Nanoparticle-Based Delivery of RNAi Therapeutics: Progress and Challenges

Abstract
:1. Introduction

{kind=link}
| Drug name | Disease | Target | Carrier | Phase | Company | Status (Clinicaltrials.gov identifier) |
|---|---|---|---|---|---|---|
| Local Delivery | ||||||
| Bevasiranib | AMD | VEG | Naked siRNA | III | Opko Health Inc. | Terminated (NCT00499590) |
| AGN-211745 Sirna027 | AMD | VEGF | Naked siRNA | II | Allergan/Sirna therapeutics | Terminated (NCT00363714) |
| PF655 | Wet AMD and DME | RTP801 | Naked siRNA | II | Quark Pharmaceuticals | Ongoing for DME (NCT01445899); Completed for AMD (NCT00713518) |
| QPI1007 | Non-arteritic ischemic optic neuropathy | Caspase 2 | Naked siRNA | I | Ongoing (NCT01064505) | |
| TD101 | Pachyonychia congenita | Keratin 6a (K6a) N171K | Naked siRNA | 1b | TransDerm | Completed (NCT00716014) |
| RXI109 | Dermal scarring | Connective tissue growth factor | Self-delivering RNAi compound (sd-RxRNA®) | I | RXi Pharmaceuticals | Initiate in 2012 |
| SYL040012 | Ocular Hypertension | ADRB2 | Naked siRNA | II | Sylentis | Ongoing (NCT01227291) |
| SYL1001 | Dye eye, ocular pain | TRPV1 | Naked siRNA | I | Ongoing (NCT01438281) | |
| Excellair | Asthma | Syk kinase | Naked siRNA | II | ZaBeCor | Ongoing |
| ALN-RSV01 | RSV infection | RSV Nucleocapsid “N” gene | Naked siRNA | II | Alnylam Pharmaceuticals | Ongoing (NCT01065935) |
| siG12D LODER | Pancreatic cancer | KRASG12D | LODER polymer | I | Silenseed | Ongoing (NCT01188785) |
| Systemic delivery | ||||||
| ALN-TTR | Transthyretin mediated amyloidosis | TTR | Lipid nanoparticles, MC3 lipid | I | Alnylam Pharmaceuticals | Ongoing (NCT01148953, ALN-TTR01; NCT01559077, ALN-TTR02) |
| ALN-PCS | Hypercholesterolemia | PCSK9 | Lipid nanoparticles, MC3 lipid | I | Ongoing (NCT01437059) | |
| ALN-VSP | Liver cancer | KSP and VEGF | Lipid nanoparticles | I | Ongoing (NCT01158079) | |
| TKM-PLK1 | Advanced sold tumor | PLK1 | Lipid nanoparticles | I | Tekmira Pharmaceuticals | Ongoing (NCT01262235) |
| KM-Ebola | Zaire Ebola or other hemorrhagic fever viruses infection | RNA polymerase L protein | Lipid nanoparticles, SNALP | I | Ongoing (NCT01518881) | |
| TKM-ApoB (PRO-040201) | Hypercholesterolemia | ApoB | Lipid nanoparticles | I | Terminated (NCT00927459) | |
| Atu027 | Advanced Solid tumor | PKN3 | Lipid nanoparticles AtuPLEX® | I | Silence Therapeutics | Ongoing (NCT00938574) |
| QPI-1002 (I5NP) | Delayed Graft Function and Acute Kidney Injury | p53 | AtuRNAi chemically modified siRNA | II for Delayed Graft Function I for acute kidney injury | Silence Therapeutics/Quark Pharmaceuticals/ Novartis Pharmaceuticals | Ongoing (NCT00802347) |
| CALAA-01 | Solid tumors | RRM2 | Cyclodextrin, PEG and Transferrin | I | Calando Pharmaceuticals | Ongoing (NCT00689065) |
2. Barriers in Systemic RNAi Delivery
2.1. Local Delivery vs. Systemic Delivery
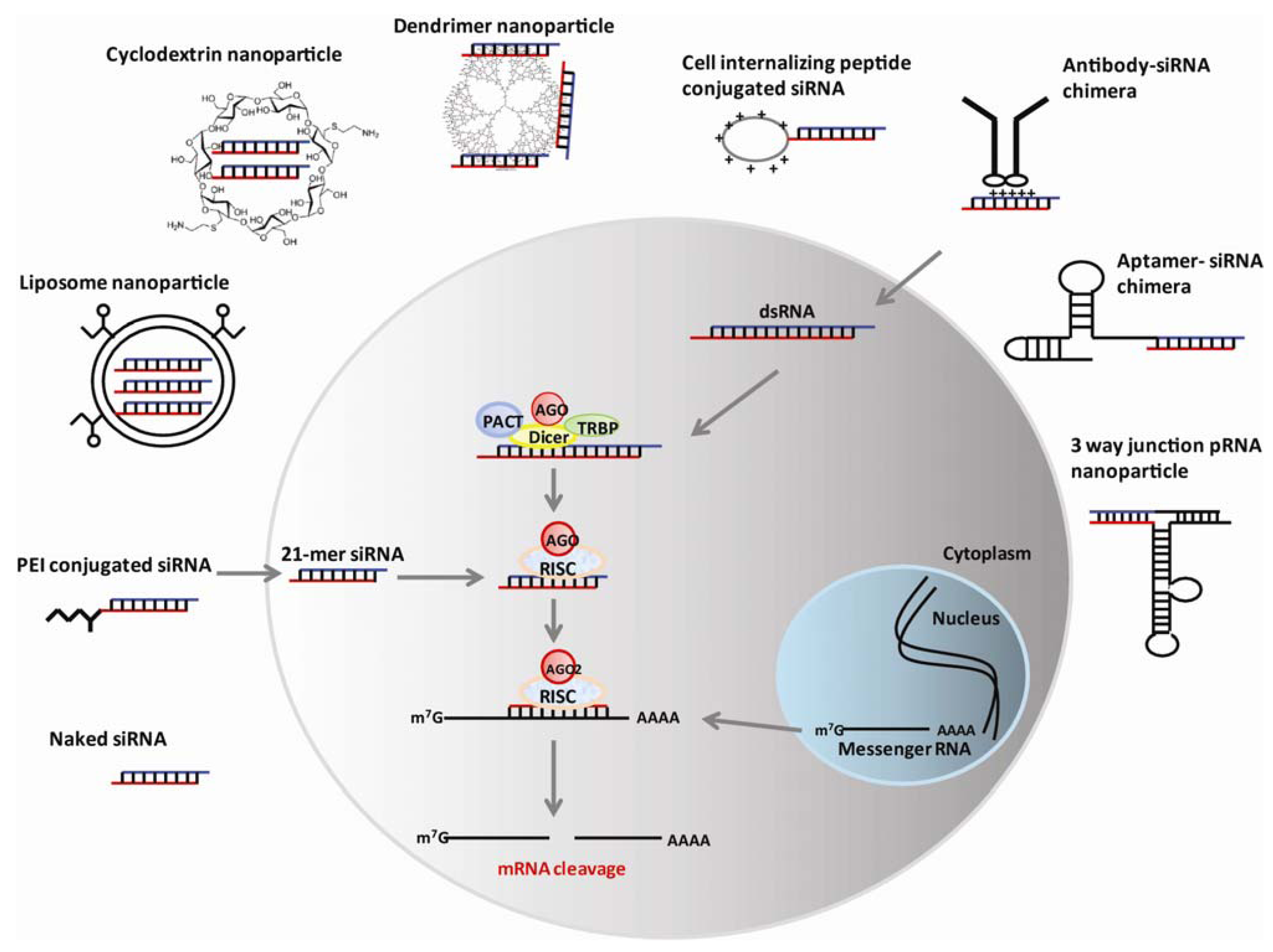
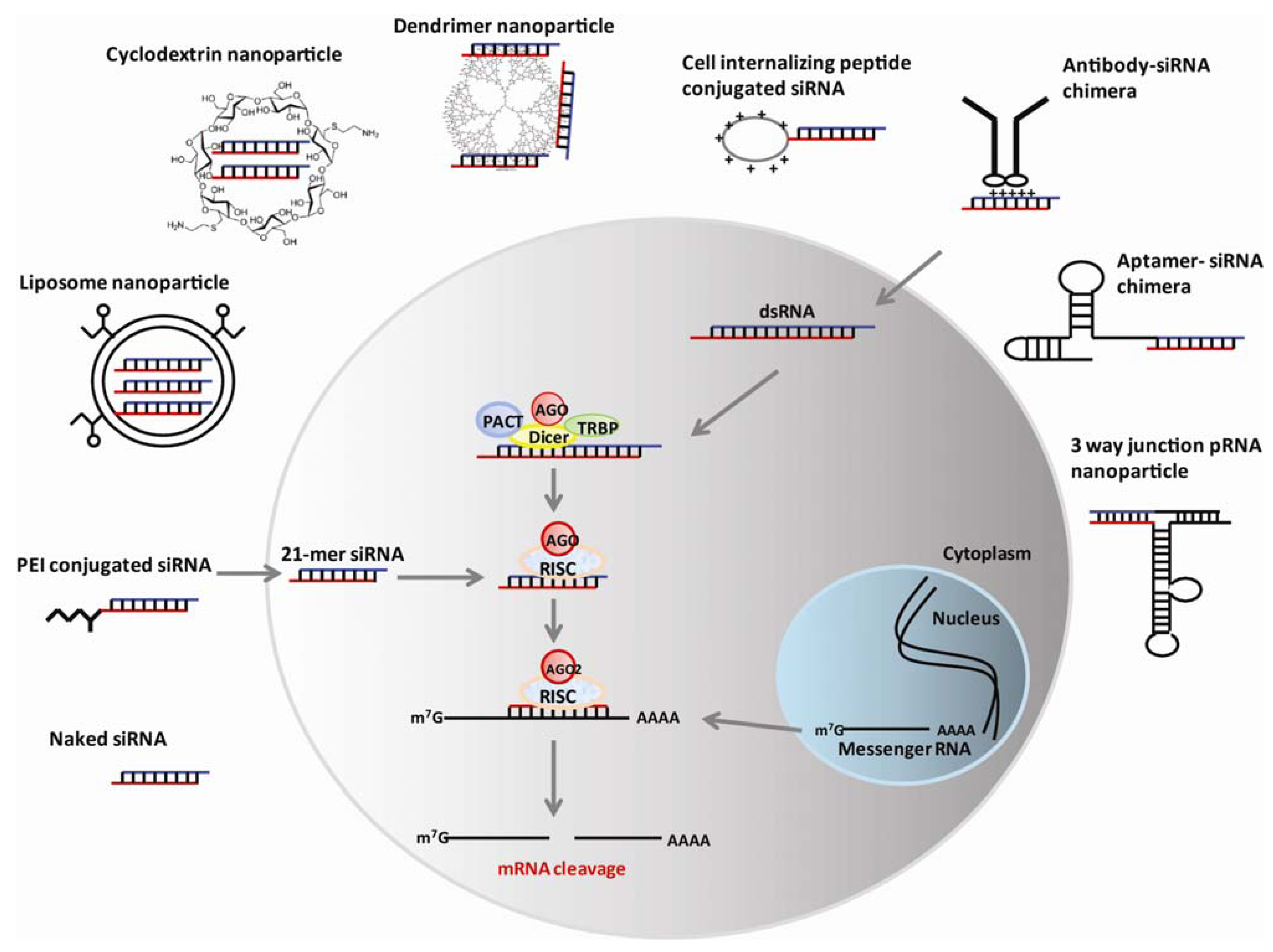
2.2. Renal Clearance
2.3. Vascular Extravasation and Diffusion
2.4. Cellular Uptake and Endosomal Escape
2.5. Cytoplasmic Location and RISC Loading of siRNAs
3. Nanotechnology-Based RNAi Delivery
3.1. Liposome-Based Nanoparticles
3.2. Cationic Dendrimers
3.3. Cyclodextrin Polymers
3.4. Polyethyleneimine (PEI)
3.5. Mesoporous Silica Nanoparticles
3.6. Protein or Peptide-Based Nanoparticles
3.7. Nucleic acid Aptamer–Based Nanoparticles
3.8. Bacteriophage phi29 Packaging RNA (pNRA)–Based Nanoparticles
4. Conclusions and Future Prospects
Acknowledgments
Conflict of Interest
References
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Zamore, P.D.; Tuschl, T.; Sharp, P.A.; Bartel, D.P. RNAi: double-stranded RNA directs the ATP-dependent cleavage of mRNA at 21 to 23 nucleotide intervals. Cell 2000, 101, 25–33. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar]
- Davidson, B.L.; McCray, P.B., Jr. Current prospects for RNA interference-based therapies. Nat. Rev. Genet. 2011, 12, 329–340. [Google Scholar] [CrossRef]
