Sphingosine-1-Phosphate-Specific G Protein-Coupled Receptors as Novel Therapeutic Targets for Atherosclerosis

{kind=link}
{kind=link}
Abstract
: Atherosclerosis is a chronic inflammatory process involving complex interactions of modified lipoproteins, monocyte-derived macrophages or foam cells, lymphocytes, endothelial cells (ECs), and vascular smooth muscle cells. Sphingosine-1-phosphate (S1P), a biologically active blood-borne lipid mediator, exerts pleiotropic effects such as cell proliferation, migration and cell-cell adhesion in a variety of cell types via five members of S1P-specific high-affinity G protein-coupled receptors (S1P1-S1P5). Among them, S1P1, S1P2 and S1P3 are major receptor subtypes which are widely expressed in various tissues. Available evidence suggest that S1P and HDL-bound S1P exert atheroprotective effects including inhibition of leukocyte adhesion and stimulation of endothelial nitric oxide synthase (eNOS) in endothelial cells (ECs) through the activation of Gi signaling pathway via S1P3 and probably S1P1, although there is still controversy. FTY720, the phosphorylation product of which is a high-affinity agonist for all S1P receptors except S1P2 and act as an immunosuppressant by downregulating S1P1 on lymphocytes, inhibits atherosclerosis in LDL receptor-null mice and apoE-null mice through the inhibition of lymphocyte and macrophage functions and probably stimulation of EC functions, without influencing plasma lipid concentrations. In contrast to S1P1 and S1P3, S1P2 facilitates atherosclerosis by activating G12/13-Rho-Rho kinase (ROCK) in apoE-null mice. S1P2 mediates transmigration of monocytes into the arterial intima, oxidized LDL accumulation and cytokine secretion in monocyte-derived macrophages, and eNOS inhibition and cytokine secretion in ECs through Rac inhibition, NF-κB activation and 3′-specific phosphoinositide phosphatase (PTEN) stimulation downstream of G12/13-Rho-ROCK. Systemic long-term administration of a selective S1P2-blocker remarkably inhibits atherosclerosis without overt toxicity. Thus, multiple S1P receptors positively and negatively regulate atherosclerosis through multitudes of mechanisms. Considering the essential and multi-faceted role of S1P2 in atherogenesis and the impact of S1P2 inactivation on atherosclerosis, S1P2 is a particularly promising therapeutic target for atherosclerosis.1. Introduction
It is increasingly recognized that atherosclerosis is a complex chronic inflammatory disease rather than a mere phenomenon of lipid deposition on the vascular wall [1,2]. In endothelial cells (ECs), proinflammatory stimuli, including hypercholesterolemia, hyperglycemia and smoking, trigger the expression of adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1) and selectins, which mediate the attachment of circulating monocytes and lymphocytes to ECs. Chemokines and other cytokines, which are produced by vascular wall cells, elicit the infiltration of adherent leukocytes into the intima. Within the intima, monocytes differentiate into macrophages and engulf modified low density-lipoprotein (LDL) through scavenger receptor-mediated endocytosis, leading to conversion of macrophages into lipid-laden macrophages, foam cells. Macrophages amplify the inflammatory responses through the release of numerous cytokines and growth factors. T cells also enter lesions and amplify the local inflammatory responses by producing proinflammatory cytokines. The cytokines and growth factors secreted by these leukocytes as well as ECs direct migration of vascular smooth muscle cells (SMCs) into lesions. In the intimal lesions, SMCs proliferate under the influence of various growth factors and release collagens and other extracellular matrices, expanding the lesions. In advanced atherosclerotic lesions, increased inflammatory activities diminish the collagen content and increase procoagulant activity, leading to plaque rupture and acute coronary thrombosis.
Sphingosine-1-phosphate (S1P) is a blood borne, lysophospholipid mediator that exerts pleiotropic activities including cell proliferation, survival, migration, cell shape and cell-cell adhesion in a variety of cell types [3-6,7]. S1P was originally shown to be released from activated platelets [8] and to be present in the plasma at around 10−7∼10−6 mol/L, largely in a form bound to plasma proteins, albumin and high density-lipoprotein (HDL) [8-10]. In agreement with this, plasma S1P levels were highly correlated with HDL concentrations [10]. A number of investigations provided evidence for the notion that HDL possesses an atheroprotective activity [11-13]. It is established that HDL plays an essential role in cholesterol efflux from cholesterol-laden macrophages as a cholesterol acceptor and cholesterol transport to the liver [13]. Besides this role of HDL, it is suggested to exert atheroprotective effects through the HDL-bound S1P [14-16]. Previous studies showed that HDL-bound S1P and other sphingolipids mediated the atheroprotective actions of HDL, including EC survival and proliferation, stimulation of endothelial nitric oxide (NO) synthase (eNOS), inhibition of endothelial expression of VCAM-1 and intracellular adhesion molecule (ICAM)-1, and inhibition of monocyte chemoattractant peptide (MCP)-1 production in SMCs [14-16]. In contrast, other studies showed that S1P exhibits proatherogenic activities. For example, the proinflammatory cytokine tumor necrosis factor-α (TNF-α) activated EC, as evaluated by the expression of adhesion molecules such as E-selectin and VCAM-1, through a sphingosine kinase (SphK), which is an S1P-synthezing enzyme (see below for details) [17]. HDL inhibited the expression of these adhesion molecules by suppressing SphK [18].
Although many reports have suggested that S1P may be involved in atherosclerosis, it remained undefined whether S1P is proatherogenic or antiatherogenic and by what mechanisms S1P modifies atherosclerosis [19]. Recent studies including ours have addressed the in vivo roles of S1P receptor subtypes in atherosclerosis using mouse models of atherosclerosis [20-23]. In this review, we will focus on the receptor subtype-specific, stimulatory and inhibitory effects of S1P on atherosclerosis and discuss the possibility of using S1P receptors as a novel therapeutic target for atherosclerosis.
2. S1P Metabolism, Receptors and Their Actions
2.1. Synthesis and Degradation of S1P
S1P is generated from sphingomyelin, an integral component of plasma membranes, by the sequential action of sphingomyelinase, ceramidase, and sphingosine kinases [24]. The SphK1 and SphK2, rate-limiting enzymes for S1P synthesis, catalyze the phosphorylation of sphingosine to produce S1P [25,26]. The SphK1 and SphK2 exhibit different expression patterns and kinetic properties and may therefore regulate different S1P-dependent processes. SphK1/SphK2 double knockout mice are embryonic lethal and virtually lack tissue S1P, indicating that S1P is produced exclusively by SphKs [27,28]. Degradation of S1P occurs by the dephosphorylation by S1P phosphatase (SPP) and the cleavage to palmitoaldehyde and phosphoethanolamine by S1P lyase [29,30]. Once synthesized, S1P is released from cells through the export across the cell membrane likely via the ATP-binding cassette (ABC) family of transporters such as ABCC1, ABCA1 and ABCG2 [31-33] and recently identified “protein two of hearts” (also known as spns2) [34,35].
The major constitutive source of plasma S1P is red blood cells with additional contribution of non-hematopoietic cells including vascular endothelial cells [36-39]. Lymphatic ECs and neural crest-derived pericytes in the thymus are constitutive sources for S1P in lymph and the thymic local milieu, respectively [40,41], while activated platelets, mast cells, macrophages and other cell types were reported to produce and secrete S1P upon stimulation [8,42,43]. S1P in plasma and locally produced by macrophages, mast cells, ECs, and other cells in lesions could be involved in atherosclerosis.
