The Potential Role of Cell Penetrating Peptides in the Intracellular Delivery of Proteins for Therapy of Erythroid Related Disorders

Abstract
:
1. Introduction
2. Genetic and Metabolic ERDs and current Treatment
2.1. Sideroblastic Anemias
2.2. Porphyrias
2.3. Hemoglobinopathies
2.3.1. Thalassemias
2.3.2. Sickle Cell Anemia
3. Protein Transduction Domain Technology for intracellular DELIVERY of Protein Therapeutics (PTs)

{kind=link}
| CPP | Origin | Aminoacid (aa) Sequence /Physicochemical nature | Length(aa) | Refs |
|---|---|---|---|---|
| TAT | HIV-1 TAT (transactivator factor of transcription) | YGRKKRRQRRR Cationic peptide | 11 | [73,81] |
| Penetratin (Antp) | Drosophila homeotic transcription factor encoded by antennapedia gene | RQIKIWFQNRRMKWKK Amphipathic pepttide | 16 | [72] |
| VP22 | herpes simplex virus VP22 transcription factor | DAATATRGRSAASRPTERPRAPARSASRPRRPVD Amphipathic peptide | 35 | [82] |
| poly- Arginines | chemically synthesized | R7, R8, R9 Cationic peptides | 9 / 8 | [83,84] |
| Transportan | galanin -mastoparan | GWTLNSAGYLLGK-INLKALAALAKKIL Chimeric - Amphipathic peptide | 27 | [85] |
| TP10 | truncated form ofTransportan | AGYLLGKINLKALAALAKKIL Chimeric - Amphipathic peptide | 21 | [86] |
| pVEC | Vascular Endothelial (Ve) - Cadherin | LLIILRRRIRKQAHAHSK Amphipathic peptide | 18 | [87] |
| Pep-1 | Trp-rich motif-SV40 NLS | KETWWETWWTEWSQPKKKRKV Chimeric - Amphipathic peptide | 21 | [88] |
| C105Y | peptide based on the residues 359-374 of alpha1-antitrypsin | CSIPPEVKFNKPFVYLI Amphipathic peptide | 17 | [89] |
| PFVYLI | derived from the synthetic peptide C105Y | PFVYLI Hydrophobic peptide | 6 | [89] |
| CADY | chemically synthesized, combining aromatic (W) and cationic (R) residues | GLWRALWRLLRSLWRLLWRA Amphipathic peptide | 20 | [90] |
| CAPHs | chemically synthesized with cationic amphiphilic polyproline helices | P11LRR to P14LRR Cationic - Amphipathic peptides | 14 to 17 | [91] |
3.1. Production of PTs via PTD Technology
3.2. Pharmacokinetic, Toxicity and Targeting of CPP-Mediated PTs
| Obstacles of CPP-mediated PTs | Possible solution of the obstacles | Refs |
|---|---|---|
| lack of tissue specificity | Development of “smart” delivery platforms | [95] |
| Use of target-specific antibodies, attached via bonds sensitive to cell environmental or external stimulus conditions | [95,104] | |
| Use of Homing peptides | [110,111] | |
| Development of tissue specific CPPs | [112] | |
| protein instability | Exchange L-amino acids of CPPs with the corresponding D-amino acids | [113] |
| Polymerization technologies (protein PEGylation) | [114] | |
| Construction of liposomes and nanoparticles | [95] | |
| immunogenicity | de-immunization by substitution of key aminoacids of T- or B-cell epitopes on CPP-mediated PT, using computer algorithms | [115,116] |
| Dosage / Route of administration (topical / i.v.) | [117] |
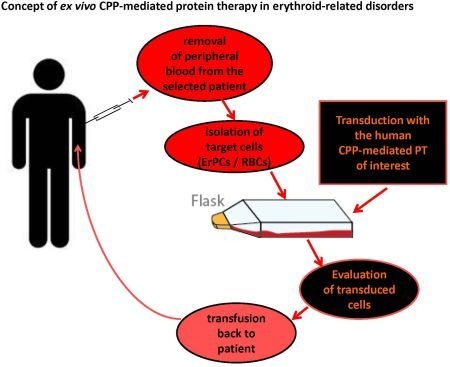
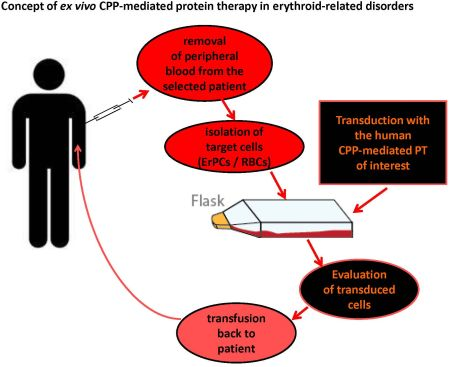
3.3. Targeted Intracellular Delivery of CPP-Mediated PTs in ErPCs and RBCs:Future ex vivo Therapy
| Biopharmaceutical Company | Product | CPP-cargo | Indication | Clinical Phase | Refs |
|---|---|---|---|---|---|
| CellGate, Inc. | PsorBan | R7-cyclosporin A | topical treatment of psoriasis | II (discontinued) | [126] |
| Revance Therapeutics, Inc. | RT-001 | TAT-botulinum toxin (TransMTS TM platform technology) | topical treatment of wrinkles and excessive sweating | IIb I | [95,124] |
| Capstone Therapeutics | AZX-100 | YARAAARQARA - HSP20 phosphopeptide | excessive dermal/keloid scarring and fibrotic disorders | II | [127] |
| KAI Pharmaceuticals | KIA-9803 KIA-1678 KIA-1455 | TAT-protein kinase Cδ inhibitor TAT-protein kinase Cε inhibitor TAT-protein kinase Cε activator | myocardial infarction pain cytoprotection ischemia | I/II | [95,124] |
| Avi Biopharma | AVI-5038 | (R-Ahx1-R)4 AhxB –PMO2 | Duchenne Muscular Dystrophy/viral infections | Pre-clinical studies in mouse | [128] |
| Centro Nazionale AIDS-Istituto Superiore di Sanità -NovartisVaccines | Trial ISS P-002 | TAT- V2-deleted Env proteins | HIV Infection | I | [129] |
| Traversa Therapeutics and Sanofi-Aventis | PTD–DRBD | PTD-DRBD3- siRNA | RNAi delivery | Pre-clinical studies | [130,131] |
