 (CH2) |
| 10 | 1.91 (q, 6.5) | 32.7 (CH2) | 1.08 (m) | 29.5–29.1 (CH2) |
| 11–16 | 1.21 (m) | 29.8–29.3 (CH2) | 1.08 (m) | 29.5–29.1 (CH2) |
| 17 | 1.26 (m) | 22.7 (CH2) | 1.08 (m) | 22.5 (CH2) |
| 18 | 0.84 (t, 6.9) | 14.1 (CH3) | 0.70 (t, 6.6) | 13.8 (CH3) |
| 1′ | 174.3 (C=O) | 174.4 (C=O) | ||
| 2′ | 2.13 (t, 7.7) | 36.6 (CH2) | 2.01 (t, 7.4) | 36.4 (CH2) |
| 3′ | 1.53 (m) | 26.0 (CH2) | 1.41 (m) | 25.8 (CH2) |
| 4′–14′ | 1.21 (m) | 29.8–29.3 (CH2) | 1.08 (m) | 29.5–29.1 (CH2) |
| 15′ | 1.26 (m) | 22.7 (CH2) | 1.08 (m) | 22.5 (CH2) |
| 16′ | 0.84 (t, 6.9) | 14.1 (CH3) | 0.70 (t, 6.6) | 13.8 (CH3) |
| 1″ | 3.10 (m) | 45.1 (CH2) | 2.96 (m) | 45.3 (CH2) |
| 2″ | 2.01 (m) | 32.0 (CH2) | 1.73 (m) | 32.3 (CH2) |
| NCH3 | 2.64 (s) | 33.1 (CH3) | 2.51 (s) | 32.7 (CH3) |
| NH | 7.23 (d, 8.4) | - | 7.32 (d, 8.4) | - |
| NH | 8.96 (br s) | - | 8.72 (br s) | - |
| Ceramide 3 | Ceramide 4 | |||
|---|---|---|---|---|
| 1HδH (mult, J in Hz) | 13CδC (type) | 1HδH (mult, J in Hz) | 13CδC (type) | |
| 1a | 4.47 (dd, 11.0, 5.3) | 62.4 (CH2) | 4.47 (dd, 11.0, 5.0) | 62.5 (CH2) |
| 1b | 4.30 (dd, 11.0, 5.3) | 4.30 (dd, 11.0, 5.0) | ||
| 2 | 4.76 (m) | 57.2 (CH) | 4.75 (m) | 57.2 (CH) |
| 3 | 4.86 (t, 6.2) | 73.6 (CH) | 4.86 (t, 6.0) | 73.6 (CH) |
| 4 | 6.07 (dd, 15.4, 6.2) | 132.9 (CH) | 6.06 (dd, 15.4, 6.3) | 132.7 (CH) |
| 5 | 6.01 (dt, 15.4, 6.2) | 131.9 (CH) | 5.99 (dt, 15.4, 6.3) | 132.6 (CH) |
| 6 | 2.18 (m) | 33.2 (CH2) | 2.10 (m) | 33.0 (CH2) |
| 7 | 2.14 (m) | 32.4 (CH2) | 1.27 (m) | 32.4 (CH2) |
| 8 | 5.50 (m) | 131.4 (CH) | 1.27 (m) | 29.9–30.3 (CH2) |
| 9 | 5.50 (m) | 130.2 (CH) | 1.27 (m) | 29.9–30.3 (CH2) |
| 10 | 2.02 (m) | 33.2 (CH2) | 1.27 (m) | 29.9–30.3 (CH2) |
| 11–16 | 1.27 (m) | 29.8–30.3 (CH2) | 1.27 (m) | 29.9–30.3 (CH2) |
| 17 | 1.27 (m) | 23.2 (CH2) | 1.27 (m) | 23.2 (CH2) |
| 18 | 0.87 (t, 5.0) | 14.6 (CH3) | 0.88 (t, 5.8) | 14.6 (CH3) |
| 1′ | - | 173.9 (C=O) | - | 173.9 (C=O) |
| 2′ | 2.48 (t, 7.7) | 37.2 (CH2) | 2.47 (t, 7.3) | 37.2(CH2) |
| 3′ | 1.84 (m) | 26.7 (CH2) | 1.84 (m) | 26.7(CH2) |
| 4′–14′ | 1.27 (m) | 29.8–30.3 (CH2) | 1.27 (m) | 29.9–30.3(CH2) |
| 15′ | 1.27 (m) | 23.2 (CH2) | 1.27 (m) | 23.2(CH2) |
| 16′ | 0.87 (t, 5.0) | 14.6 (CH3) | 0.88 (t, 5.8) | 14.6 (CH3) |
| N–H | 8.40 (d, 8.5) | - | 8.37 (d, 8.4) | - |
| Ceramide 3 | N-((2S,3R,4E,8E)-1,3-dihydroxyoctadeca-4,8-dien-2-yl)-hexadecanamide a | |||
|---|---|---|---|---|
| 1H δH (mult, J in Hz) | 13C δC (type) | 1H δH (mult, J in Hz) | 13C δC (type) | |
| 1a | 3.67 (m) b | 62.5 (CH2) | 3.70 (dd, 3.4, 11.0) | 62.5 (CH2) |
| 1b | 3.92 (m) | 3.95 (dd, 3.4, 11.0) | ||
| 2 | 3.89 (m) b | 54.8 (CH) | 3.91 (m) | 54.4 (CH) |
| 3 | 4.29 (dd, 3.6, 6.6) | 74.5 (CH) | 4.32 (br t, 4.4) | 74.7 (CH) |
