Ritonavir-Mediated Induction of Apoptosis in Pancreatic Cancer Occurs via the RB/E2F-1 and AKT Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental
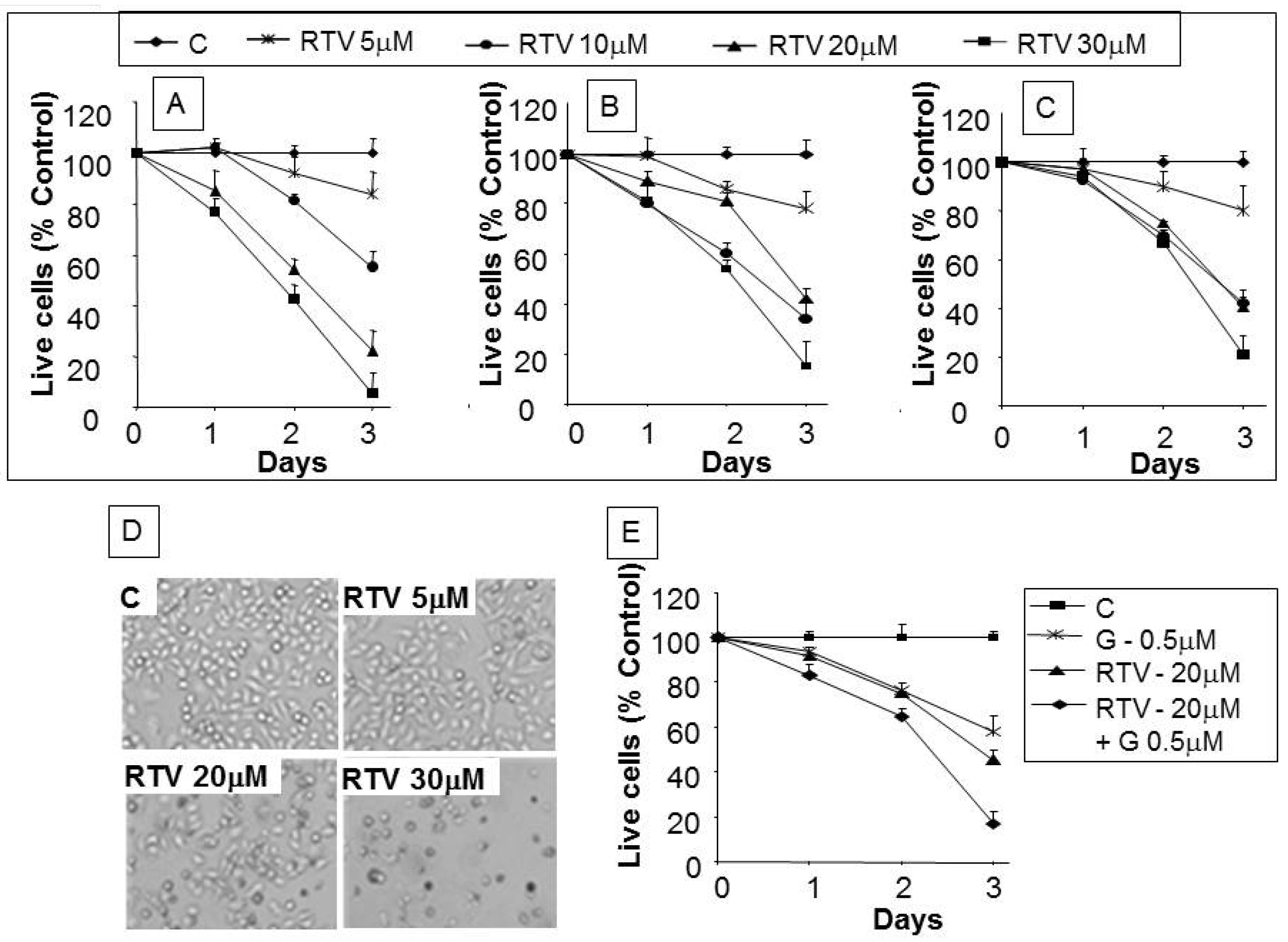
3. Results
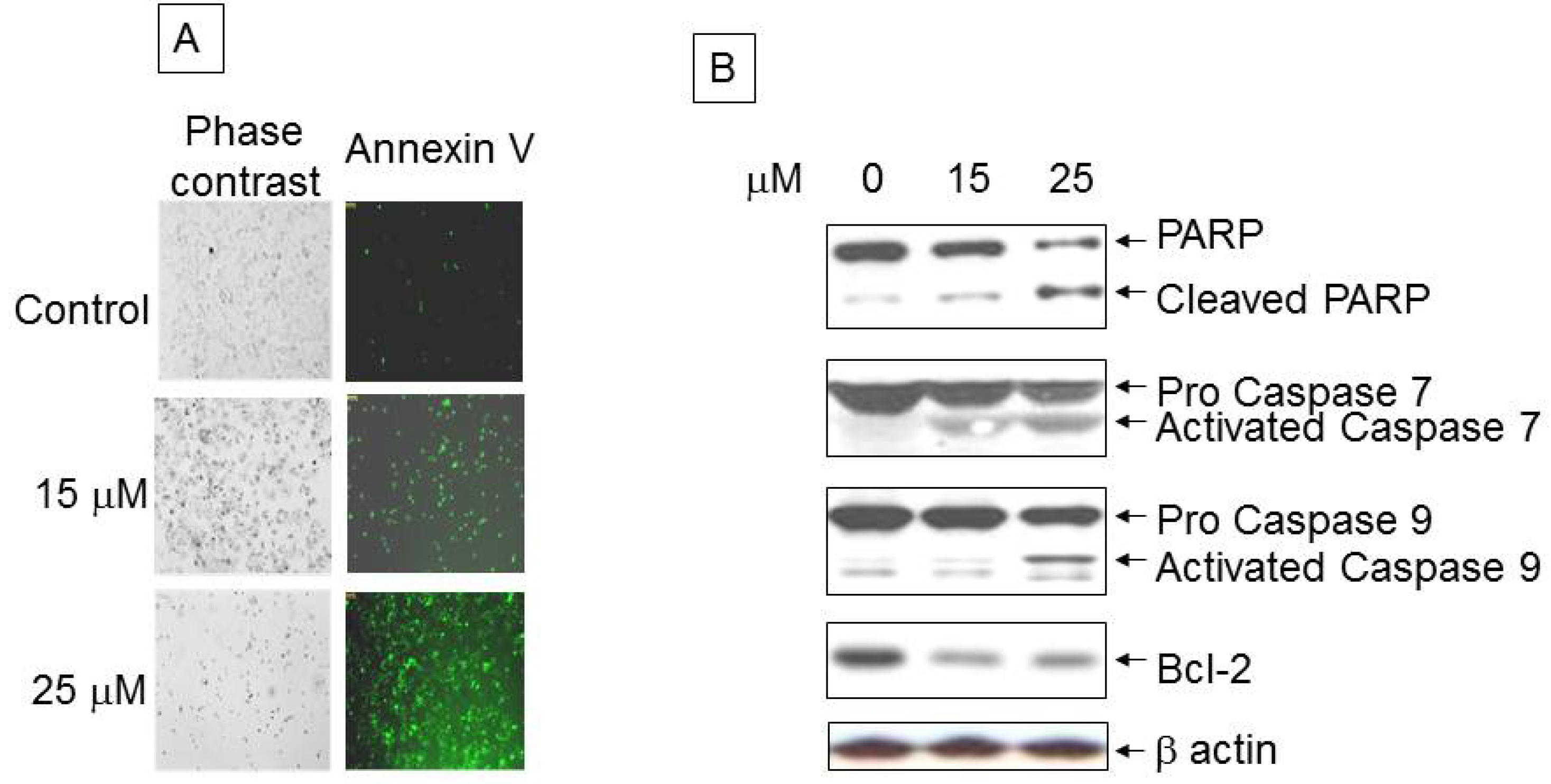
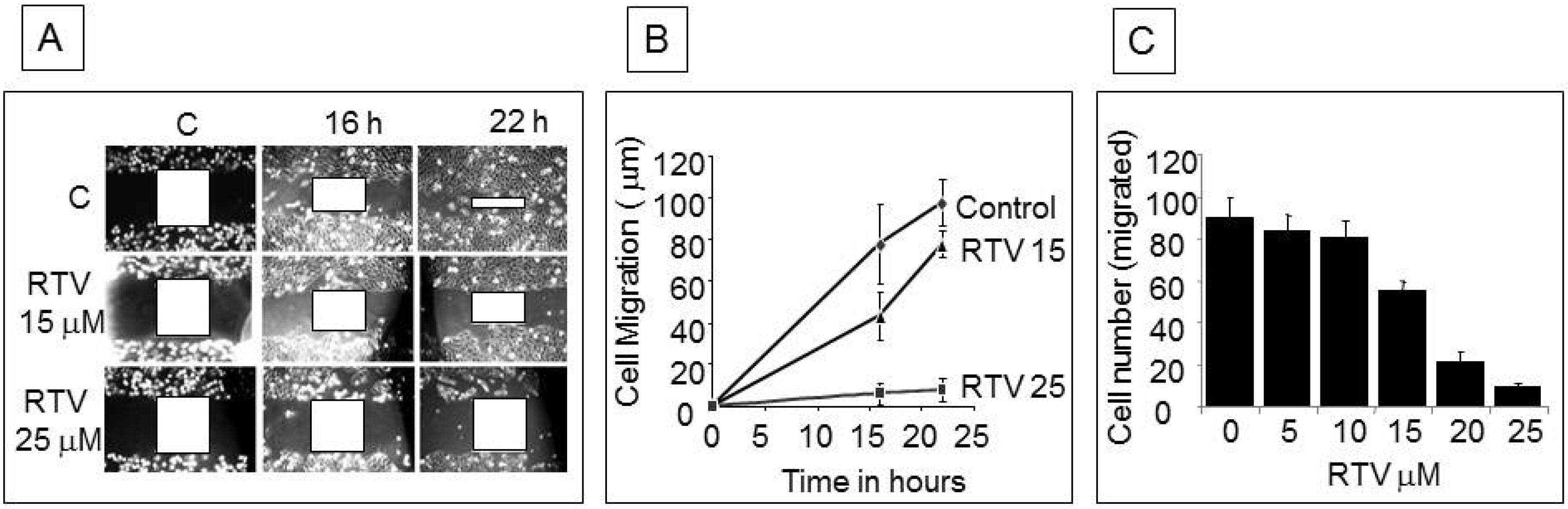
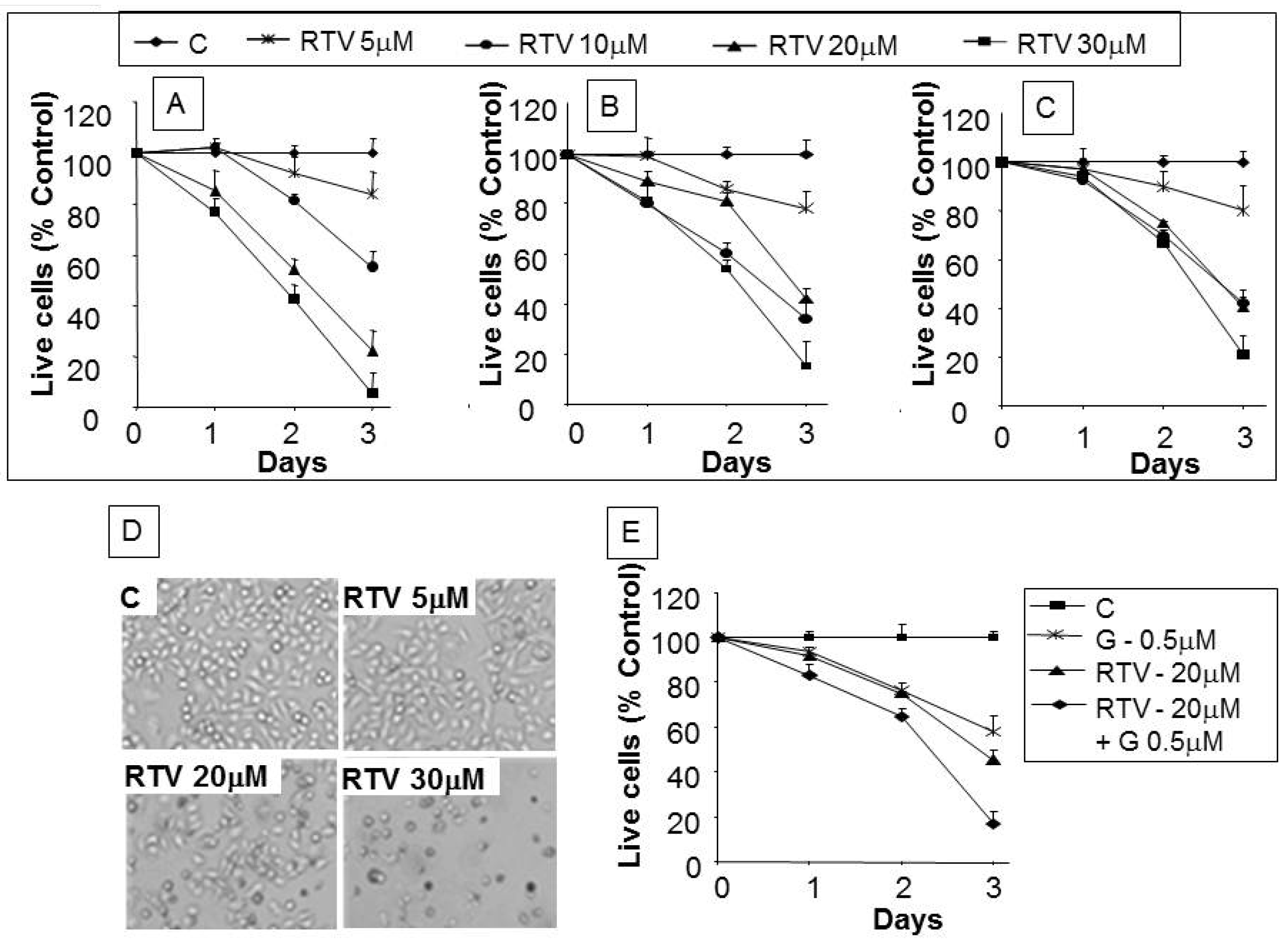
3.2. Analysis of Apoptotic Cells with Ritonavir Treatment
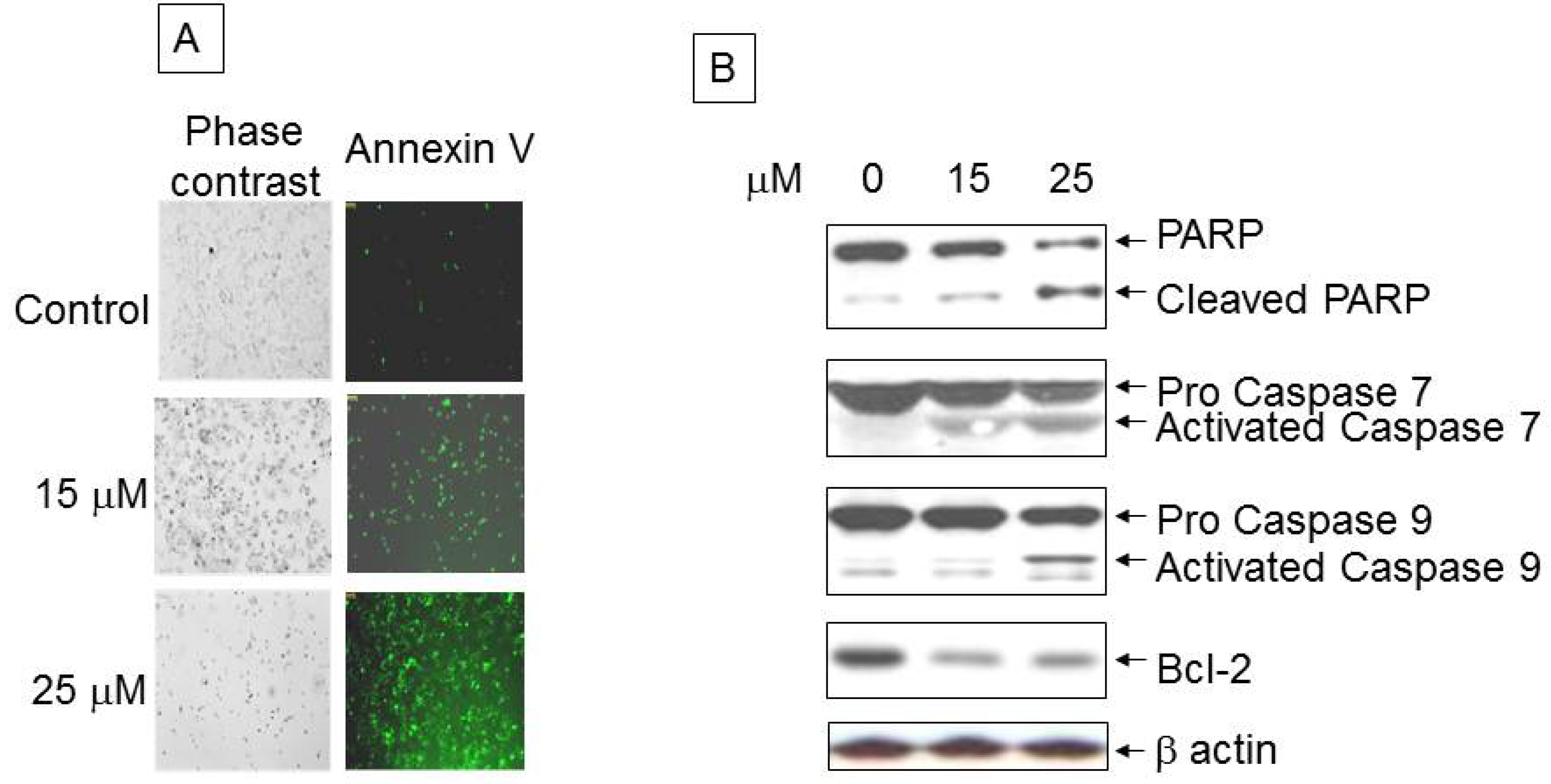
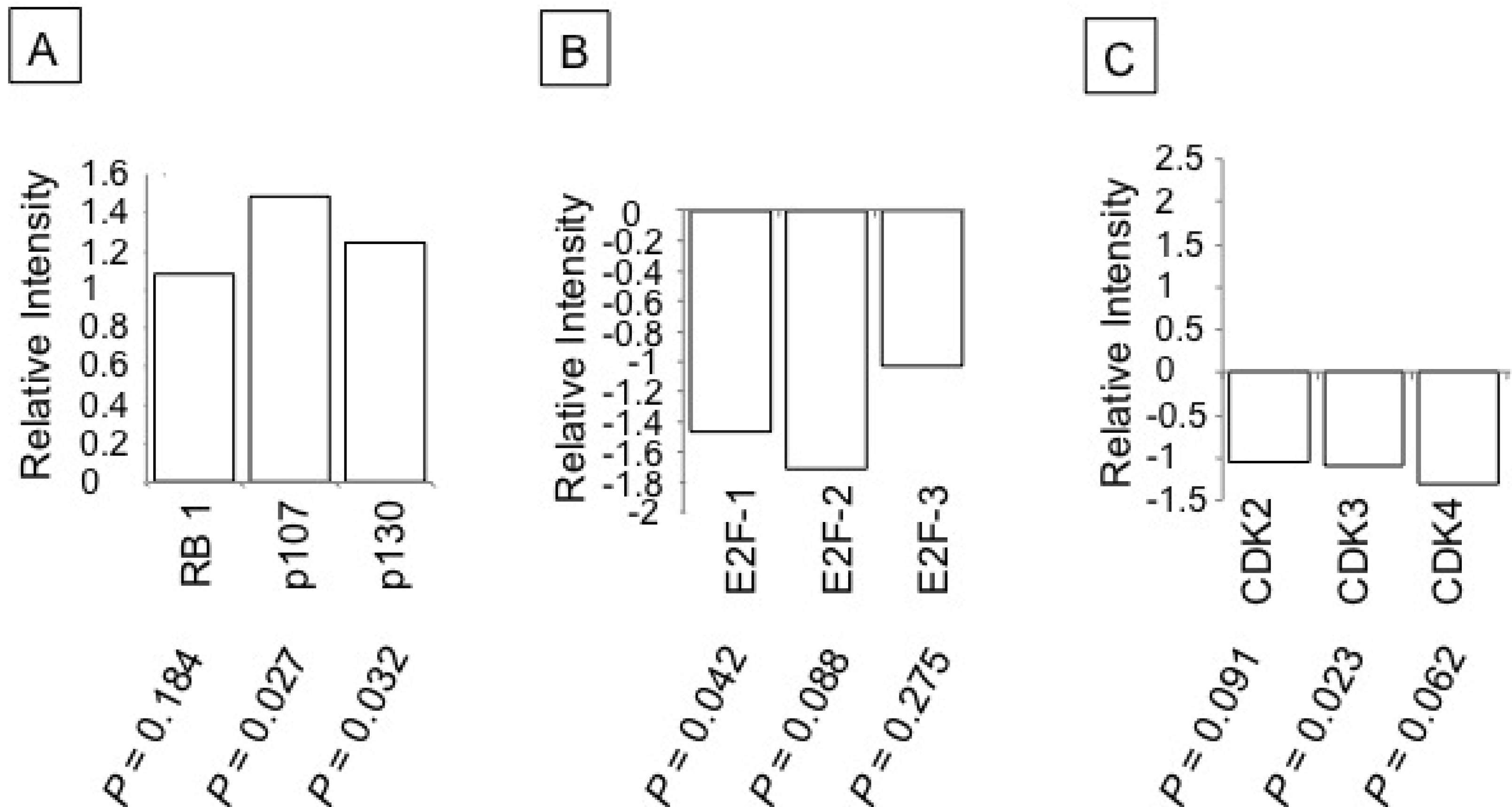
3.3. Ritonavir-Mediated Perturbations in the Expression of Cell Cycle Regulatory Genes
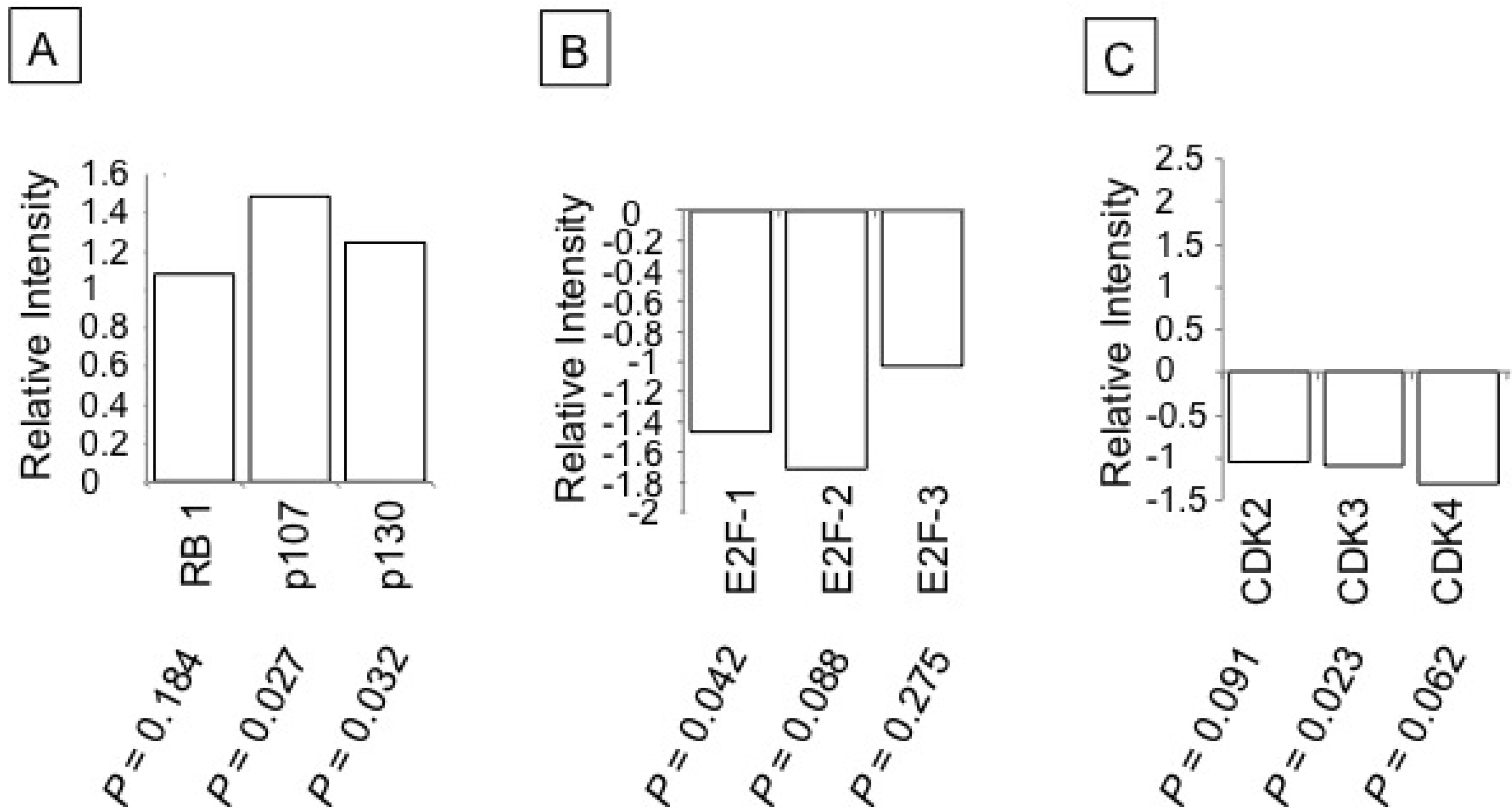
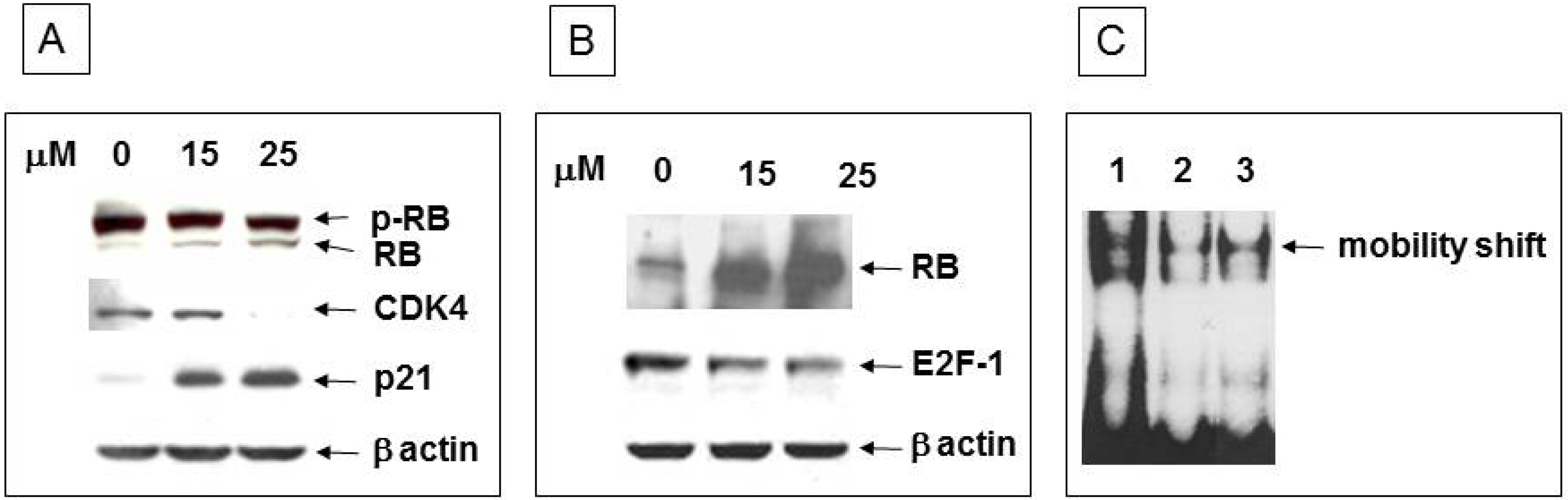
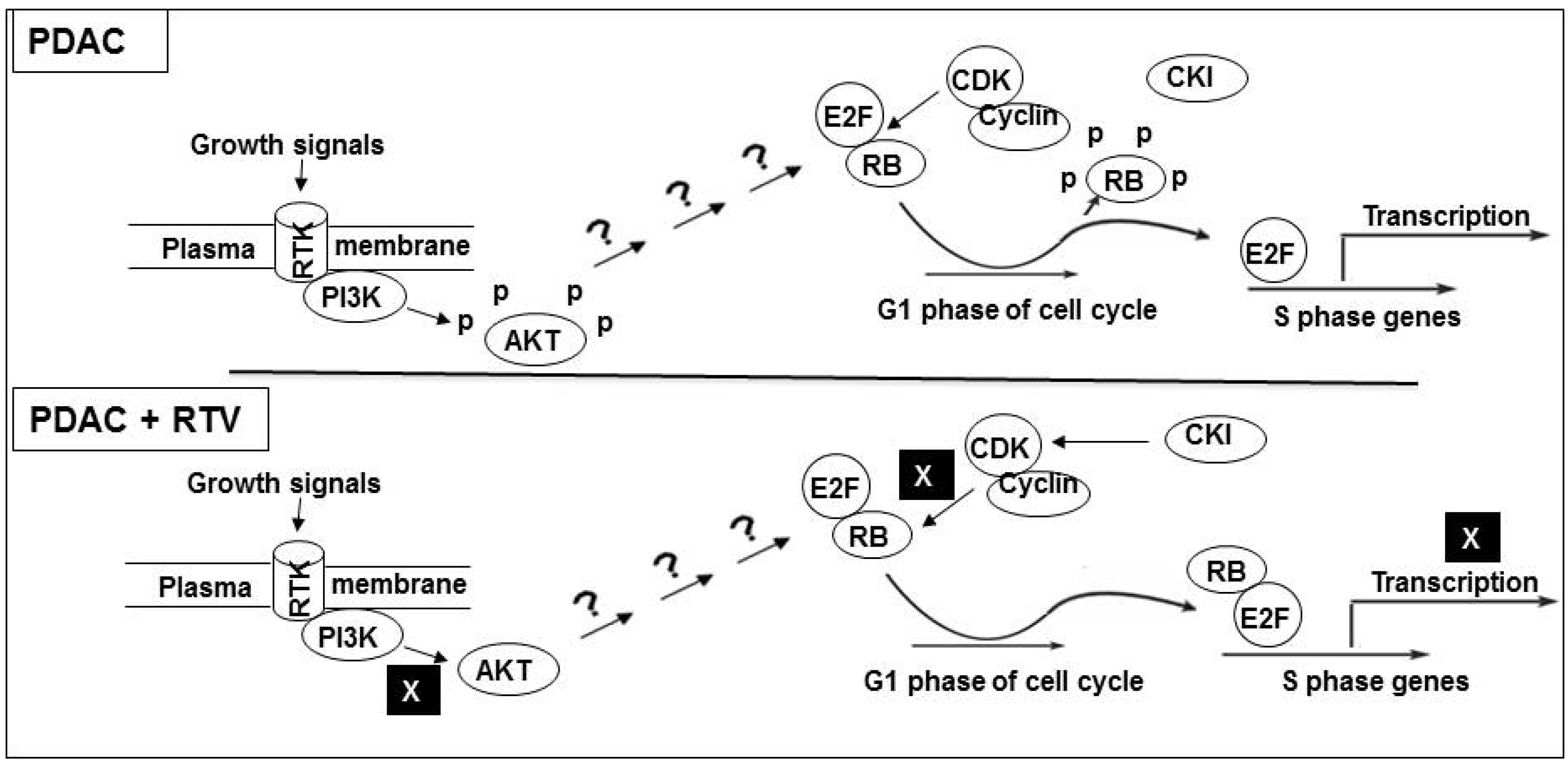
3.4. Inhibition of Cell Cycle Machinery Involved in the Phosphorylation of RB and Protection of the RB-E2F-1 Complex by Ritonavir
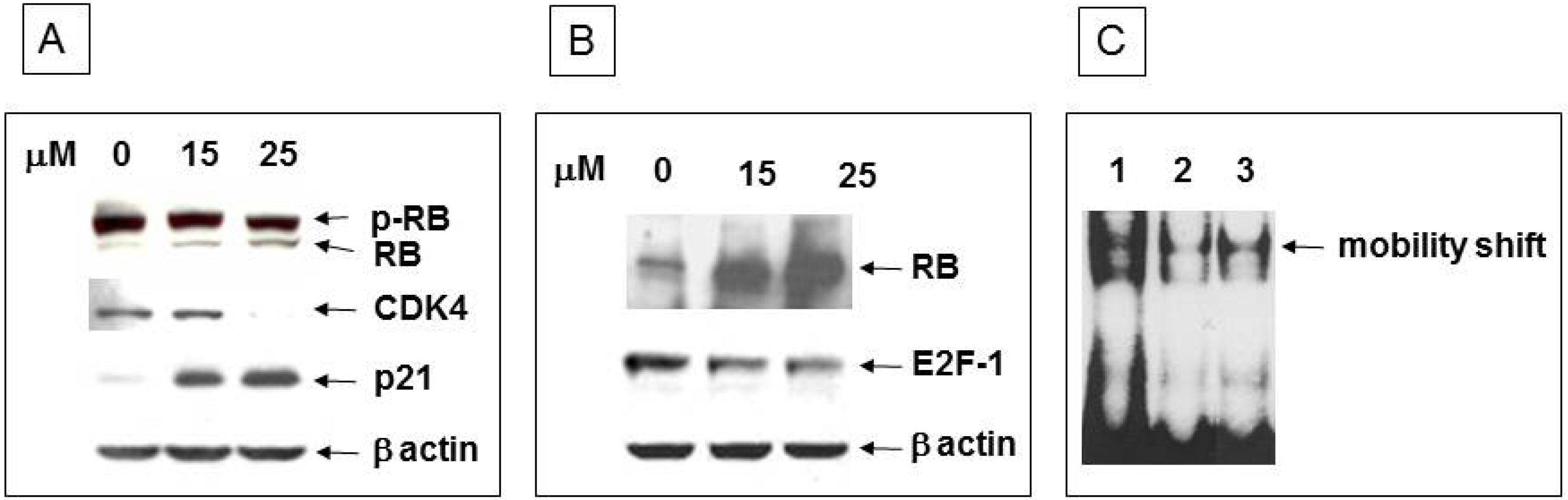
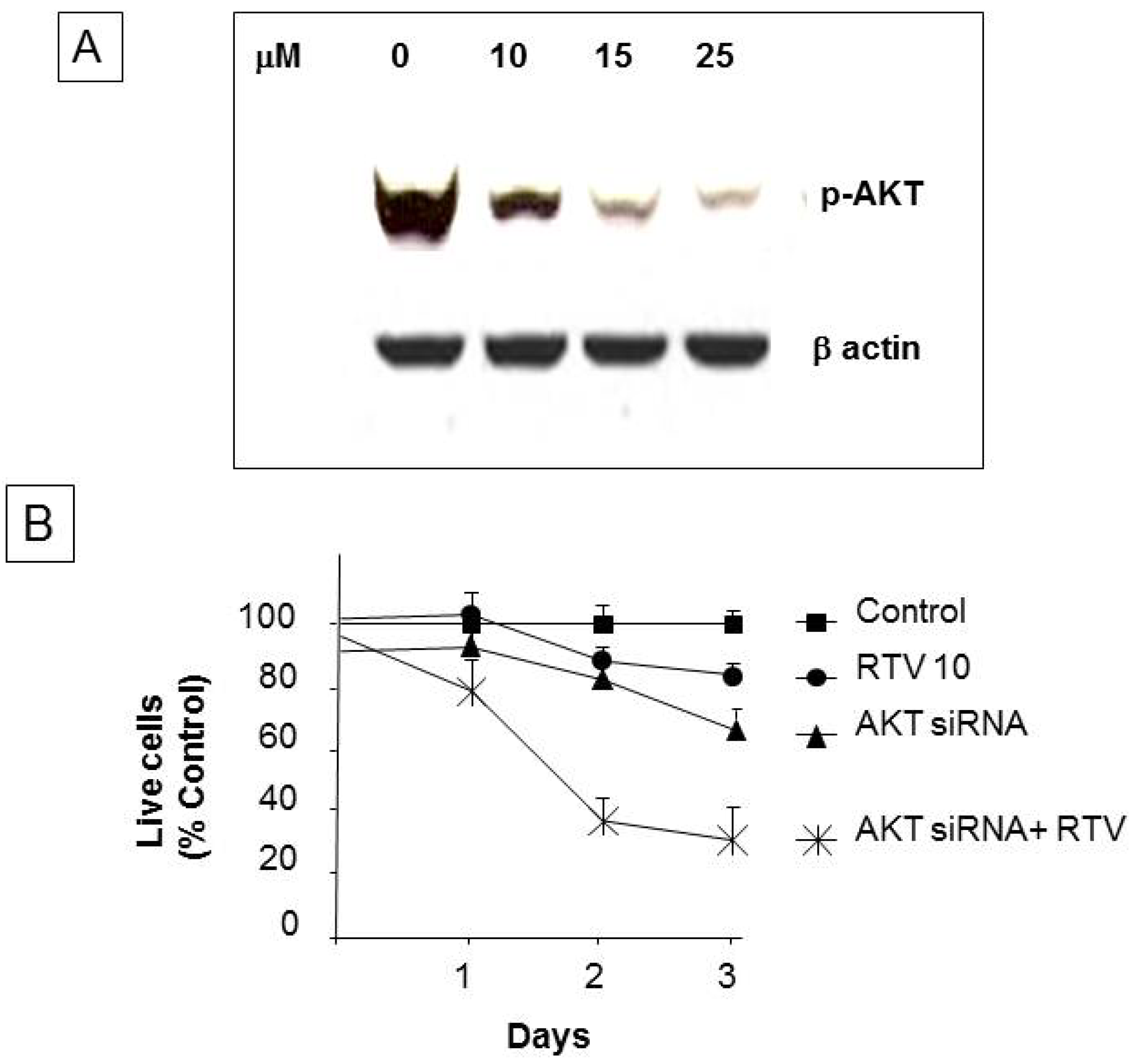
3.6. Ritonavir Inhibits AKT Pathway in PANC-1 Cells
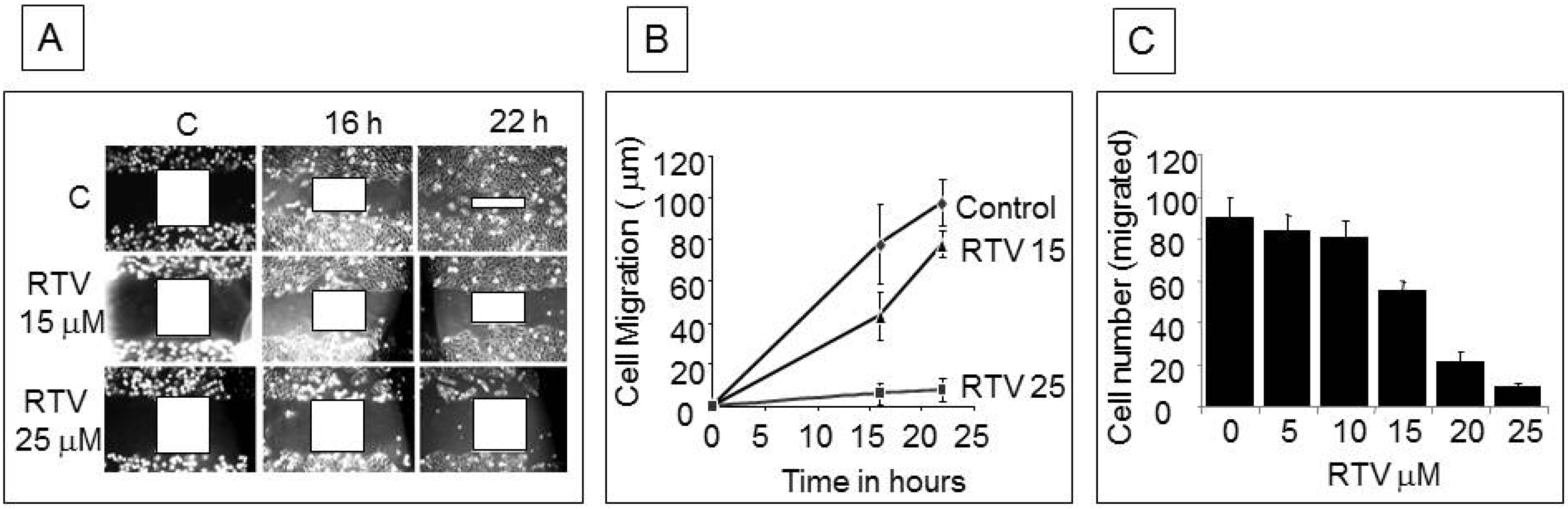