- Burnett, J.C.; Rossi, J.J. RNA-based therapeutics: current progress and future prospects. Chem. Biol. 2012, 19, 60–71. [Google Scholar] [CrossRef]
- Lares, M.R.; Rossi, J.J.; Ouellet, D.L. RNAi and small interfering RNAs in human disease therapeutic applications. Trends Biotechnol. 2010, 28, 570–579. [Google Scholar] [CrossRef]
- Dejneka, N.S.; Wan, S.; Bond, O.S.; Kornbrust, D.J.; Reich, S.J. Ocular biodistribution of bevasiranib following a single intravitreal injection to rabbit eyes. Mol. Vis. 2008, 14, 997–1005. [Google Scholar]
- Cho, W.G.; Albuquerque, R.J.; Kleinman, M.E.; Tarallo, V.; Greco, A.; Nozaki, M.; Green, M.G.; Baffi, J.Z.; Ambati, B.K.; De Falco, M.; Alexander, J.S.; Brunetti, A.; De Falco, S.; Ambati, J. Small interfering RNA-induced TLR3 activation inhibits blood and lymphatic vessel growth. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 7137–7142. [Google Scholar]
- Kleinman, M.E.; Yamada, K.; Takeda, A.; Chandrasekaran, V.; Nozaki, M.; Baffi, J.Z.; Albuquerque, R.J.; Yamasaki, S.; Itaya, M.; Pan, Y.; Appukuttan, B.; Gibbs, D.; Yang, Z.; Kariko, K.; Ambati, B.K.; Wilgus, T.A.; DiPietro, L.A.; Sakurai, E.; Zhang, K.; Smith, J.R.; Taylor, E.W.; Ambati, J. Sequence- and target-independent angiogenesis suppression by siRNA via TLR3. Nature 2008, 452, 591–597. [Google Scholar]
- Whitehead, K.A.; Langer, R.; Anderson, D.G. Knocking down barriers: advances in siRNA delivery. Nat. Rev. Drug Discov. 2009, 8, 129–138. [Google Scholar]
- Dominska, M.; Dykxhoorn, D.M. Breaking down the barriers: siRNA delivery and endosome escape. J. Cell Sci. 2010, 123, 1183–1189. [Google Scholar] [CrossRef]
- Levy-Nissenbaum, E.; Radovic-Moreno, A.F.; Wang, A.Z.; Langer, R.; Farokhzad, O.C. Nanotechnology and aptamers: applications in drug delivery. Trends Biotechnol. 2008, 26, 442–449. [Google Scholar] [CrossRef]
- Kim, S.; Kim, J.H.; Jeon, O.; Kwon, I.C.; Park, K. Engineered polymers for advanced drug delivery. Eur. J. Pharm. Biopharm. 2009, 71, 420–430. [Google Scholar] [CrossRef]
- Guo, P.; Haque, F.; Hallahan, B.; Reif, R.; Li, H. Uniqueness, advantages, challenges, solutions, and perspectives in therapeutics applying RNA nanotechnology. Nucleic Acid Ther. 2012, 22, 226–245. [Google Scholar]
- Shen, H.; Sun, T.; Ferrari, M. Nanovector delivery of siRNA for cancer therapy. Cancer Gene Ther. 2012, 19, 367–373. [Google Scholar]
- Lee, D.U.; Huang, W.; Rittenhouse, K.D.; Jessen, B. Retina Expression and Cross-Species Validation of Gene Silencing by PF-655, a Small Interfering RNA Against RTP801 for the Treatment of Ocular Disease. J. Ocul. Pharmacol. Ther. 2012. [Google Scholar]
- Sarret, P.; Dore-Savard, L.; Beaudet, N. Direct application of siRNA for in vivo pain research. Methods Mol. Biol. 2010, 623, 383–395. [Google Scholar] [CrossRef]
- Barik, S. Intranasal delivery of antiviral siRNA. Methods Mol. Biol. 2011, 721, 333–338. [Google Scholar] [CrossRef]
- DeVincenzo, J.; Lambkin-Williams, R.; Wilkinson, T.; Cehelsky, J.; Nochur, S.; Walsh, E.; Meyers, R.; Gollob, J.; Vaishnaw, A. A randomized, double-blind, placebo-controlled study of an RNAi-based therapy directed against respiratory syncytial virus. Proc. Natl. Acad. Sci. US A 2010, 107, 8800–8805. [Google Scholar]
- Juliano, R.; Alam, M.R.; Dixit, V.; Kang, H. Mechanisms and strategies for effective delivery of antisense and siRNA oligonucleotides. Nucleic Acids Res. 2008, 36, 4158–4171. [Google Scholar] [CrossRef]
- Perez-Martinez, F.C.; Guerra, J.; Posadas, I.; Cena, V. Barriers to non-viral vector-mediated gene delivery in the nervous system. Pharm. Res. 2011, 28, 1843–1858. [Google Scholar] [CrossRef]
- van de Water, F.M.; Boerman, O.C.; Wouterse, A.C.; Peters, J.G.; Russel, F.G.; Masereeuw, R. Intravenously administered short interfering RNA accumulates in the kidney and selectively suppresses gene function in renal proximal tubules. Drug Metab. Dispos. 2006, 34, 1393–1397. [Google Scholar] [CrossRef]