| 4 | 5.51 (dt, 15.5, 5.9) | 129.3 (CH) | 5.55 (dt, 15.4, 6.4) | 129.2 (CH) |
| 5 | 5.77 (dt, 16.0, 6.2) | 133.6 (CH) | 5.80 (dt, 15.4, 6.4) | 133.5 (CH) |
| 6 | 2.10 (dt, 6.6, 6.3) | 32.1 c (CH2) | 2.12 (m) | 32.1 c (CH2) |
| 7 | 2.05 (dt, 6.6, 6.3) | 32.3 c (CH2) | 2.08 (m) | 32.3 c (CH2) |
| 8 | 5.38 (dd, 17.2, 5.6) | 129.1 (CH) | 5.36 (dt, 15.2, 6.4) | 127.0 f (128.9) (CH) |
| 9 | 5.40 (dd, 17.2, 5.0) | 131.5 (CH) | 5.43 (dt, 15.2, 6.4) | 131.4 (CH) |
| 10 | 1.96 (td, 6.4, 6.8) | 32.74 c (CH2) | 1.97 (br dd, 2.2, 13.2) e | 32.6 c (CH2) |
| 11–16 | 1.25 (m) | 29.3–29.8, 32.1 d | 1.28 (m) | 29.2–29.7, 31.9 d |
| 17 | 1.25 (m) | 22.8 (CH2) | 1.28 (m) | 22.7 (CH2) |
| 18 | 0.87 (t, 6.5) | 14.2 (CH3) | 0.88 (t, 6.8) | 14.1 (CH3) |
| 1′ | - | 174.3 (C=O) | - | 174.0 (C=O) |
| 2′ | 2.21 (t, 7.6) | 37.0 (CH2) | 2.23 (t, 7.4) | 36.8 (CH2) |
| 3′ | 1.62 (p, 7.2) | 25.9 (CH2) | 1.64 (br t, 7.4) | 25.8 (CH2) |
| 4′–14′ | 1.25 (m) | 29.3–29.8, 32.1 d | 1.28 (m) | 29.2–29.7, 31.9 d |
| 15′ | 1.25 (m) | 22.8 (CH2) | 1.28 (m) | 22.7 (CH2) |
| 16′ | 0.87 (t, 6.5) | 14.2 (CH3) | 0.88 (t, 6.8) | 14.1 (CH3) |
| OH | 2.99 (bs) | - | 2.77 (bs) | - |
| N–H | 6.33 (d, 7.4) | - | 6.26 (d, 7.3) | - |
| Ceramide 4 | N-((2S,3R,4E)-1,3-dihydroxyoctadeca-4,8-dien-2-yl)-hexadecanamide a | |||
|---|---|---|---|---|
| 1H δH (mult, J in Hz) | 13C δC (type) | 1H δH (mult, J in Hz) | 13C δC (type) | |
| 1a | 3.67 (dd, 3.4, 11.3) | 62.7 (CH2) | 3.70 (dd, 3.2, 11.2) | 62.5 (CH2) |
| 1b | 3.96 (dd, 3.3, 11.3) | 3.95 (dd, 3.9, 11.2) | ||
| 2 | 3.92 (qd, 3.7, 7.5) | 54.6 (CH) | 3.90 (m) | 54.5 (CH) |
| 3 | 4.33 (dd, 4.4, 6.0) | 74.9 (CH) | 4.31 (dd, 4.4, 6.8) | 74.7 (CH) |
| 4 | 5.53 (tdd, 1.4, 6.4, 15.5) | 128.9 (CH) | 5.53 (td, 6.8, 15.5) | 128.9 (CH) |
| 5 | 5.79 (dtd, 1.2, 6.8, 15.2) | 134.5 (CH) | 5.78 (td, 6.6, 15.2) | 134.3 (CH) |
| 6 | 2.06 (m) | 32.1 (CH2) | 2.05 (m) | 32.3 (CH2) |
| 7–17 | 1.25 (m) | 22.9–32.1 (CH2) | 1.25 (m) | 22.7–31.9 (CH2) |
| 18 | 0.88 (t, 6.9) | 14.3 (CH3) | 0.87 (t, 6.6) | 14.1 (CH3) |
| 1′ | - | 174.0 (C=O) | - | 173.9 (C=O) |
| 2′ | 2.23 (dd, 7.1, 8.2) | 37.0 (CH2) | 2.22 (t, 7.8) | 36.8 (CH2) |
| 3′ | 1.62 (m) | 25.9 b (CH2) | 1.63 (m) | 25.7 b (CH2) |
| 4′–15′ | 1.25 (m) | 22.7–32.1 (CH2) | 1.25 (m) | 22.7–31.9 (CH2) |
| 16′ | 0.88 (t, 6.9) | 14.3 (CH3) | 0.87 (t, 6.6) | 14.1 (CH3) |
| OH | 2.59 (bs) | - | 2.68 (bs) | - |
| N–H | 6.22 (d, 7.6) | - | 6.24 (d, 7.3) | - |
3. Experimental Section
3.1. General Experimental Procedures
3.2. Marine Organisms
3.3. Extraction and Isolation
3.4. Methanolysis and GC-MS Analysis
3.5. Cytotoxicity Assays
4. Conclusions
Acknowledgments
Supplementary Files
References