2.2. S1P Receptors and Their Actions
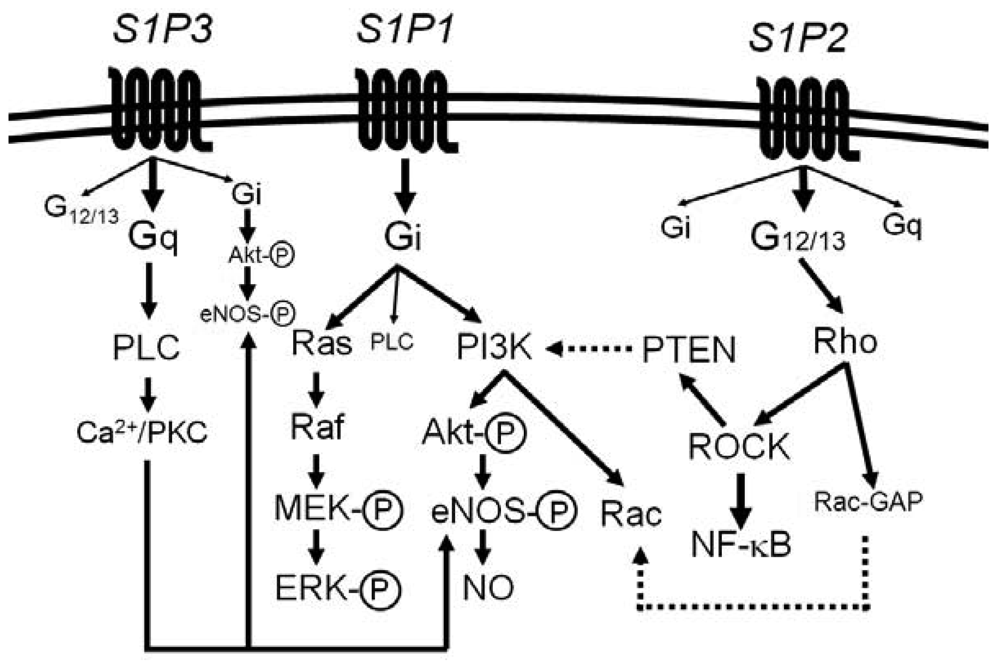
Most of the diverse biological activities of S1P are mediated by five members of S1P-specific high-affinity G protein-coupled receptors, S1P1 [or S1PR1 (gene name)] − S1P5 (or S1PR5) [44-48]. S1P1, S1P2 and S1P3 are widely expressed in various tissues and the major receptor subtypes in the vasculature [6,7,22,44,45]. The diversity of responses to S1P depends on their subtype-specific, differential coupling to various G-proteins, in combination with tissue- and cell type-specific receptor expression patterns (Figure 1) [44-46,49-52].
S1P1 couples exclusively to Gi, whereas S1P2 and S1P3 couple to multiple G proteins but the G proteins preferred by S1P2 and S1P3 are G12/13 and Gq, respectively [49-55]. Downstream of the heterotrimeric proteins, S1P1 activates phosphoinositide 3-kinase (PI3K)-Akt/Rac pathway and Ras-mitogen activated protein kinase pathway, S1P2 activates Rho-Rho kinase (ROCK) pathways including NF-κB and the 3′-specific phosphoinositide phosphatase, phosphatase and tensin homolog (PTEN), and S1P3 activates phospholipase C (PLC) pathway [49-55]. Moreover, intracellular S1P may regulate cell growth, survival and other functions in a receptor-independent manner [43,56,57]. It is unknown whether and how such intracellular mechanisms of S1P contribute to vascular physiology and diseases.
2.3. S1P Receptors in ECs
ECs, SMCs, lymphocytes and monocytes/macrophages, which are involved in atherogenesis, show distinct patterns of the expression of S1P1, S1P2 and S1P3. ECs express easily detectable levels of S1P1 and S1P3, whereas S1P2 expression appears to be relatively low [58,59]. ECs release the atheroprotective mediator NO [60]. S1P activates eNOS likely via S1P3 and probably S1P1 to stimulate NO production [61,62]. S1P also maintains endothelial barrier function via S1P1 through the facilitating effect on adherens junctional assembly [63-66].
S1P was reported to positively and negatively regulate the monocyte-EC interaction through multiple mechanisms. S1P stimulated the expression of the adhesion molecules VCAM-1 and ICAM-1 in human umbilical vein ECs (HUVECs) and monocyte adhesion in Gi- and NF-κB-dependent manner [15,67]. The down-regulation of S1P1 signaling by siRNA knockdown decreased the induction of E-selectin after tumor necrosis factor-α (TNF-α) or lipopolysaccharide (LPS) stimulation of human ECs [68]. In contrast, it was reported that S1P inhibited EC activation; S1P-containing HDL inhibited the induction of endothelial adhesion molecules by TNF-α through NO pathway in HUVECs [14,15]. In agreement with an inhibitory effect of S1P on adhesion molecules expression in ECs, S1P1 agonist SEW2871 suppressed the adherence of inflammatory mononuclear cells to TNF-α-activated aortic ECs [69,70]. In diabetic NOD mice, S1P and SEW2871 activated S1P1 to abrogate monocyte adhesion to aortic ECs in a partially NO-dependent manner and VCAM-1 expression due to their inhibitory effect on NF-κB [71]. S1P also suppressed the adhesion of monocytic cell line U937 to HUVECs via the endothelial integrins α5β1 and αvβ3, independently from the expression of adhesion molecules [72]. In addition, S1P stimulated the expression of interleukin (IL)-8 and MCP-1, chemoattractants for leukocytes, via S1P1 and S1P3 in HUVECs [73,74].
These results suggest that when the cells are exposed to exogenous S1P, the expression level of adhesion molecules and chemokines may be determined by both the NF-κB-mediated stimulatory signal and the NO-mediated inhibitory signal. The net effects of S1P on leukocyte adhesion to ECs, chemokine production, and consequent leukocyte infiltration into the intima may be affected by differences in S1P receptor expression.
2.4. S1P Receptors in SMCs
In atherosclerosis, SMCs are involved in plaque expansion and its stabilization by migrating to form a fibrous cap over the plaque and preventing it from rupture [1,2]. SMCs from adult vessels express S1P2 and S1P3, while SMCs from pups express S1P1, S1P2, and S1P3 [58,75]. S1P1 mediates migration and proliferation in response to S1P [75,76]. Our previous observations demonstrated that S1P inhibited platelet-derived growth factor (PDGF)-induced migration in adult SMCs through S1P2-G12/13-Rho-dependent Rac inhibition [52,54,77]. In agreement with inhibitory effect of S1P2 on migration of SMCs, the enhanced neointimal lesion formation was induced by ligation of the carotid artery in S1P2−/− mice, and higher rate of in vitro proliferation and migration in S1P2−/− SMCs was observed [78]. S1P2 also promoted the SMC differentiation, thereby limiting the growth potential of SMCs [79,80]. Activated SMCs are an abundant source of proatherogenic cytokines and chemokines including MCP-1 [1,2]. S1P and S1P-containing HDL inhibited NAD(P)H oxidase-dependent reactive oxygen species generation and MCP-1 production via S1P3 in SMCs [16,81].