- Wang, J.; Lu, Z.; Wientjes, M.G.; Au, J.L. Delivery of siRNA therapeutics: barriers and carriers. Aaps. J. 2010, 12, 492–503. [Google Scholar] [CrossRef]
- Danquah, M.K.; Zhang, X.A.; Mahato, R.I. Extravasation of polymeric nanomedicines across tumor vasculature. Adv. Drug Deliv. Rev. 2011, 63, 623–639. [Google Scholar] [CrossRef]
- Yuan, F.; Dellian, M.; Fukumura, D.; Leunig, M.; Berk, D.A.; Torchilin, V.P.; Jain, R.K. Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res. 1995, 55, 3752–3756. [Google Scholar]
- Moghimi, S.M.; Hunter, A.C.; Murray, J.C. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol. Rev. 2001, 53, 283–318. [Google Scholar]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Greish, K. Enhanced permeability and retention of macromolecular drugs in solid tumors: a royal gate for targeted anticancer nanomedicines. J. Drug Target. 2007, 15, 457–464. [Google Scholar] [CrossRef]
- Jain, R.K. Transport of molecules in the tumor interstitium: a review. Cancer Res. 1987, 47, 3039–3051. [Google Scholar]
- Boucher, Y.; Baxter, L.T.; Jain, R.K. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: implications for therapy. Cancer Res. 1990, 50, 4478–4484. [Google Scholar]
- Perrault, S.D.; Chan, W.C. In vivo assembly of nanoparticle components to improve targeted cancer imaging. Proc. Natl. Acad. Sci. USA 2010, 107, 11194–11199. [Google Scholar] [CrossRef]
- Netti, P.A.; Berk, D.A.; Swartz, M.A.; Grodzinsky, A.J.; Jain, R.K. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000, 60, 2497–2503. [Google Scholar]
- Wong, C.; Stylianopoulos, T.; Cui, J.; Martin, J.; Chauhan, V.P.; Jiang, W.; Popovic, Z.; Jain, R.K.; Bawendi, M.G.; Fukumura, D. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc. Natl. Acad. Sci. USA 2011, 108, 2426–2431. [Google Scholar]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Gullotti, E.; Yeo, Y. Extracellularly activated nanocarriers: a new paradigm of tumor targeted drug delivery. Mol. Pharm. 2009, 6, 1041–1051. [Google Scholar] [CrossRef]
- Davis, M.E. The first targeted delivery of siRNA in humans via a self-assembling, cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol. Pharm. 2009, 6, 659–668. [Google Scholar] [CrossRef]
- Davis, M.E.; Zuckerman, J.E.; Choi, C.H.; Seligson, D.; Tolcher, A.; Alabi, C.A.; Yen, Y.; Heidel, J.D.; Ribas, A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464, 1067–1070. [Google Scholar]
- Schroeder, A.; Levins, C.G.; Cortez, C.; Langer, R.; Anderson, D.G. Lipid-based nanotherapeutics for siRNA delivery. J. Intern. Med. 2010, 267, 9–21. [Google Scholar] [CrossRef]
- Nguyen, J.; Szoka, F.C. Nucleic acid delivery: the missing pieces of the puzzle? Acc. Chem. Res. 2012, 45, 1153–1162. [Google Scholar] [CrossRef]
- Mukherjee, S.; Ghosh, R.N.; Maxfield, F.R. Endocytosis. Physiol. Rev. 1997, 77, 759–803. [Google Scholar]
- Kumari, S.; Mg, S.; Mayor, S. Endocytosis unplugged: multiple ways to enter the cell. Cell Res. 2010, 20, 256–275. [Google Scholar] [CrossRef]
- Varkouhi, A.K.; Scholte, M.; Storm, G.; Haisma, H.J. Endosomal escape pathways for delivery of biologicals. J. Control. Release 2011, 151, 220–228. [Google Scholar] [CrossRef]
- Boussif, O.; Lezoualc'h, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar]
- Cho, Y.W.; Kim, J.D.; Park, K. Polycation gene delivery systems: escape from endosomes to cytosol. J. Pharm. Pharmacol. 2003, 55, 721–734. [Google Scholar]
- Turner, J.J.; Ivanova, G.D.; Verbeure, B.; Williams, D.; Arzumanov, A.A.; Abes, S.; Lebleu, B.; Gait, M.J. Cell-penetrating peptide conjugates of peptide nucleic acids (PNA) as inhibitors of HIV-1 Tat-dependent trans-activation in cells. Nucleic Acids Res. 2005, 33, 6837–6849. [Google Scholar] [CrossRef]