- Daly, M.; Brugler, M.R.; Cartwright, P.; Collins, A.G.; Dawson, M.N.; Fautin, D.G.; France, S.C.; McFadden, C.S.; Opresko, D.M.; Rodriguez, E.; et al. The phylum cnidarian: A review of phylogenetic patterns and diversity 300 years after Linnaeus. Zootaxa 2007, 1668, 127–182. [Google Scholar]
- Rocha, J.; Peixe, L.; Gomes, N.C.M.; Calado, R. Cnidarians as a source of new marine bioactive compounds—An overview of the last decade and future steps for bioprospecting. Mar. Drugs 2011, 9, 1860–1886. [Google Scholar] [CrossRef]
- Hu, G.P.; Yuan, J.; Sun, L.; She, Z.G.; Wu, J.H.; Lan, X.J.; Zhu, X.; Lin, Y.C.; Chen, S.P. Statistical research on marine natural products based on data obtained between 1985 and 2008. Mar. Drugs 2011, 9, 514–525. [Google Scholar] [CrossRef]
- Moore, R.E.; Scheuer, P.J. Palytoxin, a new marine toxin from a coelenterate. Science 1971, 172, 495–498. [Google Scholar]
- Uemura, D.; Hirata, Y.; Iwashita, T.; Naoki, H. Studies on palytoxins. Tetrahedron 1985, 41, 1007–1017. [Google Scholar] [CrossRef]
- Beress, L.; Zwick, J.; Kolkenbrock, H.J.; Kaul, P.N.; Wassermann, O. A method for the isolation of the Caribbean palytoxin (C-PTX) from the coelenterate (zoanthid) Palythoa caribaeorum. Toxicon 1983, 21, 285–290. [Google Scholar]
- Oku, N.; Sata, N.U.; Matsunaga, S.; Uchida, H.; Fusetani, N. Identification of palytoxin as a principle which causes morphological changes in rat 3Y1 cells in the zoanthid Palythoa aff. margaritae. Toxicon 2004, 43, 21–25. [Google Scholar] [CrossRef]
- Costa-Lotufo, L.V.; Pessoa, C.; Moraes, M.E.A.; Almeida, A.M.P.; Moraes, M.O.; Lotufo, T.M.C. Marine Organisms from Brazil as a Source of Potential Anticancer Agents. In Lead Molecules from Natural Products, 1st; Khan, M.T.H., Ather, A., Eds.; Elsevier: Amsterdam, The Netherland, 2006; pp. 181–196. [Google Scholar]
- Jimenez, P.C.; Fortier, S.C.; Lotufo, T.M.C.; Pessoa, C.; Moraes, M.E.A.; Moraes, M.O.; Costa-Lotufo, L.V. Biological activity in extracts of ascidians (Tunicata, Ascidiacea) from the northeastern Brazilian coast. J. Exp. Mar. Biol. Ecol. 2003, 1, 93–101. [Google Scholar]
- Jimenez, P.C.; Wilke, D.V.; Ferreira, E.G.; Takeara, R.; Moraes, M.O.; Silveira, E.R.; Lotufo, T.M.C.; Lopes, N.P.; Costa-Lotufo, L.V. Structure elucidation and anticancer activity of 7-oxostaurosporine derivatives from the Brazilian endemic tunicate Eudistoma vannamei. Mar. Drugs 2012, 10, 1092–1102. [Google Scholar] [CrossRef] [Green Version]
- Sousa, T.S.; Jimenez, P.C.; Ferreira, E.G.; Silveira, E.R.; Filho, R.B.; Pessoa, O.D.L.; Costa-Lotufo, L.V. Anthracyclinones from Micromonospora sp. J. Nat. Prod. 2012, 75, 489–493. [Google Scholar] [CrossRef]
- Ferreira, E.G.; Wilke, D.V.; Jimenez, P.C.; Oliveira, J.R.; Pessoa, O.D.L.; Silveira, E.R.; Viana, F.A.; Pessoa, C.; Moraes, M.O.; Hajdu, E.; et al. Guanidine alkaloids from Monanchora arbuscula: Chemistry and antitumor potential. Chem. Biodivers. 2011, 8, 1433–1445. [Google Scholar] [CrossRef]