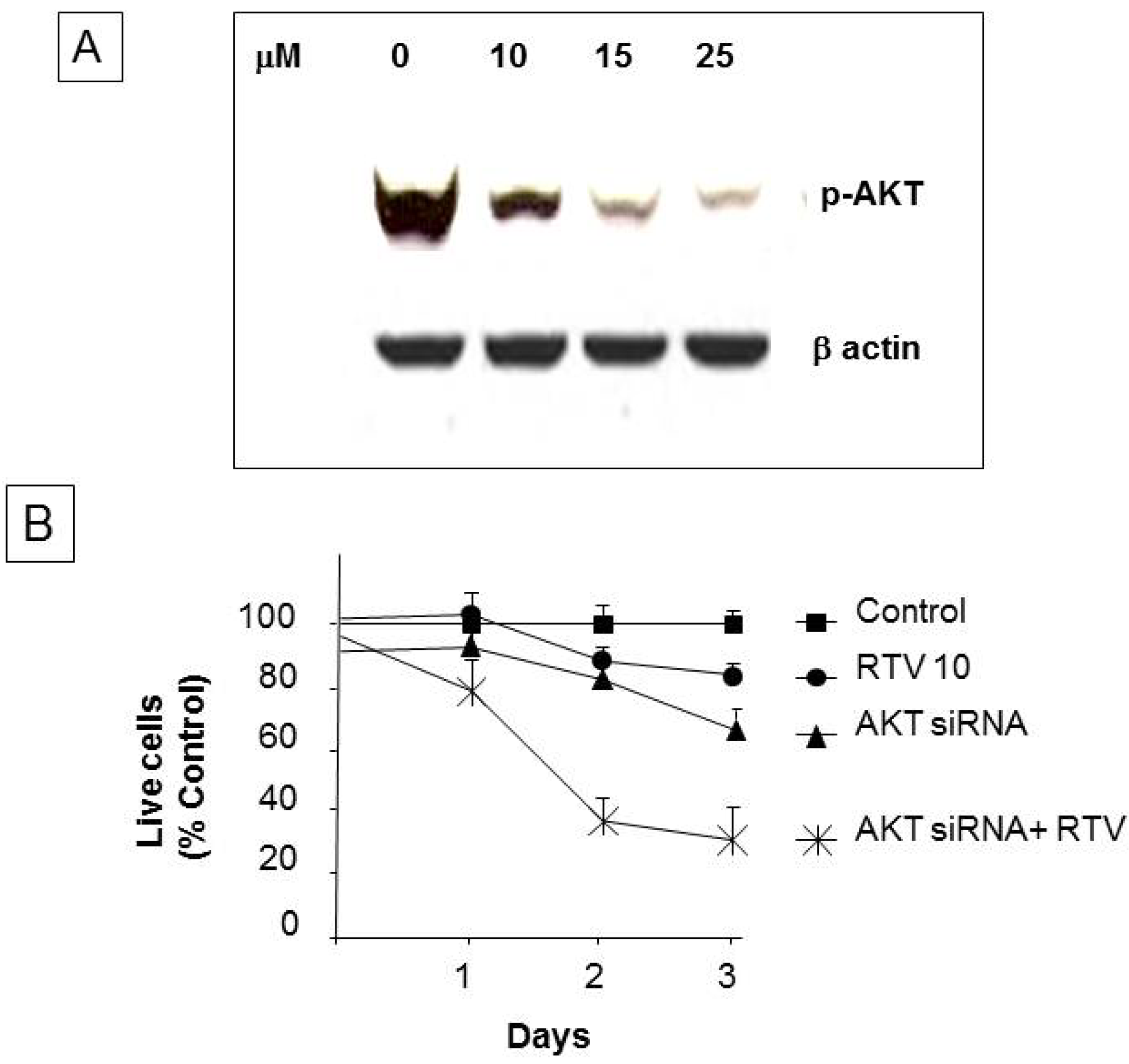
4. Discussion
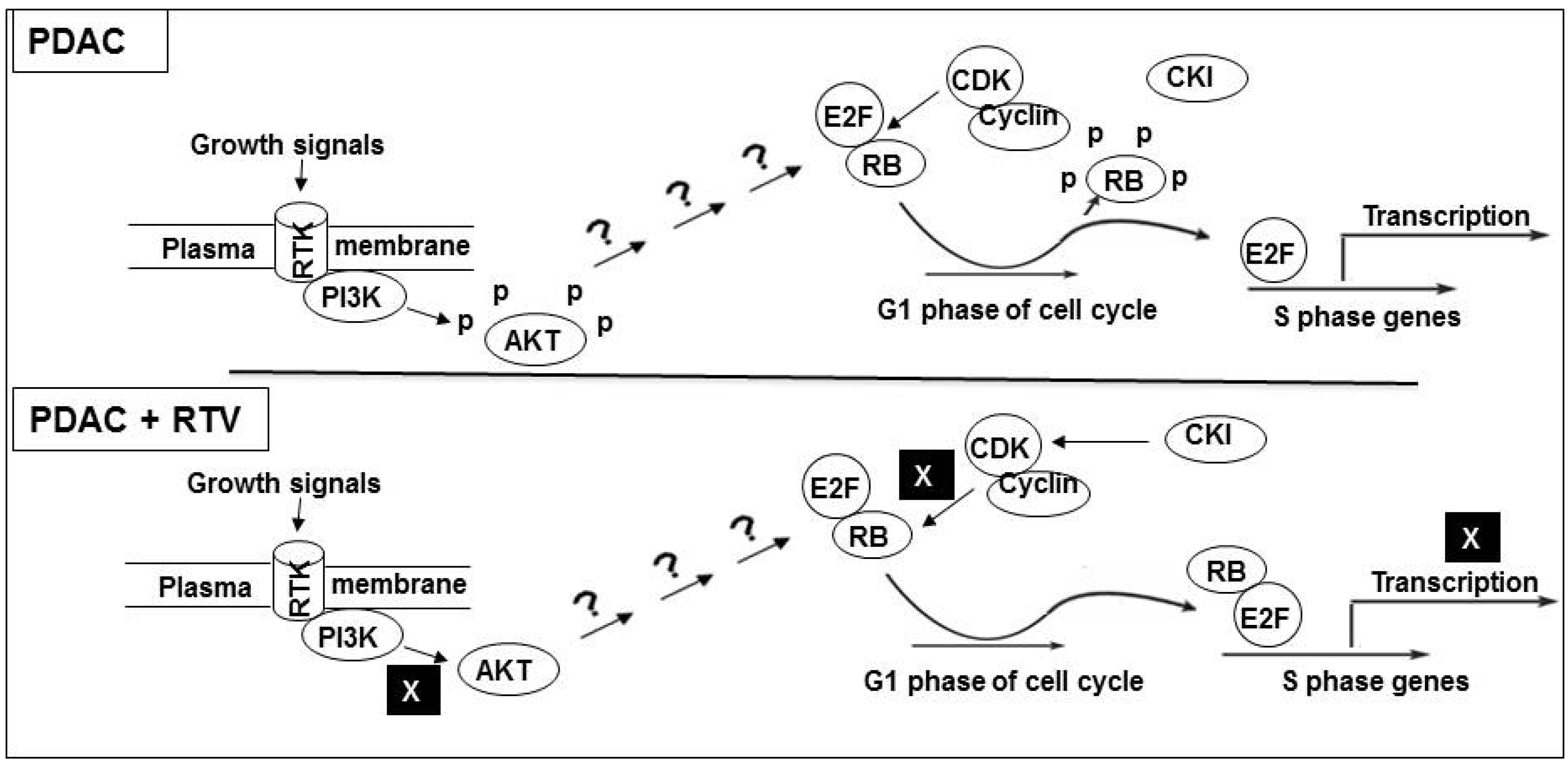
5. Conclusions
Conflicts of Interest
References
- Wang, Z.; Li, Y.; Ahmad, A.; Banerjee, S.; Azmi, A.S.; Kong, D.; Sarkar, F.H. Pancreatic cancer: Understanding and overcoming chemoresistance. Nat. Rev. Gastroenterol. Hepatol. 2010, 8, 27–33. [Google Scholar]
- Mates, J.M.; Segura, J.A.; Alonso, F.J.; Marquez, J. Anticancer antioxidant regulatory functions of phytochemicals. Curr. Med. Chem. 2011, 18, 2315–2338. [Google Scholar] [CrossRef]
- Granich, R.; Crowley, S.; Vitoria, M.; Smyth, C.; Kahn, J.G.; Bennett, R.; Lo, Y.-R.; Souteyrand, Y.; Williams, B. Highly active antiretroviral treatment as prevention of HIV transmission: Review of scientific evidence and update. Curr. Opin. HIV AIDS 2010, 5, 298–304. [Google Scholar] [CrossRef]