- Shiraishi, T.; Pankratova, S.; Nielsen, P.E. Calcium ions effectively enhance the effect of antisense peptide nucleic acids conjugated to cationic tat and oligoarginine peptides. Chem. Biol. 2005, 12, 923–929. [Google Scholar] [CrossRef]
- Abes, S.; Williams, D.; Prevot, P.; Thierry, A.; Gait, M.J.; Lebleu, B. Endosome trapping limits the efficiency of splicing correction by PNA-oligolysine conjugates. J. Control. Release 2006, 110, 595–604. [Google Scholar]
- Shiraishi, T.; Nielsen, P.E. Photochemically enhanced cellular delivery of cell penetrating peptide-PNA conjugates. FEBS Lett. 2006, 580, 1451–1456. [Google Scholar] [CrossRef]
- Matsushita-Ishiodori, Y.; Ohtsuki, T. Photoinduced RNA interference. Acc. Chem. Res. 2012, 45, 1039–1047. [Google Scholar] [CrossRef]
- Blidner, R.A.; Svoboda, K.R.; Hammer, R.P.; Monroe, W.T. Photoinduced RNA interference using DMNPE-caged 2'-deoxy-2'-fluoro substituted nucleic acids in vitro and in vivo. Mol. Biosyst. 2008, 4, 431–440. [Google Scholar] [CrossRef]
- Endoh, T.; Sisido, M.; Ohtsuki, T. Photo inducible RNA interference using cell permeable protein carrier. Nucleic Acids Symp. Ser. (Oxf.) 2007, 127–128. [Google Scholar]
- Endoh, T.; Sisido, M.; Ohtsuki, T. Cellular siRNA delivery mediated by a cell-permeant RNA-binding protein and photoinduced RNA interference. Bioconjug. Chem. 2008, 19, 1017–1024. [Google Scholar] [CrossRef]
- Endoh, T.; Sisido, M.; Ohtsuki, T. Spatial regulation of specific gene expression through photoactivation of RNAi. J. Control. Release 2009, 137, 241–245. [Google Scholar] [CrossRef]
- Braun, G.B.; Pallaoro, A.; Wu, G.; Missirlis, D.; Zasadzinski, J.A.; Tirrell, M.; Reich, N.O. Laser-Activated Gene Silencing via Gold Nanoshell-siRNA Conjugates. ACS Nano 2009, 3, 2007–2015. [Google Scholar]
- Hammond, S.M.; Boettcher, S.; Caudy, A.A.; Kobayashi, R.; Hannon, G.J. Argonaute2, a link between genetic and biochemical analyses of RNAi. Science 2001, 293, 1146–1150. [Google Scholar] [CrossRef]
- Liu, J.; Carmell, M.A.; Rivas, F.V.; Marsden, C.G.; Thomson, J.M.; Song, J.J.; Hammond, S.M.; Joshua-Tor, L.; Hannon, G.J. Argonaute2 is the catalytic engine of mammalian RNAi. Science 2004, 305, 1437–1441. [Google Scholar]
- Sakurai, K.; Amarzguioui, M.; Kim, D.H.; Alluin, J.; Heale, B.; Song, M.S.; Gatignol, A.; Behlke, M.A.; Rossi, J.J. A role for human Dicer in pre-RISC loading of siRNAs. Nucleic Acids Res. 2011, 39, 1510–1525. [Google Scholar] [CrossRef]
- Matranga, C.; Tomari, Y.; Shin, C.; Bartel, D.P.; Zamore, P.D. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell 2005, 123, 607–620. [Google Scholar] [CrossRef]
- Gibbings, D.J.; Ciaudo, C.; Erhardt, M.; Voinnet, O. Multivesicular bodies associate with components of miRNA effector complexes and modulate miRNA activity. Nat. Cell Biol. 2009, 11, 1143–1149. [Google Scholar] [CrossRef]
- Lee, Y.S.; Pressman, S.; Andress, A.P.; Kim, K.; White, J.L.; Cassidy, J.J.; Li, X.; Lubell, K.; Lim do, H.; Cho, I.S.; Nakahara, K.; Preall, J.B.; Bellare, P.; Sontheimer, E.J.; Carthew, R.W. Silencing by small RNAs is linked to endosomal trafficking. Nat. Cell Biol. 2009, 11, 1150–1156. [Google Scholar]
- Sen, G.L.; Blau, H.M. Argonaute 2/RISC resides in sites of mammalian mRNA decay known as cytoplasmic bodies. Nat. Cell Biol. 2005, 7, 633–636. [Google Scholar] [CrossRef]
- Rossi, J.J. RNAi and the P-body connection. Nat. Cell Biol. 2005, 7, 643–644. [Google Scholar] [CrossRef]
- Meister, G.; Landthaler, M.; Patkaniowska, A.; Dorsett, Y.; Teng, G.; Tuschl, T. Human Argonaute2 mediates RNA cleavage targeted by miRNAs and siRNAs. Mol. Cell 2004, 15, 185–197. [Google Scholar] [CrossRef]
- Davis, M.E.; Chen, Z.G.; Shin, D.M. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat. Rev. Drug Discov. 2008, 7, 771–782. [Google Scholar]
- Petros, R.A.; DeSimone, J.M. Strategies in the design of nanoparticles for therapeutic applications. Nat. Rev. Drug Discov. 2010, 9, 615–627. [Google Scholar]