| Xigen Pharm (Epalinges, Swiss) | XG-102 (D-JNKI-1) | TAT- JNK-inhibiting peptide | inflammatory bowel disease | Pre-clinical studies | [132] |
| Diatos – Drais Pharmaceuticals | DTS-108 | DPV10474- SN38 (activemetabolite ofirinotecan) | cancer treatment | I | [133] |
4. Conclusions and Perspectives
Abbreviations
| aa | aminoacid |
| ALAS | ALA Synthase |
| BFU-Es | burst-forming units-erythroid |
| BM | bone marrow |
| CDS | coding sequence |
| CEP | congenital erythropoietic porphyria |
| CFU-Es | colony-forming units-erythroid |
| CGAs | congenital anemias |
| CPP | Cell Penetrating Peptide |
| CPP-mediated PTs | PTs engineered via PTD Technology |
| ErPCs | Erythroid progenitor cells |
| ERDs | Erythroid related disorders |
| ERT | enzyme replacement therapy |
| EPO | erythropoietin |
| EPP | erythropoietic protoporphyria |
| FA | Fanconi anemia |
| FECH | Ferrochelatase |
| GVHD | graft versus host disease |
| HbA | adult hemoglobin |
| HbF | fetal hemoglobin |
| HbS | hemoglobin S |
| HSCs | hematopoietic stem cells |
| HSCT | HSC transplantation |
| HU | Hydroxyurea |
| iPSCs | induced Pluripotent Stem Cells |
| PEG | polyethylene glycol |
| PRT | protein replacement therapy |
| PTD | protein transduction domain |
| PTs | proteins therapeutics |
| R | arginine |
| RBC | red blood cell |
| SCD | sickle cell disease |
| α | alpha |
| β | beta |
| γ | gamma |
| δ | delta |
Conflict of Interest
References
- Tsiftsoglou, A.S.; Vizirianakis, I.S.; Strouboulis, J. Erythropoiesis: Model systems, molecular regulators, and developmental program. IUBMB Life 2009, 61, 800–830. [Google Scholar] [CrossRef]
- Jelkmann, W. Regulation of erythropoietin production. J. Physiol. 2011, 589, 1251–1258. [Google Scholar] [CrossRef]
- Schechter, A.N. Hemoglobin research and the origins of molecular medicine. Blood 2008, 112, 3927–3938. [Google Scholar] [CrossRef]
- Higgs, D.R.; Engel, J.D.; Stamatoyannopoulos, G. Thalassaemia. Lancet 2012, 379, 373–383. [Google Scholar]
- An, X.; Mohandas, N. Disorders of red cell membrane. Br. J. Haematol. 2008, 141, 367–375. [Google Scholar]
- Foller, M.; Huber, S.M.; Lang, F. Erythrocyte programmed cell death. IUBMB Life 2008, 60, 661–668. [Google Scholar] [CrossRef]
- Voon, H.P.; Vadolas, J. Controlling alpha-globin: A review of alpha-globin expression and its impact on beta-thalassemia. Haematologica 2008, 93, 1868–1876. [Google Scholar] [CrossRef]
- Komar, A.A.; Kommer, A.; Krasheninnikov, I.A.; Spirin, A.S. Cotranslational folding of globin. J. Biol. Chem. 1997, 272, 10646–10651. [Google Scholar]
- Komar, A.A.; Kommer, A.; Krasheninnikov, I.A.; Spirin, A.S. Cotranslational heme binding to nascent globin chains. FEBS Lett. 1993, 326, 261–263. [Google Scholar] [CrossRef]
- Mollan, T.L.; Yu, X.; Weiss, M.J.; Olson, J.S. The role of alpha-hemoglobin stabilizing protein in redox chemistry, denaturation, and hemoglobin assembly. Antioxid Redox Signal. 2010, 12, 219–231. [Google Scholar] [CrossRef]
- Elder, G.H. Molecular genetics of disorders of haem biosynthesis. J. Clin. Pathol. 1993, 46, 977–981. [Google Scholar] [CrossRef]
- Tanno, T.; Miller, J.L. Iron loading and overloading due to ineffective erythropoiesis. Adv. Hematol. 2010, 2010, 358283. [Google Scholar]
- Wang, S.A.; Hasserjian, R.P. Erythroid proliferations in myeloid neoplasms. Hum. Pathol. 2012, 43, 153–164. [Google Scholar]
- Kalb, R.; Neveling, K.; Nanda, I.; Schindler, D.; Hoehn, H. Fanconi anemia: Causes and consequences of genetic instability. Genome Dyn. 2006, 1, 218–242. [Google Scholar]
- Xu, X.; Qu, J.; Suzuki, K.; Li, M.; Zhang, W.; Liu, G.H.; Izpisua Belmonte, J.C. Reprogramming based gene therapy for inherited red blood cell disorders. Cell. Res. 2012, 22, 941–944. [Google Scholar] [CrossRef]
- Storb, R.F.; Lucarelli, G.; McSweeney, P.A.; Childs, R.W. Hematopoietic cell transplantation for benign hematological disorders and solid tumors. Hematology Am. Soc. Hematol. Educ. Program. 2003, 372–397. [Google Scholar]
- Snyder, E.L.; Dowdy, S.F. Recent advances in the use of protein transduction domains for the delivery of peptides, proteins and nucleic acids in vivo. Expert Opin. Drug Deliv. 2005, 2, 43–51. [Google Scholar] [CrossRef]