- Seaberg, E.C.; Wiley, D.; Martínez-Maza, O.; Chmiel, J.S.; Kingsley, L.; Tang, Y.; Margolick, J.B.; Jacobson, L.P. Cancer incidence in the multicenter aids cohort study before and during the HAART era. Cancer 2010, 116, 5507–5516. [Google Scholar] [CrossRef]
- Kumar, S.; Bryant, C.S.; Chamala, S.; Qazi, A.; Seward, S.; Pal, J.; Steffes, C.P.; Weaver, D.W.; Morris, R.; Malone, J.M. Ritonavir blocks AKT signaling, activates apoptosis and inhibits migration and invasion in ovarian cancer cells. Mol. Cancer 2009, 8. [Google Scholar] [CrossRef]
- Brunner, T.B.; Geiger, M.; Grabenbauer, G.G.; Lang-Welzenbach, M.; Mantoni, T.S.; Cavallaro, A.; Sauer, R.; Hohenberger, W.; McKenna, W.G.; Brunner, T.B.; et al. Phase I trial of the human immunodeficiency virus protease inhibitor nelfinavir and chemoradiation for locally advanced pancreatic cancer. J. Clin. Oncol. 2008, 26, 2699–2706. [Google Scholar] [CrossRef]
- DiCiommo, D.; Gallie, B.L.; Bremner, R. Retinoblastoma: The disease, gene and protein provide critical leads to understand cancer. Semin. Cancer Biol. 2000, 10, 255–269. [Google Scholar] [CrossRef]