- Arthaud, I.D.B.; Rodrigues, F.A.R.; Jimenez, P.C.; Montenegro, R.C.; Angelim, A.L.; Melo, V.M.M.; Silveira, E.R.; Freitas, H.P.S.; Sousa, T.S.; Pessoa, O.D.L.; et al. Studies on the secondary metabolites of a Pseudoalteromonas sp. isolated from sediments collected at the northeastern coast of Brazil. Chem. Biodivers. 2012, 9, 418–427. [Google Scholar] [CrossRef]
- Wilke, D.V.; Jimenez, P.C.; Pessoa, C.; Moraes, M.O.; Araújo, R.M.; Silva, W.M.B.; Silveira, E.R.; Pessoa, O.D.L.; Braz-Filho, R.; Lopes, N.P.; et al. Cytotoxic lipidic α-amino acids from the zoanthid Protopalythoa variabilis from the northeastern coast of Brazil. J. Braz. Chem. Soc. 2009, 20, 1455–1459. [Google Scholar] [CrossRef]
- Wilke, D.V.; Jimenez, P.C.; Araújo, R.M.; Da Silva, W.M.B.; Pessoa, O.D.L.; Silveira, E.R.; Pessoa, C.; Moraes, M.O.; Skwarczynski, M.; Simerska, P.; et al. Pro-Apoptotic activity of lipidic α-amino acids isolated from Protopalythoa variabilis. Bioorg. Med. Chem. 2012, 18, 7997–8004. [Google Scholar]
- Rohlfs De Macedo, C.M.R.; Belém, M.J.C. The genus Zoanthus in Brazil. 1. Characterization and anatomical revision of Zoanthus sociatus (Cnidaria, Zoanthinaria, Zoanthidae). Iheringia 1994, 77, 135–144. [Google Scholar]
- Boscolo, H.K.; Silveira, F.L. Reproductive biology of Palythoa caribaeorum and Protopalythoa variabilis (Cnidaria, Anthozoa, Zoanthidea) from the southeastern coast of Brazil. Braz. J. Biol. 2005, 65, 29–41. [Google Scholar]
- Diop, M.; Leug-Tack, D.; Braekman, J.C.; Kornprobst, J.M. Sterol composition of four Zoathidae members of the genus Palythoa from the Cape Verde Peninsula. Biochem. Syst. Ecol. 1986, 14, 151–154. [Google Scholar] [CrossRef]
- Shigemori, H.; Sato, Y.; Kagata, T.; Kobayashi, J. Palythoalones A and B, new Ecdysteroids from the marine Zoanthid Palythoa australiae. J. Nat. Prod. 1999, 62, 372–374. [Google Scholar] [CrossRef]
- Uemura, D.; Toya, Y.; Watanabe, I.; Hirata, Y. Isolation and structures of two pyrazines, palythazine and isoapalythazine from Palythoa tuberculosa. Chem. Lett. 1979, 1481–1482. [Google Scholar]
- Han, C.; Qi, J.; Shi, X.; Sagakami, Y.; Shibata, T.; Uchida, K.; Ojika, M. Prostaglandins from a Zoanthid: Paclitaxel-like neurite-degenerating and microtubule-stabilizating activities. Biosci. Biothecnol. Biochem. 2006, 70, 706–711. [Google Scholar] [CrossRef]
- Pettit, G.R.; Fujji, Y. The glycerol ethers of Palythoa liscia. J. Nat. Prod. 1982, 45, 640–646. [Google Scholar] [CrossRef]
- Bedke, D.K.; Vanderwal, C.D. Chlorosulfolipids: Structure, synthesis, and biological relevance. Nat. Prod. Rep. 2011, 28, 15–25. [Google Scholar] [CrossRef]