- Zhang, L.; Gu, F.X.; Chan, J.M.; Wang, A.Z.; Langer, R.S.; Farokhzad, O.C. Nanoparticles in medicine: therapeutic applications and developments. Clin. Pharmacol. Ther. 2008, 83, 761–769. [Google Scholar] [CrossRef]
- Gomes-da-Silva, L.C.; Fonseca, N.A.; Moura, V.; Pedroso de Lima, M.C.; Simoes, S.; Moreira, J.N. Lipid-based nanoparticles for siRNA delivery in cancer therapy: paradigms and challenges. Acc. Chem. Res. 2012, 45, 1163–1171. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Adler-Moore, J.; Proffitt, R.T. AmBisome: liposomal formulation, structure, mechanism of action and pre-clinical experience. J. Antimicrob. Chemother. 2002, 49 Suppl 1, 21–30. [Google Scholar] [CrossRef]
- Hoekstra, D.; Rejman, J.; Wasungu, L.; Shi, F.; Zuhorn, I. Gene delivery by cationic lipids: in and out of an endosome. Biochem. Soc. Trans. 2007, 35, 68–71. [Google Scholar] [CrossRef]
- Jeong, J.H.; Park, T.G.; Kim, S.H. Self-assembled and nanostructured siRNA delivery systems. Pharm. Res. 2011, 28, 2072–2085. [Google Scholar] [CrossRef]
- Uchida, E.; Mizuguchi, H.; Ishii-Watabe, A.; Hayakawa, T. Comparison of the efficiency and safety of non-viral vector-mediated gene transfer into a wide range of human cells. Biol. Pharm. Bull. 2002, 25, 891–897. [Google Scholar] [CrossRef]
- Balazs, D.A.; Godbey, W. Liposomes for use in gene delivery. J. Drug Deliv. 2011, 2011, 326497. [Google Scholar]
- Schafer, J.; Hobel, S.; Bakowsky, U.; Aigner, A. Liposome-polyethylenimine complexes for enhanced DNA and siRNA delivery. Biomaterials 2010, 31, 6892–6900. [Google Scholar] [CrossRef]
- Sakurai, Y.; Hatakeyama, H.; Sato, Y.; Akita, H.; Takayama, K.; Kobayashi, S.; Futaki, S.; Harashima, H. Endosomal escape and the knockdown efficiency of liposomal-siRNA by the fusogenic peptide shGALA. Biomaterials 2011, 32, 5733–5742. [Google Scholar] [CrossRef]
- Rossi, J.J. RNAi therapeutics: SNALPing siRNAs in vivo. Gene Ther. 2006, 13, 583–584. [Google Scholar] [CrossRef]
- Frank-Kamenetsky, M.; Grefhorst, A.; Anderson, N.N.; Racie, T.S.; Bramlage, B.; Akinc, A.; Butler, D.; Charisse, K.; Dorkin, R.; Fan, Y.; et al. Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 11915–11920. [Google Scholar]
- Landesman, Y.; Svrzikapa, N.; Cognetta, A., 3rd; Zhang, X.; Bettencourt, B.R.; Kuchimanchi, S.; Dufault, K.; Shaikh, S.; Gioia, M.; Akinc, A.; Hutabarat, R.; Meyers, R. In vivo quantification of formulated and chemically modified small interfering RNA by heating-in-Triton quantitative reverse transcription polymerase chain reaction (HIT qRT-PCR). Silence 2010, 1, 16. [Google Scholar] [CrossRef]
- Aleku, M.; Schulz, P.; Keil, O.; Santel, A.; Schaeper, U.; Dieckhoff, B.; Janke, O.; Endruschat, J.; Durieux, B.; Roder, N.; Loffler, K.; Lange, C.; Fechtner, M.; Mopert, K.; Fisch, G.; Dames, S.; Arnold, W.; Jochims, K.; Giese, K.; Wiedenmann, B.; Scholz, A.; Kaufmann, J. Atu027, a liposomal small interfering RNA formulation targeting protein kinase N3, inhibits cancer progression. Cancer Res. 2008, 68, 9788–9798. [Google Scholar]
- Strumberg, D.; Schultheis, B.; Traugott, U.; Vank, C.; Santel, A.; Keil, O.; Giese, K.; Kaufmann, J.; Drevs, J. Phase I clinical development of Atu027, a siRNA formulation targeting PKN3 in patients with advanced solid tumors. Int. J. Clin. Pharmacol. Ther. 2012, 50, 76–78. [Google Scholar]
- Boas, U.; Heegaard, P.M. Dendrimers in drug research. Chem. Soc. Rev. 2004, 33, 43–63. [Google Scholar]
- Majoros, I.J.; Williams, C.R.; Becker, A.; Baker, J.R., Jr. Methotrexate delivery via folate targeted dendrimer-based nanotherapeutic platform. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2009, 1, 502–510. [Google Scholar] [CrossRef]
- Singha, K.; Namgung, R.; Kim, W.J. Polymers in Small-Interfering RNA Delivery. Oligonucleotides 2011. [Google Scholar]
- Winkler, J. Nanomedicines based on recombinant fusion proteins for targeting therapeutic siRNA oligonucleotides. Ther. Deliv. 2011, 2, 891–905. [Google Scholar] [CrossRef]