- Gaedicke, S.; Firat-Geier, E.; Constantiniu, O.; Lucchiari-Hartz, M.; Freudenberg, M.; Galanos, C.; Niedermann, G. Antitumor effect of the human immunodeficiency virus protease inhibitor ritonavir: Induction of tumor-cell apoptosis associated with perturbation of proteasomal proteolysis. Cancer Res. 2002, 62, 6901–6908. [Google Scholar]
- Bondar, V.M.; Sweeney-Gotsch, B.; Andreeff, M.; Mills, G.B.; McConkey, D.J. Inhibition of the phosphatidylinositol 3'-kinase-AKT pathway induces apoptosis in pancreatic carcinoma cells in vitro and in vivo. Mol. Cancer Ther. 2002, 1, 989–997. [Google Scholar]
- Srirangam, A.; Mitra, R.; Wang, M.; Gorski, J.C.; Badve, S.; Baldridge, L.; Hamilton, J.; Kishimoto, H.; Hawes, J.; Li, L.; et al. Effects of HIV protease inhibitor ritonavir on Akt-regulated cell proliferation in breast cancer. Clin. Cancer Res. 2006, 12, 1883–1896. [Google Scholar] [CrossRef]
- Yang, Y.; Ikezoe, T.; Nishioka, C.; Bandobashi, K.; Takeuchi, T.; Adachi, Y.; Kobayashi, M.; Takeuchi, S.; Koeffler, H.P.; Taguchi, H. NFV, an HIV-1 protease inhibitor, induces growth arrest, reduced Akt signalling, apoptosis and docetaxel sensitisation in NSCLC cell lines. Br. J. Cancer 2006, 95, 1653–1662. [Google Scholar] [CrossRef]
- Batchu, R.B.; Shammas, M.A.; Wang, J.Y.; Freeman, J.; Rosen, N.; Munshi, N.C. Adeno-associatedvirus protects the retinoblastoma family of proteins from adenoviral-induced functional inactivation. Cancer Res. 2002, 62, 2982–2985. [Google Scholar]
- Kobayashi, H.; Suzuki, M.; Tanaka, Y.; Hirashima, Y.; Terao, T. Suppression of urokinase expression and invasiveness by urinary trypsin inhibitor is mediated through inhibition of protein kinase C- and MEK/ERK/c-Jun-dependent signaling pathways. J. Biol. Chem. 2001, 276, 2015–2022. [Google Scholar] [CrossRef]
- Altomare, D.A.; Testa, J.R. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005, 24, 7455–7464. [Google Scholar]