- Papadopoulou, L.C.; Tsiftsoglou, A.S. Transduction of human recombinant proteins into mitochondria as a protein therapeutic approach for mitochondrial disorders. Pharm. Res. 2011, 28, 2639–2656. [Google Scholar] [CrossRef]
- Suzuki, Y. Exploring transduction mechanisms of protein transduction domains (ptds) in living cells utilizing single-quantum dot tracking (sqt) technology. Sensors (Basel) 2012, 12, 549–572. [Google Scholar] [CrossRef]
- Fleming, M.D. Congenital sideroblastic anemias: Iron and heme lost in mitochondrial translation. Hematology Am. Soc. Hematol. Educ. Program. 2011, 2011, 525–531. [Google Scholar] [CrossRef]
- Ajioka, R.S.; Phillips, J.D.; Kushner, J.P. Biosynthesis of heme in mammals. Biochim. Biophys. Acta 2006, 1763, 723–736. [Google Scholar] [CrossRef]
- Harigae, H.; Furuyama, K. Hereditary sideroblastic anemia: Pathophysiology and gene mutations. Int.J. Hematol. 2010, 92, 425–431. [Google Scholar] [CrossRef]
- Bishop, D.F.; Tchaikovskii, V.; Hoffbrand, A.V.; Fraser, M.E.; Margolis, S. X-linked sideroblastic anemia due to carboxyl-terminal alas2 mutations that cause loss of binding to the beta-subunit of succinyl-coa synthetase (sucla2). J. Biol. Chem. 2012, 287, 28943–28955. [Google Scholar]
- Tsiftsoglou, A.S.; Tsamadou, A.I.; Papadopoulou, L.C. Heme as key regulator of major mammalian cellular functions: Molecular, cellular, and pharmacological aspects. Pharmacol. Ther. 2006, 111, 327–345. [Google Scholar] [CrossRef]
- Fontenay, M.; Cathelin, S.; Amiot, M.; Gyan, E.; Solary, E. Mitochondria in hematopoiesis and hematological diseases. Oncogene 2006, 25, 4757–4767. [Google Scholar] [CrossRef]
- Cuijpers, M.L.; van Spronsen, D.J.; Muus, P.; Hamel, B.C.; Swinkels, D.W. Need for early recognition and therapeutic guidelines of congenital sideroblastic anaemia. Int. J. Hematol. 2011, 94, 97–100. [Google Scholar] [CrossRef]
- Baumann Kreuziger, L.M.; Wolanskyj, A.P.; Hanson, C.A.; Steensma, D.P. Lack of efficacy of pyridoxine (vitamin b6) treatment in acquired idiopathic sideroblastic anaemia, including refractory anaemia with ring sideroblasts. Eur. J. Haematol. 2011, 86, 512–516. [Google Scholar] [CrossRef]
- Guernsey, D.L.; Jiang, H.; Campagna, D.R.; Evans, S.C.; Ferguson, M.; Kellogg, M.D.; Lachance, M.; Matsuoka, M.; Nightingale, M.; Rideout, A.; Saint-Amant, L.; Schmidt, P.J.; Orr, A.; Bottomley, S.S.; Fleming, M.D.; Ludman, M.; Dyack, S.; Fernandez, C.V.; Samuels, M.E. Mutations in mitochondrial carrier family gene slc25a38 cause nonsyndromic autosomal recessive congenital sideroblastic anemia. Nat. Genet. 2009, 41, 651–653. [Google Scholar]
- Khan, A.A.; Quigley, J.G. Control of intracellular heme levels: Heme transporters and heme oxygenases. Biochim. Biophys. Acta 2011, 1813, 668–682. [Google Scholar] [CrossRef]
- Zutz, A.; Gompf, S.; Schagger, H.; Tampe, R. Mitochondrial abc proteins in health and disease. Biochim. Biophys. Acta 2009, 1787, 681–690. [Google Scholar] [CrossRef]
- Allikmets, R.; Raskind, W.H.; Hutchinson, A.; Schueck, N.D.; Dean, M.; Koeller, D.M. Mutation of a putative mitochondrial iron transporter gene (abc7) in x-linked sideroblastic anemia and ataxia (xlsa/a). Hum. Mol. Genet. 1999, 8, 743–749. [Google Scholar] [CrossRef]
- Ye, H.; Jeong, S.Y.; Ghosh, M.C.; Kovtunovych, G.; Silvestri, L.; Ortillo, D.; Uchida, N.; Tisdale, J.; Camaschella, C.; Rouault, T.A. Glutaredoxin 5 deficiency causes sideroblastic anemia by specifically impairing heme biosynthesis and depleting cytosolic iron in human erythroblasts. J. Clin. Invest. 2010, 120, 1749–1761. [Google Scholar] [CrossRef]
- Pearson, H.A.; Lobel, J.S.; Kocoshis, S.A.; Naiman, J.L.; Windmiller, J.; Lammi, A.T.; Hoffman, R.; Marsh, J.C. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J. Pediatr. 1979, 95, 976–984. [Google Scholar] [CrossRef]
- Rawles, J.M.; Weller, R.O. Familial association of metabolic myopathy, lactic acidosis and sideroblastic anemia. Am. J. Med. 1974, 56, 891–897. [Google Scholar] [CrossRef]
- Porter, F.S.; Rogers, L.E.; Sidbury, J.B., Jr. Thiamine-responsive megaloblastic anemia. J. Pediatr. 1969, 74, 494–504. [Google Scholar] [CrossRef]
- Siegesmund, M.; van Tuyll van Serooskerken, A.M.; Poblete-Gutierrez, P.; Frank, J. The acute hepatic porphyrias: Current status and future challenges. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 593–605. [Google Scholar] [CrossRef]