- Perez-Martinez, F.C.; Ocana, A.V.; Perez-Carrion, M.D.; Cena, V. Dendrimers as vectors for genetic material delivery to the nervous system. Curr. Med. Chem. 2012, 19, 5101–5108. [Google Scholar] [CrossRef]
- Kaneshiro, T.L.; Lu, Z.R. Targeted intracellular codelivery of chemotherapeutics and nucleic acid with a well-defined dendrimer-based nanoglobular carrier. Biomaterials 2009, 30, 5660–5666. [Google Scholar] [CrossRef]
- Shen, X.C.; Zhou, J.; Liu, X.; Wu, J.; Qu, F.; Zhang, Z.L.; Pang, D.W.; Quelever, G.; Zhang, C.C.; Peng, L. Importance of size-to-charge ratio in construction of stable and uniform nanoscale RNA/dendrimer complexes. Org. Biomol. Chem. 2007, 5, 3674–3681. [Google Scholar]
- Zhou, J.; Neff, C.P.; Liu, X.; Zhang, J.; Li, H.; Smith, D.D.; Swiderski, P.; Aboellail, T.; Huang, Y.; Du, Q.; Liang, Z.; Peng, L.; Akkina, R.; Rossi, J.J. Systemic Administration of Combinatorial dsiRNAs via Nanoparticles Efficiently Suppresses HIV-1 Infection in Humanized Mice. Mol. Ther. 2011, 2228–2238. [Google Scholar]
- Agrawal, A.; Min, D.H.; Singh, N.; Zhu, H.; Birjiniuk, A.; von Maltzahn, G.; Harris, T.J.; Xing, D.; Woolfenden, S.D.; Sharp, P.A.; Charest, A.; Bhatia, S. Functional delivery of siRNA in mice using dendriworms. ACS Nano 2009, 3, 2495–2504. [Google Scholar] [CrossRef]
- Kim, S.H.; Jeong, J.H.; Lee, S.H.; Kim, S.W.; Park, T.G. LHRH receptor-mediated delivery of siRNA using polyelectrolyte complex micelles self-assembled from siRNA-PEG-LHRH conjugate and PEI. Bioconjug. Chem. 2008, 19, 2156–2162. [Google Scholar] [CrossRef]
- Yuan, Q.; Lee, E.; Yeudall, W.A.; Yang, H. Dendrimer-triglycine-EGF nanoparticles for tumor imaging and targeted nucleic acid and drug delivery. Oral Oncol. 2010, 46, 698–704. [Google Scholar] [CrossRef]
- Pun, S.H.; Bellocq, N.C.; Liu, A.; Jensen, G.; Machemer, T.; Quijano, E.; Schluep, T.; Wen, S.; Engler, H.; Heidel, J.; Davis, M.E. Cyclodextrin-modified polyethylenimine polymers for gene delivery. Bioconjug. Chem. 2004, 15, 831–840. [Google Scholar] [CrossRef]
- Davis, M.E.; Brewster, M.E. Cyclodextrin-based pharmaceutics: past, present and future. Nat. Rev. Drug Discov. 2004, 3, 1023–1035. [Google Scholar] [CrossRef]
- Davis, M.E. Design and development of IT-101, a cyclodextrin-containing polymer conjugate of camptothecin. Adv. Drug Deliv. Rev. 2009, 61, 1189–1192. [Google Scholar] [CrossRef]
- Alabi, C.; Vegas, A.; Anderson, D. Attacking the genome: emerging siRNA nanocarriers from concept to clinic. Curr. Opin. Pharmacol. 2012, 12, 427–433. [Google Scholar] [CrossRef]
- Kang, J.H.; Tachibana, Y.; Kamata, W.; Mahara, A.; Harada-Shiba, M.; Yamaoka, T. Liver-targeted siRNA delivery by polyethylenimine (PEI)-pullulan carrier. Bioorg. Med. Chem. 2010, 18, 3946–3950. [Google Scholar]
- Hobel, S.; Koburger, I.; John, M.; Czubayko, F.; Hadwiger, P.; Vornlocher, H.P.; Aigner, A. Polyethylenimine/small interfering RNA-mediated knockdown of vascular endothelial growth factor in vivo exerts anti-tumor effects synergistically with Bevacizumab. J. Gene Med. 2010, 12, 287–300. [Google Scholar]
- Schiffelers, R.M.; Ansari, A.; Xu, J.; Zhou, Q.; Tang, Q.; Storm, G.; Molema, G.; Lu, P.Y.; Scaria, P.V.; Woodle, M.C. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res. 2004, 32, e149. [Google Scholar]
- Biswal, B.K.; Debata, N.B.; Verma, R.S. Development of a targeted siRNA delivery system using FOL-PEG-PEI conjugate. Mol. Biol. Rep. 2010, 37, 2919–2926. [Google Scholar] [CrossRef]
- Nie, C.; Liu, C.; Chen, G.; Dai, J.; Li, H.; Shuai, X. Hepatocyte-targeted psiRNA delivery mediated by galactosylated poly(ethylene glycol)-graft-polyethylenimine in vitro. J. Biomater. Appl. 2011, 26, 255–275. [Google Scholar] [CrossRef]
- Moghimi, S.M.; Symonds, P.; Murray, J.C.; Hunter, A.C.; Debska, G.; Szewczyk, A. A two-stage poly(ethylenimine)-mediated cytotoxicity: implications for gene transfer/therapy. Mol. Ther. 2005, 11, 990–995. [Google Scholar] [CrossRef]