- Tsantoulis, P.K.; Gorgoulis, V.G. Involvement of E2F transcription factor family in cancer. Eur. J. Cancer 2005, 41, 2403–2414. [Google Scholar] [CrossRef]
- Kariya, R.; Taura, M.; Suzu, S.; Kai, H.; Katano, H.; Okada, S. HIV protease inhibitor Lopinavir induces apoptosis of primary effusion lymphoma cells via suppression of NF-kappaB pathway. Cancer Lett. 2014, 342, 52–59. [Google Scholar] [CrossRef]
- Ikezoe, T.; Hisatake, Y.; Takeuchi, T.; Ohtsuki, Y.; Yang, Y.; Said, J.W.; Taguchi, H.; Koeffler, H.P. HIV-1 protease inhibitor, ritonavir: A potent inhibitor of CYP3A4, enhanced the anticancer effects of docetaxel in androgen-independent prostate cancer cells in vitro and in vivo. Cancer Res. 2004, 64, 7426–7431. [Google Scholar] [CrossRef]
- Sato, A.; Asano, T.; Ito, K.; Asano, T. 17-Allylamino-17-demethoxygeldanamycin and ritonavir inhibit renal cancer growth by inhibiting the expression of heat shock factor-1. Int. J. Oncol. 2012, 41, 46–52. [Google Scholar]