2.5. S1P Receptors in Monocytes/Macrophages
Circulating monocytes and monocyte-derived macrophages play a crucial role in atherosclerosis by adhering to activated ECs, transmigrating into the intima, and differentiating into macrophages and lipid-laden foam cells [1,2,82]. Monocytes and macrophages express multiple S1P receptors [83-86], but show species-specific difference. Human monocytes express S1P1, S1P2 and S1P4, and human macrophages express S1P1-S1P4 [83], while murine bone marrow (BM)-derived macrophages mainly express S1P1 and S1P2 [22,84-86]. S1P2 mediates inhibition of C5a-induced migration of murine primary macrophages in vitro, and macrophages isolated from S1P2-knockout (S1P2−/−) mice displayed enhanced recruitment during thioglycollate-induced peritonitis [87]. S1P may positively and negatively regulate the transmigration of monocytes and macrophages into the intima via S1P1 and S1P2.
Recent studies indicated that SphK activation is involved in inflammatory responses via the action of the intracellular S1P in macrophages [56,57], whereas extracellular S1P predominantly triggers anti-inflammatory responses via binding to cell surface S1P receptors [88]. S1P selectively attenuates Toll-like receptor 2 signaling via S1P1/2-mediated negative cross-talk in murine macrophages, thus preventing macrophage activation [89]. S1P also promoted the conversion of macrophages from the proinflammatory (M1) to anti-inflammatory (M2) phenotype with S1P1-mediated inhibition of LPS-induced secretion of TNF-α, MCP-1 and IL-12 in murine peritoneal macrophages [86]. These results suggest that S1P facilitates the anti-inflammatory signal generation in macrophages via S1P1.
Oxidized LDL exerts cytotoxic effects to induce apoptosis in macrophages [90]. The environment within atherosclerotic lesions is extremely proapoptotic. As for other cells, S1P protects macrophages against apoptosis [91,92]. S1P, which is derived from apoptotic cells, activates PI3K, ERK and Ca2+ signaling in macrophages to protect them against apoptosis through the heme oxygenase 1-dependent upregulation of the anti-apoptotic proteins Bcl-2 and Bcl-xL [91,92].
2.6. S1P Receptors in Lymphocytes
The circulation of mature lymphocytes between blood and secondary lymphoid tissues is a central process in the immune surveillance. The immunosuppressant FTY720 (fingolimod), a structural analogue of sphingosine, which is phosphorylated in vivo by SphK2 [93]. The phosphorylated product of FTY720 (FTY720-P) is a high-affinity agonist for S1P1, S1P3, S1P4 and S1P5 but not S1P2, depletes lymphocyte from the blood by binding to and downregulating S1P1 on both T and B lymphocytes [38,94-96]. FTY720 is called a functional antagonist after this action. FTY720 has recently been officially approved as an orally available therapeutics for multiple sclerosis in U.S.A. and Russia [97].
3. Effects of FTY720 on Atherosclerosis
FTY720-P acts on S1P1 and induces immunosuppression by sequestering lymphocytes, particularly T lymphocytes, in secondary lymphoid organs and decreasing circulating lymphocytes. T lymphocytes are involved in the initiation and progression of atherosclerosis [1,2]. EC-derived NO is an atheroprotective mediator [60] and upregulated by endothelial S1P3 and probably S1P1 [61,62]. Therefore, it is rational to hypothesize that FTY720 may have an impact on atherosclerosis through its immunosuppressing and eNOS-stimulating effects.
Two groups tested the pharmacological actions of FTY720 on the initiation and progression of atherosclerosis in atherosclerotic models, apoE−/− mice and LDLR−/− mice [20,21]. Keul et al. demonstrated that oral FTY720 administration (1.25 mg/kg body weight/day) for 20 weeks resulted in more than a 50% reduction of atherosclerotic lesion volumes in apoE−/− mice fed a high fat (Western) diet without any influence on plasma lipid concentrations [21]. The reduction of atherosclerotic lesions was accompanied by decreases in macrophage density and collagen deposition in the lesions but not changes in the density of CD3+ T lymphocytes or SMCs. As expected, FTY720 administration induced lymphopenia, indicating that FTY720 induced immunosuppression by effectively downregulating lymphocyte S1P1. In contrast, S1P3-mediated, NO-dependent vasodilator response of aortae isolated from FTY720-administered mice to acute FTY720 challenge remained intact, suggesting that chronic FTY720 administration did not downregulate endothelial S1P3 or compromise S1P3-mediated eNOS activation. In addition, they showed that FTY720-P treatment of isolated aortic segments and cultured SMCs potently inhibited thrombin-induced MCP-1 release. Thrombin-induced MCP-1 response was abolished in tissues and cells from S1P3-null mice. The gene expression of cytokines including IL-12, IL-10, IL-4 or IFN-γ in isolated peritoneal macrophages was not different between mice receiving FTY720 and vehicle. Based on these observations, they suggested that FTY720 inhibited atherosclerosis mainly by suppressing monocyte/macrophage recruitment to atherosclerotic lesions through mechanisms involving S1P3-mediated, probably NO-dependent inhibition of MCP-1 production.
Nofer et al. has demonstrated in high cholesterol diet (HCD)-fed LDLR−/− mice that intraperitoneal injection of FTY720 three times a week (0.04 or 0.4 mg/kg body weight/day) for 16 weeks reduced atherosclerotic lesions in a dose-dependent manner [20]. FTY720 substantially decreased CD3+ T lymphocytes in lesions but not affect macrophage density, smooth muscle density or collagen content. Moreover, FTY720 inhibited necrotic core formation. FTY720 only at the high dose lowered the peripheral blood lymphocyte with a preferential decrease in T cells, particularly CD4+ helper T cell subset. The plasma level of the T cell cytokine IFN-γ was reduced with diminished concanavarin A-induced in vitro mitogenesis of lymphocytes from mice receiving either the low or high dose of FTY720, suggesting that FTY720 attenuated Th1 immune responses. Analogous to lymphocytes, the plasma levels of the macrophage-derived cytokines, TNF-α, IL-6 and IL12, were reduced in mice receiving FTY720 although macrophage accumulation in plaques was not altered in FTY720-treated mice. These observations collectively suggested that chronic administration of FTY720 attenuates development of atherosclerosis through the inhibition of functions of T cells and macrophages.
Recently, it was shown that in apoE−/− mice on a normal diet, hypercholesterolemia is induced by treatment with a relatively higher dose of FTY720 (3 mg/kg/day) for 12 weeks, which possibly counteracts its anti-atherogenic effect on immune cell distribution [98].
These studies indicate that FTY720 effectively inhibits atherosclerosis, without affecting blood lipid profiles, at the doses which inhibit T cells activity as evaluated with circulating lymphocyte numbers and T cell-specific cytokine production as markers [20,21]. Moreover, the reduced plasma levels of the cytokines, which are abundantly produced by macrophages, suggest that FTY720 directly or indirectly via the regulation of lymphocytes inhibits macrophage activity. In addition, FTY720 may inhibit mobilization of monocytes/macrophages into lesions through mechanisms involving the attenuation of chemokine production. Accumulated evidence indicates that FTY720-induced sequestration of T cells in lymphoid organs and resultant lymphopenia occurs as a result of downregulation of lymphocyte S1P1, i.e. the functional antagonism of S1P1 [38,94-96]. It remains unclear whether chronic administration of FTY720 similarly downregulates macrophage S1P1 in vivo and consequently induces significant functional changes of macrophages. Besides the immune cells as targets of FTY720, this compound may have non-immune cell targets, including ECs and SMCs, in inhibiting atherosclerosis. FTY720-P activates eNOS to stimulate NO production in ECs via S1P3 and probably S1P1. In this respect, it is noted that chronic FTY720 administration did not impair FTY720-P-induced vasodilation, which suggest that S1P3 and/or S1P1 in ECs were not downregulated [21]. It is likely that FTY720-P activates endothelial S1P3 and S1P1 as a functional agonist, leading to increased release of NO. Likewise, S1P3 and S1P1 may mediate inhibition of the expression of cytokines including the monocyte chemoattractant MCP-1, leading to inhibition of monocytic infiltration into lesions.