- Li, X.; Chen, Y.; Wang, M.; Ma, Y.; Xia, W.; Gu, H. A mesoporous silica nanoparticle - PEI - Fusogenic peptide system for siRNA delivery in cancer therapy. Biomaterials 2013, 34, 1391–1401. [Google Scholar] [CrossRef]
- Hom, C.; Lu, J.; Liong, M.; Luo, H.; Li, Z.; Zink, J.I.; Tamanoi, F. Mesoporous silica nanoparticles facilitate delivery of siRNA to shutdown signaling pathways in mammalian cells. Small 2010, 6, 1185–1190. [Google Scholar] [CrossRef]
- Breccia, M.; Lo-Coco, F. Gemtuzumab ozogamicin for the treatment of acute promyelocytic leukemia: mechanisms of action and resistance, safety and efficacy. Expert Opin. Biol. Ther. 2011, 11, 225–234. [Google Scholar] [CrossRef]
- Yao, Y.D.; Sun, T.M.; Huang, S.Y.; Dou, S.; Lin, L.; Chen, J.N.; Ruan, J.B.; Mao, C.Q.; Yu, F.Y.; Zeng, M.S.; Zang, J.Y.; Liu, Q.; Su, F.X.; Zhang, P.; Lieberman, J.; Wang, J.; Song, E. Targeted Delivery of PLK1-siRNA by ScFv Suppresses Her2+ Breast Cancer Growth and Metastasis. Sci. Transl. Med. 2012, 4, 130ra148. [Google Scholar]
- Song, E.; Zhu, P.; Lee, S.K.; Chowdhury, D.; Kussman, S.; Dykxhoorn, D.M.; Feng, Y.; Palliser, D.; Weiner, D.B.; Shankar, P.; Marasco, W.A.; Lieberman, J. Antibody mediated in vivo delivery of small interfering RNAs via cell-surface receptors. Nat. Biotechnol. 2005, 23, 709–717. [Google Scholar]
- Kim, S.S.; Subramanya, S.; Peer, D.; Shimaoka, M.; Shankar, P. Antibody-mediated delivery of siRNAs for anti-HIV therapy. Methods Mol. Biol. 2011, 721, 339–353. [Google Scholar]
- Kumar, P.; Ban, H.S.; Kim, S.S.; Wu, H.; Pearson, T.; Greiner, D.L.; Laouar, A.; Yao, J.; Haridas, V.; Habiro, K.; Yang, Y.G.; Jeong, J.H.; Lee, K.Y.; Kim, Y.H.; Kim, S.W.; Peipp, M.; Fey, G.H.; Manjunath, N.; Shultz, L.D.; Lee, S.K.; Shankar, P. T cell-specific siRNA delivery suppresses HIV-1 infection in humanized mice. Cell 2008, 134, 577–586. [Google Scholar]
- Gump, J.M.; Dowdy, S.F. TAT transduction: the molecular mechanism and therapeutic prospects. Trends Mol. Med. 2007, 13, 443–448. [Google Scholar] [CrossRef]
- Lundberg, P.; El-Andaloussi, S.; Sutlu, T.; Johansson, H.; Langel, U. Delivery of short interfering RNA using endosomolytic cell-penetrating peptides. FASEB J. 2007, 21, 2664–2671. [Google Scholar] [CrossRef]
- Sugahara, K.N.; Teesalu, T.; Karmali, P.P.; Kotamraju, V.R.; Agemy, L.; Girard, O.M.; Hanahan, D.; Mattrey, R.F.; Ruoslahti, E. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell 2009, 16, 510–520. [Google Scholar] [CrossRef]
- Feron, O. Tumor-penetrating peptides: a shift from magic bullets to magic guns. Sci. Transl. Med. 2010, 2, 34ps26. [Google Scholar] [CrossRef]
- Dupont, E.; Prochiantz, A.; Joliot, A. Penetratin story: an overview. Methods Mol. Biol. 2011, 683, 21–29. [Google Scholar] [CrossRef]
- Berry, C.C. Intracellular delivery of nanoparticles via the HIV-1 tat peptide. Nanomedicine (Lond.) 2008, 3, 357–365. [Google Scholar] [CrossRef]
- Endoh, T.; Ohtsuki, T. Cellular siRNA delivery using cell-penetrating peptides modified for endosomal escape. Adv. Drug Deliv. Rev. 2009, 61, 704–709. [Google Scholar] [CrossRef]
- Xiong, X.B.; Lavasanifar, A. Traceable multifunctional micellar nanocarriers for cancer-targeted co-delivery of MDR-1 siRNA and doxorubicin. ACS Nano 2011, 5, 5202–5213. [Google Scholar] [CrossRef]
- Leng, Q.; Scaria, P.; Zhu, J.; Ambulos, N.; Campbell, P.; Mixson, A.J. Highly branched HK peptides are effective carriers of siRNA. J. Gene Med. 2005, 7, 977–986. [Google Scholar] [CrossRef]
- Zhou, J.; Rossi, J.J. Aptamer-targeted cell-specific RNA interference. Silence 2010, 1, 4. [Google Scholar] [CrossRef]
- Tuerk, C.; MacDougal, S.; Gold, L. RNA pseudoknots that inhibit human immunodeficiency virus type 1 reverse transcriptase. Proc. Natl. Acad. Sci. USA 1992, 89, 6988–6992. [Google Scholar] [CrossRef]
- Famulok, M.; Mayer, G. Aptamers as tools in molecular biology and immunology. Curr. Top Microbiol. Immunol. 1999, 243, 123–136. [Google Scholar]