- Hift, R.J.; Thunell, S.; Brun, A. Drugs in porphyria: From observation to a modern algorithm-based system for the prediction of porphyrogenicity. Pharmacol.Ther. 2011, 132, 158–169. [Google Scholar] [CrossRef]
- Cappellini, M.D.; Brancaleoni, V.; Graziadei, G.; Tavazzi, D.; Di Pierro, E. Porphyrias at a glance: Diagnosis and treatment. Intern. Emerg. Med. 2010, 5 (Suppl. 1), S73–80. [Google Scholar]
- Hunter, G.A.; Ferreira, G.C. Molecular enzymology of 5-aminolevulinate synthase, the gatekeeper of heme biosynthesis. Biochim. Biophys. Acta 2011, 1814, 1467–1473. [Google Scholar] [CrossRef]
- Schmitt, C.; Ducamp, S.; Gouya, L.; Deybach, J.C.; Puy, H. [inheritance in erythropoietic protoporphyria]. Pathol. Biol. (Paris) 2010, 58, 372–380. [Google Scholar] [CrossRef]
- Phillips, J.D.; Steensma, D.P.; Pulsipher, M.A.; Spangrude, G.J.; Kushner, J.P. Congenital erythropoietic porphyria due to a mutation in gata1: The first trans-acting mutation causative for a human porphyria. Blood 2007, 109, 2618–2621. [Google Scholar] [CrossRef]
- May, B.K.; Dogra, S.C.; Sadlon, T.J.; Bhasker, C.R.; Cox, T.C.; Bottomley, S.S. Molecular regulation of heme biosynthesis in higher vertebrates. Prog. Nucleic. Acid. Res. Mol. Biol. 1995, 51, 1–51. [Google Scholar] [CrossRef]
- Simon, N.G.; Herkes, G.K. The neurologic manifestations of the acute porphyrias. J. Clin. Neurosci. 2011, 18, 1147–1153. [Google Scholar] [CrossRef]
- Perutz, M.F.; Lehmann, H. Molecular pathology of human haemoglobin. Nature 1968, 219, 902–909. [Google Scholar] [CrossRef]
- Angastiniotis, M.; Modell, B. Global epidemiology of hemoglobin disorders. Ann. N Y Acad. Sci. 1998, 850, 251–269. [Google Scholar] [CrossRef]
- Birgens, H.; Ljung, R. The thalassaemia syndromes. Scand. J. Clin. Lab. Invest. 2007, 67, 11–25. [Google Scholar] [CrossRef]
- Weatherall, D.J. Thalassemia as a global health problem: Recent progress toward its control in the developing countries. Ann. N Y Acad. Sci. 2010, 1202, 17–23. [Google Scholar] [CrossRef]
- Weatherall, D.J. Genetic variation and susceptibility to infection: The red cell and malaria. Br. J. Haematol. 2008, 141, 276–286. [Google Scholar] [CrossRef]
- Giardine, B.; van Baal, S.; Kaimakis, P.; Riemer, C.; Miller, W.; Samara, M.; Kollia, P.; Anagnou, N.P.; Chui, D.H.; Wajcman, H.; Hardison, R.C.; Patrinos, G.P. Hbvar database of human hemoglobin variants and thalassemia mutations: 2007 update. Hum. Mutat. 2007, 28, 206. [Google Scholar]
- Urbinati, F.; Madigan, C.; Malik, P. Pathophysiology and therapy for haemoglobinopathies. Part ii: Thalassaemias. Expert Rev. Mol. Med. 2006, 8, 1–26. [Google Scholar]
- Muncie, H.L., Jr.; Campbell, J. Alpha and beta thalassemia. Am. Fam. Physician 2009, 80, 339–344. [Google Scholar]
- Harteveld, C.L.; Higgs, D.R. Alpha-thalassaemia. Orphanet. J. Rare Dis. 2010, 5, 13. [Google Scholar] [CrossRef]
- Bourantas, K.; Economou, G.; Georgiou, J. Administration of high doses of recombinant human erythropoietin to patients with beta-thalassemia intermedia: A preliminary trial. Eur. J. Haematol. 1997, 58, 22–25. [Google Scholar]
- Gambari, R. Alternative options for DNA-based experimental therapy of beta-thalassemia. Expert Opin. Biol. Ther. 2012, 12, 443–462. [Google Scholar] [CrossRef]
- Stamatoyannopoulos, G. Control of globin gene expression during development and erythroid differentiation. Exp. Hematol. 2005, 33, 259–271. [Google Scholar] [CrossRef]
- Hankins, J.; Aygun, B. Pharmacotherapy in sickle cell disease--state of the art and future prospects. Br. J. Haematol 2009, 145, 296–308. [Google Scholar] [CrossRef]
- Ataga, K.I. Novel therapies in sickle cell disease. Hematology Am. Soc. Hematol. Educ. Program. 2009, 54–61. [Google Scholar] [CrossRef]
- Elborai, Y.; Uwumugambi, A.; Lehmann, L. Hematopoietic stem cell transplantation for thalassemia. Immunotherapy 2012, 4, 947–956. [Google Scholar] [CrossRef]
- Mogul, M.J. Unrelated cord blood transplantation vs matched unrelated donor bone marrow transplantation: The risks and benefits of each choice. Bone Marrow Transplant. 2000, 25 Suppl 2, S58–60. [Google Scholar] [CrossRef]