- Vos, J.P.; Lopes-Cardozo, M.; Gadella, B.N. Metabolic and functional aspects of sulfogalactolipids. Biochim. Biophys. Acta 1994, 1211, 125–149. [Google Scholar] [CrossRef]
- Ishizuka, I. Chemistry and functional distribution of sulfoglycolipids. Prog. Lipid Res. 1997, 36, 245–319. [Google Scholar] [CrossRef]
- Benson, A.A. The plant sulfolipid. Adv. Lipid Res. 1963, 1, 387–394. [Google Scholar]
- Rhoades, E.R.; Streeter, C.; Turk, J.; Hsu, F.F. Characterization of sulfolipids of Mycobacterium tuberculosis H37Rv by multiple-stage linear ion-trap high-resolution mass spectrometry with electrospray ionization reveals that the family of sulfolipid II predominates. Biochemistry 2011, 50, 9135–9147. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Horrocks, L.A. On the role of sulfolipids in mammalian metabolism. Mol. Cell. Biochem. 1985, 66, 87–95. [Google Scholar]
- Shimojima, M. Biosynthesis and functions of the plant sulfolipid. Prog. Lipid Res. 2011, 50, 234–249. [Google Scholar]
- Haines, T.H. Halogen- and sulfur-containing lipids of Ochromonas. Ann. Rev. Microbiol. 1973, 27, 403–412. [Google Scholar] [CrossRef]
- Benning, C. Biosynthesis and function of the sulfolipid sulfoquinovosyl diacylglycerol. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1998, 49, 53–75. [Google Scholar] [CrossRef]
- Elovson, J.; Vagelos, P.R. A new class of lipids: Chlorosulfolipids. Proc. Natl. Acad. Sci. USA 1969, 62, 957–963. [Google Scholar]
- Nilewski, C.; Geisser, R.W.; Carreira, E.M. Total synthesis of a chlorosulpholipid cytotoxin associated with seafood poisoning. Nature 2009, 457, 573–576. [Google Scholar]
- Nilewski, C.; Geisser, R.W.; Ebert, M.O.; Carreira, E.M. Conformational and configurational analysis in the study and synthesis of chlorinated natural products. J. Am. Chem. Soc. 2009, 131, 15866–15876. [Google Scholar]
- Nilewski, C.; Deprez, N.R.; Fessard, T.C.; Li, D.B.; Geisser, R.W.; Carreira, E.M. Synthesis of undecachlorosulfolipid A: Re-evaluation of the nominal structure. Angew. Chem. Int. Ed. Engl. 2011, 50, 7940–7943. [Google Scholar]
- Inagaki, M.; Isobe, R.; Kawano, Y.; Miyamoto, T.; Komori, T.; Higuchi, R. Isolation and structure of three new ceramides from the starfish Acanthaster planci. Eur. J. Org. Chem. 1998, 1, 129–131. [Google Scholar]
- Shioiri, T.; Irako, N. An efficient synthesis of sulfobacin A (flavocristamide B), sulfobacin B, and flavocristamide A. Tetrahedron 2000, 56, 9129–9142. [Google Scholar] [CrossRef]
- Zaed, A.M.; Sutherland, A. Total synthesis of clavaminol A, C and H. Org. Biomol. Chem. 2011, 9, 8030–8037. [Google Scholar]