- Dassie, J.P.; Liu, X.Y.; Thomas, G.S.; Whitaker, R.M.; Thiel, K.W.; Stockdale, K.R.; Meyerholz, D.K.; McCaffrey, A.P.; McNamara, J.O., 2nd; Giangrande, P.H. Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat. Biotechnol. 2009, 27, 839–849. [Google Scholar]
- Zhou, J.; Li, H.; Li, S.; Zaia, J.; Rossi, J.J. Novel dual inhibitory function aptamer-siRNA delivery system for HIV-1 therapy. Mol. Ther. 2008, 16, 1481–1489. [Google Scholar] [CrossRef]
- Zhou, J.; Swiderski, P.; Li, H.; Zhang, J.; Neff, C.P.; Akkina, R.; Rossi, J.J. Selection, characterization and application of new RNA HIV gp 120 aptamers for facile delivery of Dicer substrate siRNAs into HIV infected cells. Nucleic Acids Res. 2009, 37, 3094–3109. [Google Scholar]
- Thiel, K.W.; Hernandez, L.I.; Dassie, J.P.; Thiel, W.H.; Liu, X.; Stockdale, K.R.; Rothman, A.M.; Hernandez, F.J.; McNamara, J.O., 2nd; Giangrande, P.H. Delivery of chemo-sensitizing siRNAs to HER2+-breast cancer cells using RNA aptamers. Nucleic Acids Res. 2012, 40, 6319–6337. [Google Scholar]
- McNamara, J.O., 2nd; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef]
- Zhao, N.; Bagaria, H.G.; Wong, M.S.; Zu, Y. A nanocomplex that is both tumor cell-selective and cancer gene-specific for anaplastic large cell lymphoma. J. Nanobiotechnology 2011, 9, 2. [Google Scholar]
- Kim, E.; Jung, Y.; Choi, H.; Yang, J.; Suh, J.S.; Huh, Y.M.; Kim, K.; Haam, S. Prostate cancer cell death produced by the co-delivery of Bcl-xL shRNA and doxorubicin using an aptamer-conjugated polyplex. Biomaterials 2010, 31, 4592–4599. [Google Scholar] [CrossRef]
- Bagalkot, V.; Gao, X. siRNA-aptamer chimeras on nanoparticles: preserving targeting functionality for effective gene silencing. ACS Nano 2011, 5, 8131–8139. [Google Scholar] [CrossRef]
- Ye, X.; Hemida, M.; Zhang, H.M.; Hanson, P.; Ye, Q.; Yang, D. Current advances in Phi29 pRNA biology and its application in drug delivery. Wiley Interdiscip. Rev. RNA 2012. [Google Scholar]
- Guo, P. The emerging field of RNA nanotechnology. Nat. Nanotechnol. 2010, 5, 833–842. [Google Scholar] [CrossRef]
- Tarapore, P.; Shu, Y.; Guo, P.; Ho, S.M. Application of phi29 motor pRNA for targeted therapeutic delivery of siRNA silencing metallothionein-IIA and survivin in ovarian cancers. Mol. Ther. 2011, 19, 386–394. [Google Scholar] [CrossRef]
- Liu, J.; Guo, S.; Cinier, M.; Shlyakhtenko, L.S.; Shu, Y.; Chen, C.; Shen, G.; Guo, P. Fabrication of stable and RNase-resistant RNA nanoparticles active in gearing the nanomotors for viral DNA packaging. ACS Nano 2011, 5, 237–246. [Google Scholar] [CrossRef]
- Zhou, J.; Shu, Y.; Guo, P.; Smith, D.D.; Rossi, J.J. Dual functional RNA nanoparticles containing phi29 motor pRNA and anti-gp120 aptamer for cell-type specific delivery and HIV-1 inhibition. Methods 2011, 54, 284–294. [Google Scholar] [CrossRef]
- Shu, D.; Shu, Y.; Haque, F.; Abdelmawla, S.; Guo, P. Thermodynamically stable RNA three-way junction for constructing multifunctional nanoparticles for delivery of therapeutics. Nat. Nanotechnol. 2011, 6, 658–667. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhou, J.; Shum, K.-T.; Burnett, J.C.; Rossi, J.J. Nanoparticle-Based Delivery of RNAi Therapeutics: Progress and Challenges. Pharmaceuticals 2013, 6, 85-107. https://doi.org/10.3390/ph6010085
Zhou J, Shum K-T, Burnett JC, Rossi JJ. Nanoparticle-Based Delivery of RNAi Therapeutics: Progress and Challenges. Pharmaceuticals. 2013; 6(1):85-107. https://doi.org/10.3390/ph6010085
Chicago/Turabian StyleZhou, Jiehua, Ka-To Shum, John C. Burnett, and John J. Rossi. 2013. "Nanoparticle-Based Delivery of RNAi Therapeutics: Progress and Challenges" Pharmaceuticals 6, no. 1: 85-107. https://doi.org/10.3390/ph6010085
APA StyleZhou, J., Shum, K.-T., Burnett, J. C., & Rossi, J. J. (2013). Nanoparticle-Based Delivery of RNAi Therapeutics: Progress and Challenges. Pharmaceuticals, 6(1), 85-107. https://doi.org/10.3390/ph6010085