- Quek, L.; Thein, S.L. Molecular therapies in beta-thalassaemia. Br. J. Haematol. 2007, 136, 353–365. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Payen, E.; Negre, O.; Wang, G.; Hehir, K.; Fusil, F.; Down, J.; Denaro, M.; Brady, T.; Westerman, K.; Cavallesco, R.; Gillet-Legrand, B.; Caccavelli, L.; Sgarra, R.; Maouche-Chretien, L.; Bernaudin, F.; Girot, R.; Dorazio, R.; Mulder, G.J.; Polack, A.; Bank, A.; Soulier, J.; Larghero, J.; Kabbara, N.; Dalle, B.; Gourmel, B.; Socie, G.; Chretien, S.; Cartier, N.; Aubourg, P.; Fischer, A.; Cornetta, K.; Galacteros, F.; Beuzard, Y.; Gluckman, E.; Bushman, F.; Hacein-Bey-Abina, S.; Leboulch, P. Transfusion independence and hmga2 activation after gene therapy of human beta-thalassaemia. Nature 2010, 467, 318–322. [Google Scholar]
- Sarakul, O.; Vattanaviboon, P.; Wilairat, P.; Fucharoen, S.; Abe, Y.; Muta, K. Inhibition of alpha-globin gene expression by rnai. Biochem. Biophys. Res. Commun. 2008, 369, 935–938. [Google Scholar] [CrossRef]
- Vadolas, J.; Wardan, H.; Orford, M.; Williamson, R.; Ioannou, P.A. Cellular genomic reporter assays for screening and evaluation of inducers of fetal hemoglobin. Hum. Mol. Genet. 2004, 13, 223–233. [Google Scholar]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef]
- Boosalis, M.S.; Castaneda, S.A.; Trudel, M.; Mabaera, R.; White, G.L.; Lowrey, C.H.; Emery, D.W.; Mpollo, M.S.; Shen, L.; Wargin, W.A.; Bohacek, R.; Faller, D.V.; Perrine, S.P. Novel therapeutic candidates, identified by molecular modeling, induce gamma-globin gene expression in vivo. Blood Cells Mol. Dis. 2011, 47, 107–116. [Google Scholar] [CrossRef]
- Samakoglu, S.; Lisowski, L.; Budak-Alpdogan, T.; Usachenko, Y.; Acuto, S.; Di Marzo, R.; Maggio, A.; Zhu, P.; Tisdale, J.F.; Riviere, I.; Sadelain, M. A genetic strategy to treat sickle cell anemia by coregulating globin transgene expression and rna interference. Nat. Biotechnol. 2006, 24, 89–94. [Google Scholar] [CrossRef]
- Hanna, J.; Wernig, M.; Markoulaki, S.; Sun, C.W.; Meissner, A.; Cassady, J.P.; Beard, C.; Brambrink, T.; Wu, L.C.; Townes, T.M.; Jaenisch, R. Treatment of sickle cell anemia mouse model with ips cells generated from autologous skin. Science 2007, 318, 1920–1923. [Google Scholar] [CrossRef]
- Douay, L. In vitro generation of red blood cells for transfusion: A model for regenerative medicine. Regen. Med. 2012, 7, 1–2. [Google Scholar] [CrossRef]
- Migliaccio, A.R.; Masselli, E.; Varricchio, L.; Whitsett, C. Ex-vivo expansion of red blood cells: How real for transfusion in humans? Blood Rev. 2012, 26, 81–95. [Google Scholar] [CrossRef]
- Stephens, D.J.; Pepperkok, R. The many ways to cross the plasma membrane. Proc. Natl. Acad. Sci. USA 2001, 98, 4295–4298. [Google Scholar] [CrossRef]
- Fawell, S.; Seery, J.; Daikh, Y.; Moore, C.; Chen, L.L.; Pepinsky, B.; Barsoum, J. Tat-mediated delivery of heterologous proteins into cells. Proc. Natl. Acad. Sci. USA 1994, 91, 664–668. [Google Scholar] [CrossRef]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Brasseur, R.; Divita, G. Happy birthday cell penetrating peptides: Already 20years. Biochim. Biophys. Acta 2010, 1798, 2177–2181. [Google Scholar] [CrossRef]
- Vives, E. Present and future of cell-penetrating peptide mediated delivery systems: "Is the trojan horse too wild to go only to troy?". J. Control. Release 2005, 109, 77–85. [Google Scholar] [CrossRef]
- Snyder, E.L.; Dowdy, S.F. Cell penetrating peptides in drug delivery. Pharm Res. 2004, 21, 389–393. [Google Scholar] [CrossRef]
- Eguchi, A.; Dowdy, S.F. Sirna delivery using peptide transduction domains. Trends Pharmacol. Sci. 2009, 30, 341–345. [Google Scholar] [CrossRef]
- El Andaloussi, S.A.; Hammond, S.M.; Mager, I.; Wood, M.J. Use of cell-penetrating-peptides in oligonucleotide splice switching therapy. Curr. Gene Ther. 2012, 12, 161–178. [Google Scholar] [CrossRef]
- Gautam, A.; Singh, H.; Tyagi, A.; Chaudhary, K.; Kumar, R.; Kapoor, P.; Raghava, G.P. Cppsite: A curated database of cell penetrating peptides. Database (Oxford) 2012, 2012, bas015. [Google Scholar]
- Milletti, F. Cell-penetrating peptides: Classes, origin, and current landscape. Drug Discov. Today 2012, 17, 850–860. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In vivo protein transduction: Delivery of a biologically active protein into the mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef]