Systemic administration of non-selective immunosuppressive drugs will probably not be useful for the treatment of atherosclerosis because of adverse effects including serious infections [99]. Although FTY720 at a low dose did not reduce circulating lymphocytes [20], it decreased the plasma levels of T cell-specific cytokines, which suggest that FTY720 at the low dose might be accompanied by immune suppression. It is necessary to fully dissect the molecular mechanisms underlying the anti-atherogenic effect of FTY720 at various doses.
4. S1P2 as a New Target for Treatment of Atherosclerosis
Three major S1P receptors, S1P1, S1P2 and S1P3, are expressed in ECs, SMCs and monocytes/macrophages. Among these, S1P2 but not S1P1 or S1P3 in SMCs and ECs mediate inhibition of chemoattractant-directed cell migration [54,58,100]. This unique functional property of S1P2 can be accounted for by the distinct signaling capacity of S1P2; differently from S1P1 and S1P3, which are Gi-coupled receptors, S1P2 couples mainly to G12/13 to result in Rho activation, Rho-dependent Rac inhibition and PTEN stimulation, leading to chemorepulsion. Our recent observations [22,101] showed that ECs express a significant level of S1P2 in vivo although commonly employed HUVECs do not express an easily detectable level of S1P2. S1P1 and S1P3 stimulate eNOS in ECs and inhibit leukocyte adhesion to ECs. In contrast, the Rho- ROCK pathway, which is activated preferentially by S1P2, is reported to participate in an inflammatory response [55,102]. In addition, S1P2 mediates the opposite effect on migration of monocytes/macrophages to that of S1P1 and S1P3. These observations raised an intriguing possibility that S1P2 may have a distinct role in atherosclerosis from S1P1 and S1P3.
We have studied the role of S1P2 in atherosclerosis by using S1P2-deleted and non-deleted apoE−/− mice [22]. The en face plaque area in spread aortae was dramatically reduced (approximately70%) in homozygous knockout (S1P2−/−) mice compared with S1P2+/+ mice after 16 weeks of HCD. The plaque area in heterozygous knockout (S1P2+/−) mice was intermediate between S1P2−/− and S1P2+/+ mice, indicating that S1P2 has a gene dose-dependent proatherogenic effect. In the plaques of S1P2−/− mice, the macrophage density was decreased compared with S1P2+/+ mice whereas SMC density in the plaques was increased in S1P2−/− mice. Consistent with our data, Hla and colleagues very recently showed that S1P2-deficiency markedly inhibited atherosclerosis in apoE−/− mice [23]. The mRNA expression levels of the proinflammatory cytokines TNF-α, IL-6, IFN-γ and MCP-1, and the adhesion molecule VCAM-1 were reduced in the aortae of HCD-fed S1P2−/− mice compared with S1P2+/+ mice, whereas the phosphorylation of eNOS was increased in the aorta of S1P2−/− mice [22]. The mRNA expression of S1P1, S1P3, and the S1P synthesizing and degradation enzymes including SphK-1, SphK-2, SPL and SPP1 in the aorta was not different between S1P2+/+ and S1P2−/− mice. Thus, the atherosclerotic lesion is reduced in S1P2−/− mice with the diminished inflammatory activity and the increased atheroprotective NO activity.
Our previous study using β-galactosidase (LacZ)-knockin mice at the S1P2 locus, in which LacZ gene expression is driven by endogenous S1P2 promoter, showed that S1P2 is expressed in ECs and SMCs of normal blood vessels in a variety of organs and the bone marrow (BM) [100]. In the atherosclerotic lesion in the aortic sinus of LacZ-knockin apoE−/− mice fed HCD, macrophages, ECs, and intimal and medial SMCs were found to express S1P2 [22]. The role of S1P2 in BM-derived cells for atherosclerosis was studied by analyzing BM-chimera mice [22]. The deletion of S1P2 in BM cells markedly reduced atherosclerotic lesions compared with control. Thus, S1P2 in BM-derived cells, most likely monocytes and macrophages, play the critical role in atherosclerosis. The study by Skoura et al. [23] supported the importance of S1P2 in BM-derived cells in atherosclerosis.
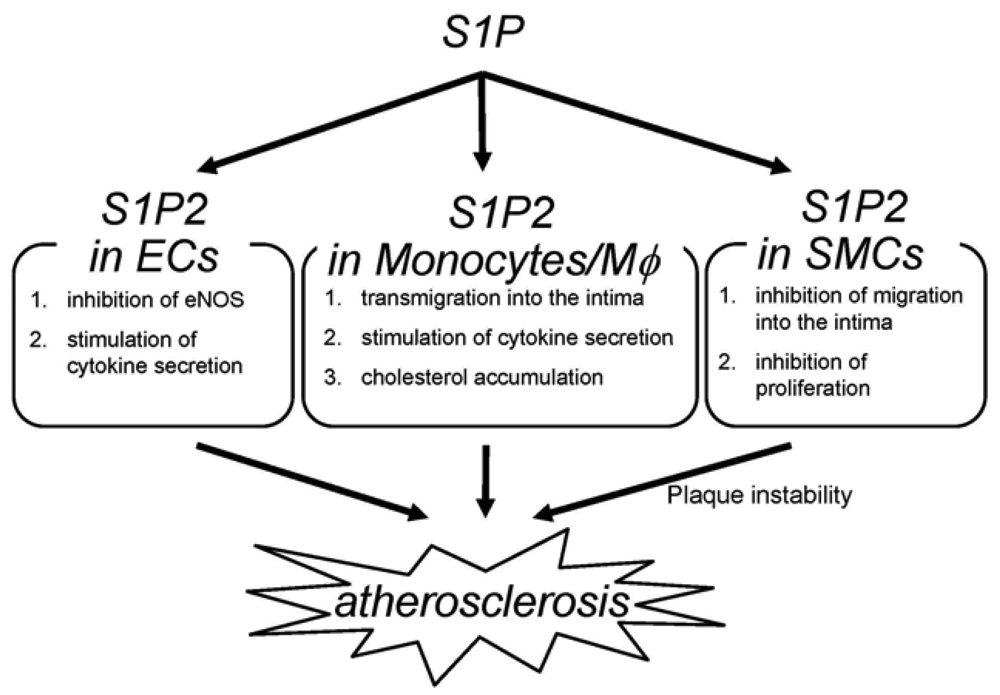
The deletion of S1P2 has a substantial impact on macrophage functions including cholesterol accumulation, cytokine production and migration (Figure 2) [22]; deletion of S1P2 in macrophages markedly inhibits accumulation of modified LDL through both a substantial decrease in uptake of oxidized LDLs and a modest increase in cholesterol efflux. These effects are accompanied by decreases in scavenger receptor expression (CD36 and scavenger receptor-A) and increases in cholesterol efflux transporter (ABCA1 and ABCG1) expression. These effects of S1P2-deficiency lead to inhibition of foam cell formation and reductions in atherosclerotic lesions. Second, deletion of S1P2 in macrophages inhibits the proinflammatory responses by suppressing Rho-ROCK-NF-κB signaling pathway, which is essential for the expression of the proatherogenic gene products including cytokines such as TNF-α and the scavenger receptor CD36. The ABCA1 mRNA expression is negatively regulated by ROCK through mechanisms involving LXR downregulation. Third, S1P2 possesses a profound influence on transmigration of monocytes/macrophages into atherosclerotic lesions. S1P2 mediates inhibition of macrophage migration toward a higher concentration of S1P (chemorepulsion) whereas S1P1 mediates stimulation of migration toward a higher concentration of S1P (chemotaxis). The blood S1P concentration is estimated to be much higher than that in tissues. The chemokines including MCP-1, which are produced in atherosclerotic lesions, attract monocytes to lesions. Therefore, S1P2 in monocytes very likely promotes macrophage transmigration into lesions. In fact, intravenously infused S1P2+/+ macrophages more robustly transmigrated into the vascular wall compared with S1P2−/− macrophages. The number of total monocytes and activated monocytes (CD11b+Ly6Chi) in the peripheral blood did not differ between S1P2+/+ and S1P2−/− mice, suggesting that S1P2-deficiency did not affect mobilization of monocytes to peripheral blood or their activation. These stimulatory effects of S1P2 on modified LDL accumulation, cytokine production, and transendothelial migration underlie the proatherogenic action of macrophage S1P2. Besides monocytes/macrophages, bone marrow-derived mast cells express S1P2, which stimulates degranulation [42]. Mast cells are implicated in plaque progression and destabilization, and therefore may contribute to the proatherogenic effect of S1P2 [103]. In our study, activated mast cells were reduced in the aortic wall of S1P2−/− mice. The role of mast cell S1P2 in atherogenesis remains to be clarified.