- Steeg, P.S.; Steeg, P.S. Tumor metastasis: Mechanistic insights and clinical challenges. Nat. Med. 2006, 12, 895–904. [Google Scholar] [CrossRef]
- Pati, S.; Pelser, C.B.; Dufraine, J.; Bryant, J.L.; Reitz, M.S., Jr.; Weichold, F.F. Antitumorigenic effects of HIV protease inhibitor ritonavir: Inhibition of Kaposi sarcoma. Blood 2002, 99, 3771–3779. [Google Scholar] [CrossRef]
- Yamamoto, S.; Tomita, Y.; Hoshida, Y.; Morooka, T.; Nagano, H.; Dono, K.; Umeshita, K.; Sakon, M.; Ishikawa, O.; Ohigashi, H.; et al. Prognostic significance of activated Akt expression in pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2004, 10, 2846–2850. [Google Scholar] [CrossRef]
- Gatti, G.; di Biagio, A.; Casazza, R.; de Pascalis, C.; Bassetti, M.; Cruciani, M.; Vella, S.; Bassetti, D. The relationship between ritonavir plasma levels and side-effects: Implications for therapeutic drug monitoring. AIDS 1999, 13, 2083–2089. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Batchu, R.B.; Gruzdyn, O.V.; Bryant, C.S.; Qazi, A.M.; Kumar, S.; Chamala, S.; Kung, S.T.; Sanka, R.S.; Puttagunta, U.S.; Weaver, D.W.; et al. Ritonavir-Mediated Induction of Apoptosis in Pancreatic Cancer Occurs via the RB/E2F-1 and AKT Pathways. Pharmaceuticals 2014, 7, 46-57. https://doi.org/10.3390/ph7010046
Batchu RB, Gruzdyn OV, Bryant CS, Qazi AM, Kumar S, Chamala S, Kung ST, Sanka RS, Puttagunta US, Weaver DW, et al. Ritonavir-Mediated Induction of Apoptosis in Pancreatic Cancer Occurs via the RB/E2F-1 and AKT Pathways. Pharmaceuticals. 2014; 7(1):46-57. https://doi.org/10.3390/ph7010046
Chicago/Turabian StyleBatchu, Ramesh B., Oksana V. Gruzdyn, Christopher S. Bryant, Aamer M. Qazi, Sanjeev Kumar, Sreedhar Chamala, Shu T. Kung, Ramana S. Sanka, Udaya S. Puttagunta, Donald W. Weaver, and et al. 2014. "Ritonavir-Mediated Induction of Apoptosis in Pancreatic Cancer Occurs via the RB/E2F-1 and AKT Pathways" Pharmaceuticals 7, no. 1: 46-57. https://doi.org/10.3390/ph7010046