- Elliott, G.; O'Hare, P. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell 1997, 88, 223–233. [Google Scholar] [CrossRef]
- Futaki, S.; Suzuki, T.; Ohashi, W.; Yagami, T.; Tanaka, S.; Ueda, K.; Sugiura, Y. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 2001, 276, 5836–5840. [Google Scholar]
- Wender, P.A.; Cooley, C.B.; Geihe, E.I. Beyond cell penetrating peptides: Designed molecular transporters. Drug Discov. Today Technol. 2012, 9, e49–e55. [Google Scholar] [CrossRef]
- Lindgren, M.; Gallet, X.; Soomets, U.; Hallbrink, M.; Brakenhielm, E.; Pooga, M.; Brasseur, R.; Langel, U. Translocation properties of novel cell penetrating transportan and penetratin analogues. Bioconjug. Chem. 2000, 11, 619–626. [Google Scholar] [CrossRef]
- Soomets, U.; Lindgren, M.; Gallet, X.; Hallbrink, M.; Elmquist, A.; Balaspiri, L.; Zorko, M.; Pooga, M.; Brasseur, R.; Langel, U. Deletion analogues of transportan. Biochim. Biophys. Acta 2000, 1467, 165–176. [Google Scholar] [CrossRef]
- Elmquist, A.; Lindgren, M.; Bartfai, T.; Langel, U. Ve-cadherin-derived cell-penetrating peptide, pvec, with carrier function. Exp. Cell. Res. 2001, 269, 237–244. [Google Scholar] [CrossRef]
- Gros, E.; Deshayes, S.; Morris, M.C.; Aldrian-Herrada, G.; Depollier, J.; Heitz, F.; Divita, G. A non-covalent peptide-based strategy for protein and peptide nucleic acid transduction. Biochim. Biophys. Acta 2006, 1758, 384–393. [Google Scholar] [CrossRef]
- Rhee, M.; Davis, P. Mechanism of uptake of c105y, a novel cell-penetrating peptide. J. Biol. Chem. 2006, 281, 1233–1240. [Google Scholar] [CrossRef]
- Crombez, L.; Aldrian-Herrada, G.; Konate, K.; Nguyen, Q.N.; McMaster, G.K.; Brasseur, R.; Heitz, F.; Divita, G. A new potent secondary amphipathic cell-penetrating peptide for sirna delivery into mammalian cells. Mol. Ther. 2009, 17, 95–103. [Google Scholar] [CrossRef]
- Kalafut, D.; Anderson, T.N.; Chmielewski, J. Mitochondrial targeting of a cationic amphiphilic polyproline helix. Bioorg Med. Chem. Lett. 2012, 22, 561–563. [Google Scholar]
- Green, I.; Christison, R.; Voyce, C.J.; Bundell, K.R.; Lindsay, M.A. Protein transduction domains: Are they delivering? Trends Pharmacol. Sci. 2003, 24, 213–215. [Google Scholar] [CrossRef]
- Fischer, R.; Fotin-Mleczek, M.; Hufnagel, H.; Brock, R. Break on through to the other side-biophysics and cell biology shed light on cell-penetrating peptides. Chembiochem 2005, 6, 2126–2142. [Google Scholar] [CrossRef]
- Mueller, J.; Kretzschmar, I.; Volkmer, R.; Boisguerin, P. Comparison of cellular uptake using 22 cpps in 4 different cell lines. Bioconjug. Chem. 2008, 19, 2363–2374. [Google Scholar] [CrossRef]
- Koren, E.; Torchilin, V.P. Cell-penetrating peptides: Breaking through to the other side. Trends Mol. Med. 2012, 18, 385–393. [Google Scholar] [CrossRef]
- Jones, A.T.; Sayers, E.J. Cell entry of cell penetrating peptides: Tales of tails wagging dogs. J. Control. Release 2012, 161, 582–591. [Google Scholar] [CrossRef]
- Sung, M.; Poon, G.M.; Gariepy, J. The importance of valency in enhancing the import and cell routing potential of protein transduction domain-containing molecules. Biochim Biophys Acta 2006, 1758, 355–363. [Google Scholar] [CrossRef]
- Vocero-Akbani, A.; Lissy, N.A.; Dowdy, S.F. Transduction of full-length tat fusion proteins directly into mammalian cells: Analysis of t cell receptor activation-induced cell death. Methods Enzymol. 2000, 322, 508–521. [Google Scholar] [CrossRef]
- Foltopoulou, P.F.; Tsiftsoglou, A.S.; Bonovolias, I.D.; Ingendoh, A.T.; Papadopoulou, L.C. Intracellular delivery of full length recombinant human mitochondrial l-sco2 protein into the mitochondria of permanent cell lines and sco2 deficient patient's primary cells. Biochim. Biophys. Acta 2010, 1802, 497–508. [Google Scholar] [CrossRef]
- Flinterman, M.; Farzaneh, F.; Habib, N.; Malik, F.; Gaken, J.; Tavassoli, M. Delivery of therapeutic proteins as secretable tat fusion products. Mol. Ther. 2009, 17, 334–342. [Google Scholar] [CrossRef]
- Chen, X.; Bai, Y.; Zaro, J.L.; Shen, W.C. Design of an in vivo cleavable disulfide linker in recombinant fusion proteins. Biotechniques 2010, 49, 513–518. [Google Scholar] [CrossRef]