S1P2−/− ECs display altered phenotypes compared with wild-type ECs (Figure 2) [22,100]. eNOS and its product NO have atheroprotective properties. Consistent with the observation that eNOS phosphorylation is increased in the aortae of S1P2−/− mice compared with S1P2+/+ mice, S1P2−/− ECs show stimulation of eNOS phosphorylation in response to S1P stimulation whereas S1P2+/+ ECs exhibits a decrease in eNOS phosphorylation in response to S1P. In S1P2−/− ECs, S1P3 and probably S1P1 mediates eNOS phosphorylation most likely through stimulation of the well known eNOS activating protein kinase Akt. In contrast, S1P induces inhibition of Akt in S1P2+/+ ECs through S1P2-mediated, ROCK-dependent PTEN stimulation [55], which results in inhibition of Akt and consequently eNOS [22]. S1P2−/− ECs also showed suppression of the expression of the proinflammatory cytokines including MCP-1 and GM-CSF [22], as in the aortae [104,105]. Because both MCP-1 and GM-CSF, powerful chemoattractants for monocytes, are the NF-κB target genes, the inhibited cytokine response to S1P in S1P2−/− ECs is due to diminished ROCK-dependent NF-κB activation. Thus, S1P2 in ECs could participate in atherosclerosis by regulating adhesion molecule expression, cytokine production and consequently monocyte/macrophage flux, platelet activation and thrombus formation, and intimal cell proliferation through Rho-ROCK-PTEN-mediated Akt-eNOS regulation and Rho-ROCK-NF-κB-mediated regulation of proinflammatory gene expression.
S1P2 deletion induces alterations of the phenotypes in SMCs (Figure 2) [22]; S1P2−/− SMCs show enhanced proliferation in the presence of serum. This could be mediated probably at least partially by Akt stimulation due to loss of ROCK-mediated PTEN stimulation in S1P2−/− SMCs. S1P2−/− SMCs also exhibit loss of chemorepulsion to S1P. These phenotypes of S1P2-deficient SMCs might bring about a higher SMC density in atherosclerotic lesions of S1P2−/− mice.
The lesions in S1P2−/− mice may be stabilized compared with S1P2+/+ mice because of a lower macrophage density and higher SMC density in lesions of S1P2−/− mice. S1P2−/− macrophages display resistance to apoptosis induced by TNF-α and cycloheximide. Stimulated survival of S1P2−/− macrophages may also favor plaque stabilization although the relationship between macrophage apoptosis and atherogenesis is also complex [106].
These observations in S1P2-deleted mice raised the intriguing possibility that pharmacological S1P2 blockade could afford therapeutic efficacy for atherosclerosis. JTE-K1 is a selective S1P2 antagonist [107]. We tested the effect of the systemic administration of JTE-K1 into HCD-fed S1P2+/+apoE−/− mice for eight weeks. Oral administration of JTE-K1 (12.5 mg/kg twice daily) by gavage reduced the en face plaque area approximately by 60% with the lower density of macrophages and higher density of SMCs in atherosclerotic lesions compared with the vehicle control, thus recapitulating the phenotypes of S1P2−/− mice [22]. The treatment of isolated macrophages with JTE-K1 suppressed uptake of DiI-acLDL and stimulated cholesterol efflux, confirming the effectiveness of S1P2 blockade at the cellular level. The administration of JTE-K1 did not affect food intake or body weight gain in mice over eight weeks. Mice receiving JTE-K1 did not exhibit ataxia or tilting of the trunk due to vestibular dysfunction that had been reported in S1P2−/− mice, or any other discernible abnormality.
5. Concluding Remarks
Current therapy for human atherosclerotic lesions focuses on reducing the concentration of plasma LDL-associated cholesterol in the blood mainly by administering HMG CoA reductase inhibitors and other drugs [108-110]. Lowering blood cholesterol concentration leads to inhibition of the accumulation of modified LDL in the subendothelial layer, and consequently inhibition of atherosclerotic lesion formation. Although statins also exert plaque stabilizing and anti-inflammatory effects [99,108-110], no therapy to directly target foam cell formation in the face of elevated circulating LDL is currently available.
In genetic mice models for atherosclerosis, pharmacological blockade of chemoattractant receptors that are expressed in monocytes, including leukotriene B4 receptor and RANTES receptors, effectively inhibited plaque formation [111,112]. However, pharmacological blockage of leukocytes recruitment to inflammatory sites may be associated with the side effect of diminished defence mechanisms against infectious pathogens. HDL-associated S1P and the sphingosine mimetic FTY720 seem to exert atheroprotective actions via S1P1 and S1P3. In the case of FTY720, it might be necessary to separate the favorable EC-protective effect from the immunosuppressive effect.
In contrast to S1P1 and S1P3, S1P2 is a clearly proatherogenic receptor to act on the three cell types, macrophages, ECs and SMCs [22]. The activity of S1P2 seems to be diverse compared with chemokine receptors; S1P2 is involved in modified LDL uptake, cytokine production, migration, eNOS activation, and apoptosis in these cells. Systemic long-term administration of a selective S1P2-blocker can recapitulate the favorable effects of S1P2-deficiency without overt toxicity. As human monocytes and macrophages express S1P2 as in mice [83], S1P2 likely plays a similar role in human atherosclerotic lesion formation. Therefore, a selective S1P2-blocker could have clinical benefit as a new therapeutics for atherosclerosis. The combination of an S1P2-blocker and statins may be of great use. Thus, S1P receptors, particularly S1P2, are promising therapeutic targets for atherosclerosis.