- Smurnyy, Y.D.; Blinov, K.A.; Churanova, T.S.; Elyashberg, M.E.; Williams, A.J. Toward more reliable 13C and 1H chemical shift prediction: A systematic comparison of neural-network and least-squares regression based approaches. J. Chem. Inf. Model. 2008, 48, 128–134. [Google Scholar] [CrossRef]
- Kleinpeter, E.; Seidl, P.R. The γ- and the δ-effects in 13C NMR spectroscopy in terms of nuclear chemical shielding (NCS) analysis. J. Phys. Org. Chem. 2004, 17, 680–685. [Google Scholar]
- Han, A.-R.; Song, J.-I.; Jang, D.S.; Min, H.-Y.; Lee, S.K.; Seo, E.-K. Cytotoxic constituents of the octocoral Dendronephthya gigantea. Arch. Pharm. Res. 2005, 28, 290–293. [Google Scholar] [CrossRef]
- Chakrabarty, M.; Batabyal, A.; Barua, A.K. New ceramides from the hypotensive extract of a sea aneome, Paracondylactis indicus. J. Nat. Prod. 1994, 57, 393–395. [Google Scholar] [CrossRef]
- Shin, J.; Seo, Y. Isolation of new ceramides from the gorgonian Acabaria undulata. J. Nat. Prod. 1995, 58, 948–953. [Google Scholar] [CrossRef]
- Tan, R.X.; Chen, J.H. The cerebrosides. Nat. Prod. Rep. 2003, 20, 509–534. [Google Scholar] [CrossRef]
- Chang, Y.T.; Choi, J.; Ding, S.; Prieschl, E.E.; Baumruker, T.; Lee, J.M.; Chung, S.K.; Schultz, P.G. The synthesis and biological characterization of a ceramide library. J. Am. Chem. Soc. 2002, 124, 1856–1857. [Google Scholar]
- Park, J.; Li, Q.; Chang, Y.-T.; Kim, S.T. Inhibitory activity of a ceramide library on interleukin-4 production from activated T cells. Bioorg. Med. Chem. 2005, 13, 2589–2595. [Google Scholar] [CrossRef]
- Zeng, X.; Xiang, L.; Li, C.-Y.; Wang, Y.; Qiu, G.; Zhag, Z.-X.; He, X. Cytotoxic ceramides and glycerides from the roots of Livistona chinensis. Fitoterapia 2012, 83, 609–616. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Almeida, J.G.L.; Maia, A.I.V.; Wilke, D.V.; Silveira, E.R.; Braz-Filho, R.; La Clair, J.J.; Costa-Lotufo, L.V.; Pessoa, O.D.L. Palyosulfonoceramides A and B: Unique Sulfonylated Ceramides from the Brazilian Zoanthids Palythoa caribaeorum and Protopalyhtoa variabilis. Mar. Drugs 2012, 10, 2846-2860. https://doi.org/10.3390/md10122846
Almeida JGL, Maia AIV, Wilke DV, Silveira ER, Braz-Filho R, La Clair JJ, Costa-Lotufo LV, Pessoa ODL. Palyosulfonoceramides A and B: Unique Sulfonylated Ceramides from the Brazilian Zoanthids Palythoa caribaeorum and Protopalyhtoa variabilis. Marine Drugs. 2012; 10(12):2846-2860. https://doi.org/10.3390/md10122846
Chicago/Turabian StyleAlmeida, Jose Gustavo L., Ana Isabel V. Maia, Diego V. Wilke, Edilberto R. Silveira, Raimundo Braz-Filho, James J. La Clair, Leticia V. Costa-Lotufo, and Otília Deusdenia L. Pessoa. 2012. "Palyosulfonoceramides A and B: Unique Sulfonylated Ceramides from the Brazilian Zoanthids Palythoa caribaeorum and Protopalyhtoa variabilis" Marine Drugs 10, no. 12: 2846-2860. https://doi.org/10.3390/md10122846