- Zaro, J.L.; Fei, L.; Shen, W.C. Recombinant peptide constructs for targeted cell penetrating peptide-mediated delivery. J. Control. Release 2012, 158, 357–361. [Google Scholar] [CrossRef]
- Sawant, R.M.; Hurley, J.P.; Salmaso, S.; Kale, A.; Tolcheva, E.; Levchenko, T.S.; Torchilin, V.P. "Smart" drug delivery systems: Double-targeted ph-responsive pharmaceutical nanocarriers. Bioconjug. Chem. 2006, 17, 943–949. [Google Scholar] [CrossRef]
- Sawant, R.R.; Jhaveri, A.M.; Torchilin, V.P. Immunomicelles for advancing personalized therapy. Adv. Drug Deliv. Rev. 2012, 64, 1436–1446. [Google Scholar] [CrossRef]
- Aubry, S.; Burlina, F.; Dupont, E.; Delaroche, D.; Joliot, A.; Lavielle, S.; Chassaing, G.; Sagan, S. Cell-surface thiols affect cell entry of disulfide-conjugated peptides. FASEB J. 2009, 23, 2956–2967. [Google Scholar] [CrossRef]
- Kale, A.A.; Torchilin, V.P. "Smart" drug carriers: Pegylated tatp-modified ph-sensitive liposomes. J. Liposome Res. 2007, 17, 197–203. [Google Scholar] [CrossRef]
- Khafagy el, S.; Morishita, M. Oral biodrug delivery using cell-penetrating peptide. Adv. Drug Deliv. Rev. 2012, 64, 531–539. [Google Scholar] [CrossRef]
- Liu, B.R.; Li, J.F.; Lu, S.W.; Leel, H.J.; Huang, Y.W.; Shannon, K.B.; Aronstam, R.S. Cellular internalization of quantum dots noncovalently conjugated with arginine-rich cell-penetrating peptides. J. Nanosci. Nanotechnol. 2010, 10, 6534–6543. [Google Scholar] [CrossRef]
- Hu, J.W.; Liu, B.R.; Wu, C.Y.; Lu, S.W.; Lee, H.J. Protein transport in human cells mediated by covalently and noncovalently conjugated arginine-rich intracellular delivery peptides. Peptides 2009, 30, 1669–1678. [Google Scholar] [CrossRef]
- Jarver, P.; Mager, I.; Langel, U. In vivo biodistribution and efficacy of peptide mediated delivery. Trends Pharmacol. Sci. 2010, 31, 528–535. [Google Scholar] [CrossRef]
- Snyder, E.L.; Saenz, C.C.; Denicourt, C.; Meade, B.R.; Cui, X.S.; Kaplan, I.M.; Dowdy, S.F. Enhanced targeting and killing of tumor cells expressing the cxc chemokine receptor 4 by transducible anticancer peptides. Cancer Res. 2005, 65, 10646–10650. [Google Scholar] [CrossRef]
- Heffernan, C.; Sumer, H.; Guillemin, G.J.; Manuelpillai, U.; Verma, P.J. Design and screening of a glial cell-specific, cell penetrating peptide for therapeutic applications in multiple sclerosis. PLoS One 2012, 7, e45501. [Google Scholar]
- Verdurmen, W.P.; Bovee-Geurts, P.H.; Wadhwani, P.; Ulrich, A.S.; Hallbrink, M.; van Kuppevelt, T.H.; Brock, R. Preferential uptake of l-versus d-amino acid cell-penetrating peptides in a cell type-dependent manner. Chem. Biol. 2011, 18, 1000–1010. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, S.; Liao, Z.; Wang, C.; Liu, Y.; Feng, S.; Jiang, X.; Chang, J. Peglated magnetic polymeric liposome anchored with tat for delivery of drugs across the blood-spinal cord barrier. Biomaterials 2010, 31, 6589–6596. [Google Scholar] [CrossRef]
- Weber, C.A.; Mehta, P.J.; Ardito, M.; Moise, L.; Martin, B.; De Groot, A.S. T cell epitope: Friend or foe? Immunogenicity of biologics in context. Adv. Drug Deliv. Rev. 2009, 61, 965–976. [Google Scholar] [CrossRef]
- Bryson, C.J.; Jones, T.D.; Baker, M.P. Prediction of immunogenicity of therapeutic proteins: Validity of computational tools. BioDrugs 2010, 24, 1–8. [Google Scholar]
- Schellekens, H. Bioequivalence and the immunogenicity of biopharmaceuticals. Nat. Rev. Drug Discov. 2002, 1, 457–462. [Google Scholar] [CrossRef]
- Sarko, D.; Beijer, B.; Boy, R.G.; Nothelfer, E.M.; Leotta, K.; Eisenhut, M.; Altmann, A.; Haberkorn, U.; Mier, W. The pharmacokinetics of cell-penetrating peptides. Mol. Pharm. 2010, 7, 2224–2231. [Google Scholar] [CrossRef]
- D'Souza, G.G.; Weissig, V. Subcellular targeting: A new frontier for drug-loaded pharmaceutical nanocarriers and the concept of the magic bullet. Expert Opin. Drug Deliv. 2009, 6, 1135–1148. [Google Scholar] [CrossRef]
- Mossalam, M.; Dixon, A.S.; Lim, C.S. Controlling subcellular delivery to optimize therapeutic effect. Ther. Deliv. 2010, 1, 169–193. [Google Scholar] [CrossRef]
- Davis, J.R.; Kakar, M.; Lim, C.S. Controlling protein compartmentalization to overcome disease. Pharm. Res. 2007, 24, 17–27. [Google Scholar]