Acknowledgments
This work was supported by grants from the Ministry of Education, Science, Sports and Culture of Japan and the Japan Society for the Promotion of Science, and funds from the Kanazawa University Strategic Research Development Program, the Institute of Seizon and Life Sciences and AstraZeneca.
References
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar]
- Zhang, H.; Desai, N.N.; Olivera, A.; Seki, T.; Brooker, G.; Spiegel, S. Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J. Cell Biol. 1991, 144, 155–167. [Google Scholar]
- Bornfeldt, K.E.; Graves, L.M.; Raines, E.W.; Igarashi, Y.; Wayman, G.; Yamamura, S.; Yatomi, Y.; Sidhu, J.S.; Krebs, E.G.; Hakomori, S.; Ross, R. Sphingosine-1-phosphate inhibits PDGF-induced chemotaxis of human arterial smooth muscle cells: spatial and temporal modulation of PDGF chemotactic signal transduction. J. Cell Biol. 1995, 130, 193–206. [Google Scholar]
- Postma, F.R.; Jalink, K.; Hengeveld, T.; Moolenaar, W.H. Sphingosine-1-phosphate rapidly induces Rho-dependent neurite retraction: action through a specific cell surface receptor. EMBO J. 1996, 15, 2388–2392. [Google Scholar]
- Lee, M.J.; Thangada, S.; Claffery, K.P.; Ancellin, N.; Liu, C.H.; Kluk, M.; Volpi, M.; Sha'afi, R.I.; Hla, T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 1999, 99, 301–312. [Google Scholar]
- Takuwa, Y. Sphingosine-1-phosphate signaling and biological activities in the cardiovascular system. Biochim. Biophys. Acta 2008, 1781, 483–488. [Google Scholar]
- Yatomi, Y.; Igarashi, Y.; Yang, L.; Hisano, N.; Qi, R.; Asazuma, N.; Satoh, K.; Ozaki, Y.; Kume, S. Sphingosine 1-phosphate, a bioactive sphingolipid abundantly stored in platelets, is a normal constituent of human plasma and serum. J. Biochem. 1997, 121, 969–973. [Google Scholar]
- Sachinidis, A.; Kettenhofen, R.; Seewald, S.; Gouni-Berthold, I.; Schmitz, U.; Seul, C.; Ko, Y.; Vetter, H. Evidence that lipoproteins are carriers of bioactive factors. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2412–2421. [Google Scholar]
- Kimura, T.; Sato, K.; Kuwabara, A.; Tomura, H.; Ishiwara, M.; Kobayashi, I.; Ui, M.; Okajima, F. Sphingosine 1-phosphate may be a major component of plasma lipoproteins responsible for the cytoprotective actions in human umbilical vein endothelial cells. J. Biol. Chem. 2001, 276, 31780–31785. [Google Scholar]
- Cockerill, G.W.; Rye, K.A.; Gamble, J.R.; Vadas, M.A.; Barter, P.J. High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1987–1994. [Google Scholar]
- Watson, A.D.; Berliner, J.A.; Hama, S.Y.; La Du, B.N.; Faull, K.F.; Fogelman, A.M.; Navab, M. Protective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J. Clin. Invest. 1995, 96, 2882–2891. [Google Scholar]
- Wang, X.; Collins, H.L.; Ranalletta, M.; Fuki, I.V.; Billheimer, J.T.; Rothblat, G.H.; Tall, A.R.; Rader, D.J. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J. Clin. Invest. 2007, 117, 2216–2224. [Google Scholar]
- Kimura, T.; Tomura, H.; Mogi, C.; Kuwabara, A.; Damirin, A.; Ishizuka, T.; Sekiguchi, A.; Ishiwara, M.; Im, D.S.; Sato, K.; Murakami, M.; Okajima, F. Role of scavenger receptor class B type I and sphingosine 1-phosphate receptors in high density lipoprotein-induced inhibition of adhesion molecule expression in endothelial cells. J. Biol. Chem. 2006, 281, 37457–37467. [Google Scholar]
- Zhang, B.; Tomura, H.; Kuwabara, A.; Kimura, T.; Miura, S.; Noda, K.; Okajima, F.; Saku, K. Correlation of high density lipoprotein (HDL)-associated sphingosine 1-phosphate with serum levels of HDL-cholesterol and apolipoproteins. Atherosclerosis 2005, 178, 199–205. [Google Scholar]
- Tölle, M.; Pawlak, A.; Schuchardt, M.; Kawamura, A.; Tietge, U.J.; Lorkowski, S.; Keul, P.; Assman, G.; Chun, J.; Levkau, B.; van der Giet, M.; Nofer, J.R. HDL-associated lysosphingolipids inhibit NAD(P)H oxidase-dependent monocyte chemoattractant protein-1 production. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1542–1548. [Google Scholar]
- Xia, P.; Gamble, J.R.; Rye, K.A.; Wang, L.; Hu, C.S.T.; Cockerill, P.; Khew-Goodall, Y.; Bert, A.G.; Barter, P.J.; Vadas, M.A. Tumor necrosis factor-a induces adhesion molecule expression through the sphingosine kinase pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 14196–14201. [Google Scholar]
- Xia, P.; Vadas, M.A.; Rye, K.A.; Barter, P.J.; Gamble, J.R. High density lipoproteins (HDL) interrupt the sphingosine kinase signaling pathway. J. Biol. Chem. 1999, 274, 33143–33147. [Google Scholar]
- Tölle, M.; Levkau, B.; Kleuser, B.; van der Giet, M. Sphingosine-1-phosphate and FTY720 as anti-atherosclerotic lipid compounds. Eur. J. Clin. Invest. 2007, 37, 171–179. [Google Scholar]
- Nofer, J.R.; Bot, M.; Brodde, M.; Taylor, P.J.; Salm, P.; Brinkmann, V.; van Berkel, T.; Assmann, G.; Biessen, E.A.L. FTY720, a synthetic sphingosine 1 phosphate analogue, inhibits development of atherosclerosis in low-density lipoprotein receptor deficient mice. Circulation 2007, 115, 501–508. [Google Scholar]
- Keul, P.; Tölle, M.; Lucke, S.; von Wnuck Lipinski, K.; Heusch, G.; Schuchardt, M.; van der Giet, M.; Levkau, B. The sphingosine-1-phosphate analogue FTY720 reduces atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 607–613. [Google Scholar]
- Wang, F.; Okamoto, Y.; Inoki, I.; Yoshioka, K.; Du, W.; Qi, X.; Takuwa, N.; Gonda, K.; Yamamoto, Y.; Ohkawa, R.; Nishiuchi, T.; Sugimoto, N.; Yatomi, Y.; Mitsumori, K.; Asano, M.; Kinoshita, M.; Takuwa, Y. Sphingosine-1-phosphate receptor-2 deficiency leads to inhibition of macrophage proinflammatory activities and atherosclerosis in apoE-deficient mice. J. Clin. Invest. 2010, 120, 3979–3995. [Google Scholar]
- Skoura, A.; Michoud, J.; Im, D.S.; Thangada, S.; Xiong, Y.; Smith, J.; Hla, T. Sphingosine-1-phosphate receptor-2 function I myeloid cells regulates vascular inflammation and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010. [Google Scholar] [CrossRef]