- Rapoport, M.; Salman, L.; Sabag, O.; Patel, M.S.; Lorberboum-Galski, H. Successful tat-mediated enzyme replacement therapy in a mouse model of mitochondrial e3 deficiency. J. Mol. Med. 2011, 89, 161–170. [Google Scholar] [CrossRef]
- Macewan, S.R.; Chilkoti, A. Harnessing the power of cell-penetrating peptides: Activatable carriers for targeting systemic delivery of cancer therapeutics and imaging agents. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2012. [Google Scholar] [CrossRef]
- Johnson, R.M.; Harrison, S.D.; Maclean, D. Therapeutic applications of cell-penetrating peptides. Methods Mol. Biol. 2010, 683, 535–551. [Google Scholar]
- van den Berg, A.; Dowdy, S.F. Protein transduction domain delivery of therapeutic macromolecules. Curr. Opin. Biotechnol. 2011, 22, 888–893. [Google Scholar] [CrossRef]
- Rothbard, J.B.; Garlington, S.; Lin, Q.; Kirschberg, T.; Kreider, E.; McGrane, P.L.; Wender, P.A.; Khavari, P.A. Conjugation of arginine oligomers to cyclosporin a facilitates topical delivery and inhibition of inflammation. Nat. Med. 2000, 6, 1253–1257. [Google Scholar]
- Lopes, L.B.; Furnish, E.J.; Komalavilas, P.; Flynn, C.R.; Ashby, P.; Hansen, A.; Ly, D.P.; Yang, G.P.; Longaker, M.T.; Panitch, A.; Brophy, C.M. Cell permeant peptide analogues of the small heat shock protein, hsp20, reduce tgf-beta1-induced ctgf expression in keloid fibroblasts. J. Invest. Dermatol. 2009, 129, 590–598. [Google Scholar] [CrossRef]
- Lebleu, B.; Moulton, H.M.; Abes, R.; Ivanova, G.D.; Abes, S.; Stein, D.A.; Iversen, P.L.; Arzumanov, A.A.; Gait, M.J. Cell penetrating peptide conjugates of steric block oligonucleotides. Adv. Drug Deliv. Rev. 2008, 60, 517–529. [Google Scholar] [CrossRef]
- Ensoli, B.; Bellino, S.; Tripiciano, A.; Longo, O.; Francavilla, V.; Marcotullio, S.; Cafaro, A.; Picconi, O.; Paniccia, G.; Scoglio, A.; Arancio, A.; Ariola, C.; Ruiz Alvarez, M.J.; Campagna, M.; Scaramuzzi, D.; Iori, C.; Esposito, R.; Mussini, C.; Ghinelli, F.; Sighinolfi, L.; Palamara, G.; Latini, A.; Angarano, G.; Ladisa, N.; Soscia, F.; Mercurio, V.S.; Lazzarin, A.; Tambussi, G.; Visintini, R.; Mazzotta, F.; Di Pietro, M.; Galli, M.; Rusconi, S.; Carosi, G.; Torti, C.; Di Perri, G.; Bonora, S.; Ensoli, F.; Garaci, E. Therapeutic immunization with Hiv-1 tat reduces immune activation and loss of regulatory t-cells and improves immune function in subjects on haart. PLoS One 2010, 5, e13540. [Google Scholar]
- Eguchi, A.; Dowdy, S.F. Efficient sirna delivery by novel ptd-drbd fusion proteins. Cell. Cycle 2010, 9, 424–425. [Google Scholar] [CrossRef]
- Palm-Apergi, C.; Eguchi, A.; Dowdy, S.F. Ptd-drbd sirna delivery. Methods Mol. Biol. 2011, 683, 339–347. [Google Scholar] [CrossRef]
- Reinecke, K.; Eminel, S.; Dierck, F.; Roessner, W.; Kersting, S.; Chromik, A.M.; Gavrilova, O.; Laukevicience, A.; Leuschner, I.; Waetzig, V.; Rosenstiel, P.; Herdegen, T.; Sina, C. The jnk inhibitor xg-102 protects against tnbs-induced colitis. PLoS One 2012, 7, e30985. [Google Scholar]
- Meyer-Losic, F.; Nicolazzi, C.; Quinonero, J.; Ribes, F.; Michel, M.; Dubois, V.; de Coupade, C.; Boukaissi, M.; Chene, A.S.; Tranchant, I.; Arranz, V.; Zoubaa, I.; Fruchart, J.S.; Ravel, D.; Kearsey, J. Dts-108, a novel peptidic prodrug of sn38: In vivo efficacy and toxicokinetic studies. Clin. Cancer Res. 2008, 14, 2145–2153. [Google Scholar]
- Drakopoulou, E.; Papanikolaou, E.; Anagnou, N.P. The ongoing challenge of hematopoietic stem cell-based gene therapy for beta-thalassemia. Stem Cells Int. 2011, 987980. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Papadopoulou, L.C.; Tsiftsoglou, A.S. The Potential Role of Cell Penetrating Peptides in the Intracellular Delivery of Proteins for Therapy of Erythroid Related Disorders. Pharmaceuticals 2013, 6, 32-53. https://doi.org/10.3390/ph6010032
Papadopoulou LC, Tsiftsoglou AS. The Potential Role of Cell Penetrating Peptides in the Intracellular Delivery of Proteins for Therapy of Erythroid Related Disorders. Pharmaceuticals. 2013; 6(1):32-53. https://doi.org/10.3390/ph6010032
Chicago/Turabian StylePapadopoulou, Lefkothea C., and Asterios S. Tsiftsoglou. 2013. "The Potential Role of Cell Penetrating Peptides in the Intracellular Delivery of Proteins for Therapy of Erythroid Related Disorders" Pharmaceuticals 6, no. 1: 32-53. https://doi.org/10.3390/ph6010032