- Augé, N.; Nikolova-Karakashian, M.; Carpentier, S.; Parthasarathy, S.; Négre-Salvayre, R.; Merril, A.H., Jr.; Levade, T. Role of sphingosine 1-phosphate in the mitogenesis induced by oxidized low density lipoprotein in smooth muscle cells via activation of sphingomyelinase, ceramidase, and sphingosine kinase. J. Biol. Chem. 1999, 274, 21533–21538. [Google Scholar]
- Kohama, T.; Olivera, A.; Edsall, L.; Nagiec, M.M.; Dickson, R.; Spiegel, S. Molecular cloning and functional characterization of murine sphingosine kinase. J. Biol. Chem. 1998, 273, 23722–23728. [Google Scholar]
- Liu, H.; Sugiura, M; Nava, V.E.; Edsall, L.C.; Kono, K.; Poulton, S.; Milstien, S.; Kohama, T.; Spiegel, S. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J. Biol. Chem. 2000, 275, 19513–19520. [Google Scholar]
- Mizugishi, K.; Yamashita, T.; Olivera, A.; Miller, G.F.; Spiegel, S.; Proia, R.L. Essential role for sphingosine kinases in neural and vascular development. Mol. Cell. Biol. 2005, 25, 11113–11121. [Google Scholar]
- Mizugishi, K.; Li, C.; Olivera, A.; Bielawski, J.; Bielawska, A.; Deng, C.X.; Proia, R.L. Maternal disturbance in activated sphingolipid metabolism causes pregnancy loss in mice. J. Clin. Invest. 2007, 117, 2993–3006. [Google Scholar]
- Mandala, S.M.; Thornton, R.; Galve-Roperh, I.; Poulton, S.; Peterson, C.; Olivera, A.; Bergstrom, J.; Kurtz, M.B.; Spiegel, S. Molecular cloning and characterization of a lipid phosphohydrolase that degrades sphingosine-1-phosphate and induces cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 7859–7864. [Google Scholar]
- Zhou, J.; Saba, J.D. Identification of the first mammalian sphingosine phosphate lyase gene and its functional expression in yeast. Biochem. Biophys. Res. Commun. 1998, 242, 502–507. [Google Scholar]
- Mitra, P.; Oskeritzian, C.A.; Payne, S.G.; Beaven, M.A.; Milstien, S.; Spiegel, S. Role of ABCC1 in export of sphingosine 1-phosphate from mast cells. Proc. Natl. Acad. Sci. USA 2006, 103, 16394–16399. [Google Scholar]
- Sato, K.; Malchikhuu, E.; Horiuchi, Y.; Mogi, C.; Tomura, H.; Tosaka, M.; Kuwabara, A.; Okajima, F. Critical role of ABCA1 transporter in sphingosine 1-phosphate release from astrocytes. J. Neurochem. 2007, 103, 2610–2619. [Google Scholar]
- Takabe, K.; Kim, R.H.; Allegood, J.C.; Mitra, P.; Ramachandran, S.; Nagahashi, M.; Harikumar, K.B.; Hait, N.C.; Milstien, S.; Spiegel, S. Estradial induces export of sphingosine-1-phosphate from breast cancer cells via ABCC1 and ABCG2. J. Biol. Chem. 2010, 285, 10477–10486. [Google Scholar]
- Osborne, N.; Brand-Arzamendi, K.; Ober, E.A.; Jin, S.W.; Verkade, H.; Holtzman, N.G.; Yelon, D.; Stainier, D.Y. The spinster homolog, two of hearts, is required for sphingosine 1-phosphate signaling in zebrafish. Curr. Biol. 2008, 18, 1882–1888. [Google Scholar]
- Kawahara, A.; Nishi, T.; Hisano, Y.; Fukui, H.; Yamaguchi, A.; Mochizuki, N. The sphingolipid transporter spns2 functions in migration of zebrafish myocardial precursors. Science 2009, 323, 524–527. [Google Scholar]
- Venkataraman, K.; Thangada, S.; Michaud, J.; Oo, M.L.; Ai, Y.; Lee, Y.M.; Wu, M.; Parikh, N.S.; Khan, F.; Proia, R.L.; Hla, T. Extracellular export of sphingosine kinase-1a contributes to the vascular S1P gradient. Biochem. J. 2006, 397, 461–471. [Google Scholar]
- Venkataraman, K.; Lee, Y.M.; Michaud, J.; Thangada, S.; Ai, Y.; Bonkovsky, H.L.; Parikh, N.S.; Habrukowich, C.; Hla, T. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ. Res. 2008, 102, 630–632. [Google Scholar]
- Pappu, R.; Schwab, S.R.; Cornelissen, I.; Pereira, J.P.; Regard, J.B.; Xu, Y.; Camerer, E.; Zheng, Y.W.; Huang, Y.; Cyster, J.G.; Coughlin, S.R. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 2007, 316, 295–298. [Google Scholar]
- Hänel, P.; Andréani, P.; Gräler, M.H. Erythrocytes store and release sphingosine 1-phosphate in blood. FASEB J. 2007, 21, 1202–1209. [Google Scholar]
- Pham, T.H.; Baluk, P.; Grigorova, I.; Bankovich, A.J.; Pappu, R.; Coughlin, S.R.; McDonald, D.M.; Schwab, S.R.; Cyster, J.G. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J. Exp. Med. 2010, 207, 17–27. [Google Scholar]
- Zachariah, M.A.; Cyster, J.G. Neural crest-derived pericytes promote egress of mature thymocytes at the corticomedullary junction. Science 2010, 328, 1129–1135. [Google Scholar]
- Olivera, A.; Mizugishi, K.; Tikhonova, A.; Ciaccia, L.; Odom, S.; Proia, R.L.; Rivera, J. The sphingosine kinase-sphingosine-1-phosphate axis is a determinant of mast cell function and anaphylaxis. Immunity 2007, 26, 287–297. [Google Scholar]
- Puneet, P.; Yap, C.T.; Wong, L.; Lam, Y.; Koh, D.R.; Moochhala, S.; Pfeilscifter, J.; Huwiler, A.; Melendez, A.J. SphK1 regulates proinflammatory responses associated with endotoxin and polymicrobial sepsis. Science 2010, 328, 1290–1294. [Google Scholar]
- Hla, T.; Macig, T. Anabundant transcript induced in differentiating human endothelial cells encodes a polypeptide with a structural similarities to G-protein-coupled receptors. J. Biol. Chem. 1990, 265, 9308–9313. [Google Scholar]
- Okazaki, T.; Ishizuka, N.; Sakurai, T.; Kurokawa, L.; Goto, K.; Kumada, M.; Takuwa, Y. Molecular cloning of a novel putative G protein-coupled receptor expressed in the cardiovascular system. Biochem. Biophys. Res. Commun. 1993, 190, 1104–1109. [Google Scholar]
- Yamaguchi, F.; Tokuda, M.; Hatase, O.; Brenner, S. Molecular cloning of the novel human G protein-coupled receptor (GPCR) gene mapped on chromosome 9. Biochem. Biophys. Res. Commun. 1996, 227, 608–614. [Google Scholar]
- An, S.; Bleu, T.; Hallmark, O.G.; Goetzl, E.J. Characterizationof a novel subtype of human G protein-coupled receptor for lysophosphatidic acid. J. Biol. Chem. 1998, 273, 7906–7910. [Google Scholar]
- Im, D.S.; Clemens, J.; Macdonald, T.L.; Lynchu, K.R. Characterization of the human and mouse sphingosine 1-phosphate receptor, S1P5 (Edg-8): structure-activity relationship of sphingosine 1-phosphate receptors. Biochemistry 2001, 40, 14053–14060. [Google Scholar]
- Okamoto, H.; Takuwa, N.; Gonda, K.; Okazaki, H.; Chang, K.; Yatomi, Y.; Shigematsu, H.; Takuwa, Y. EDG1 is a functional sphingosine-1-phosphate receptor that is linked via a Gi/o to multiple signaling pathways, including phospholipase C activation, Ca2+ mobilization, Ras-mitogen-activated protein kinase activation, and adenylate cyclase inhibition. J. Biol. Chem. 1998, 273, 27104–27110. [Google Scholar]
- Gonda, K.; Okmaoto, H.; Takuwa, N.; Yatomi, Y.; Okazaki, H.; Kimura, S.; Sillard, R.; Harii, K.; Takuwa, Y. The novel sphingosine 1-phosphate receptor AGR16 is coupled via pertussis toxin-sensitive and –insensitive G-proteins to multiple signaling pathways. Biochem. J. 1999, 337, 67–75. [Google Scholar]
- Okamoto, H.; Takuwa, N.; Yatomi, Y.; Gonda, K.; Shigematsu, H.; Takuwa, Y. EDG3 is a functional receptor specific for sphingosine 1-phosphate and sphingosylphosphocholine with signaling characteristics distinct from EDG1 and AGR16. Biochem. Biophys. Res. Commun. 1999, 260, 203–208. [Google Scholar]
- Okamoto, H.; Takuwa, N.; Yokomizo, T.; Sugimoto, N.; Sakurada, S.; Shigematsu, H.; Takuwa, Y. Inhibitory regulation of Rac activation, membrane ruffling, and cell migration by the G protein-coupled sphingosine-1-phosphate receptor EDG5 but not EDG1 or EDG3. Mol. Cell. Biol. 2000, 20, 9247–9261. [Google Scholar]
- Ishii, I.; Friedman, B.; Ye, X.; Kawamura, S.; McGiffert, C.; Contos, J.J.; Kingsbury, M.A.; Zhang, G.; Brown, J.H.; Chun, J. Selective loss of sphingosine 1-phosphate signaling with no obvious phenotypic abnormality in mice lacking its G protein-coupled receptor, LP(B3)/EDG-3. J. Biol. Chem. 2001, 276, 33697–33704. [Google Scholar]
- Sugimoto, N.; Takuwa, N.; Okamoto, H.; Sakurada, S.; Takuwa, Y. Inhibitory and stimulatory regulation of Rac and cell motility by the G12/13-Rho and Gi pathways integrated downstream of a single G protein-coupled sphingosine-1-phosphate receptor isoform. Mol. Cell. Biol. 2003, 23, 1534–1545. [Google Scholar]
- Sanchez, T.; Skoura, A.; Wu, M.T.; Casserly, B.; Harrington, E.O.; Hla, T. Induction of vascular permeability by the sphingsine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1312–1318. [Google Scholar]
- Hait, N.C.; Allegood, J.; Maceyka, M.; Strub, G.M.; Harikumar, K.B.; Singh, S.K.; Luo, C.; Marmorstein, R.; Kordula, T.; Milstien, S.; Spiegel, S. Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 2009, 325, 1254–1257. [Google Scholar]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; Milstien, S.; Spiegel, S. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar]
- Ryu, Y.; Takuwa, N.; Sugimoto, N.; Sakurada, S.; Usui, S.; Okamoto, H.; Matsui, O.; Takuwa, Y. Sphingosine-1-phosphate, a platelet-derived lysophospholipid mediator, negatively regulates cellular Rac activity and cell migration in vascular smooth muscle cells. Circ. Res. 2002, 90, 335–332. [Google Scholar]
- Inoki, I.; Takuwa, N.; Sugimoto, N.; Yoshioka, K.; Takata, S.; Kaneko, S.; Takuwa, Y. Negative regulation of endothelial morphogenesis and angiogenesis by S1P2 receptor. Biochem. Biophys. Res. Commun. 2006, 346, 293–300. [Google Scholar]
- Kuhlencordt, P.J.; Gyurko, R.; Han, F.; Scherrer-Crosbie, M.; Aretz, T.H.; Hajiar, R.; Picard, M.H.; Huang, P.L. Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease in apolipoprotein E/endothelial nitric oxide synthase double-knockout mice. Circulation 2001, 104, 448–454. [Google Scholar]
- Nofer, J.R.; van der Giet, M.; Tölle, M.; Wolinska, I.; von Wnuck Lipinski, K.; Baba, H.A.; Tietge, U.J.; Godecke, A.; Ishii, I.; Kleuser, B.; Schäfers, M.; Fobker, M.; Zidek, W.; Assmann, G.; Chun, J.; Levkau, B. HDL induces NO-dependent vasorelaxation via the lysophospholipid receptor S1P3. J. Clin. Invest. 2004, 113, 569–581. [Google Scholar]
- Tölle, M.; Levkau, B.; Keul, P.; Brinkmann, V.; Giebing, G.; Schonfelder, G.; Schäfers, M.; von Wnuck Lipinski, K.; Jankowski, J.; Jankowski, V.; Chun, J.; Zidek, W.; van der Giet, M. Immunomodulator FTY720 induced eNOS-dependent arterial vasodilatation via the lysophopholipid receptor S1P3. Circ. Res. 2005, 96, 913–920. [Google Scholar]
- Peng, X.; Hassoun, P.M.; Sammani, S.; McVerry, B.J.; Burne, M.J.; Rabb, H.; Pearse, D.; Tuder, R.M.; Garcia, J.G. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am. J. Respir. Crit. Care Med. 2004, 169, 1245–1251. [Google Scholar]
- Lee, J.F.; Zeng, Q.; Ozaki, H.; Wang, L.; Hand, A.R.; Hla, T.; Wang, E.; Lee, J. Dual roles of tight junction-associated protein, zonula occludens-1, in sphingosine 1-phosphate-mediated endothelial chemotaxis and barrier integrity. J. Biol. Chem. 2006, 281, 29190–29200. [Google Scholar]
- Camerer, E.; Regard, J.B.; Cornelissen, I.; Srinivasan, Y.; Duong, D.N.; Palmer, D.; Pham, T.H.; Wong, J.S.; Pappu, R.; Coughlin, S.R. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J. Clin. Invest. 2009, 119, 1871–1879. [Google Scholar]
- Marsolais, D.; Rosen, H. Chemical modulators of sphingosine-1-phosphate receptors as barrier-oriented therapeutic molecules. Nat. Rev. Drug Discov. 2009, 8, 297–307. [Google Scholar]
- Lee, H.; Lin, C.I.; Liao, J.J.; Lee, Y.W.; Yang, H.Y.; Lee, C.Y.; Hsu, H.Y.; Wu, H.L. Lysophopholipids increase ICAM-1 expression in HUVEC through a Gi- and NF-kappaB-dependent mechanism. Am. J. Physiol. Cell Physiol. 2004, 287, C1657–C1666. [Google Scholar]
- Krump-Konvalinkova, V.; Yasuda, S.; Rubic, T.; Makarova, N.; Mages, J.; Eri, W.; Vosseler, C.; Kirkpatrick, C.J.; Tigyi, G.; Siess, W. Stable knock-down of the sphingosine 1-phosphate receptor S1P1 influences multiple functions of human endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 546–552. [Google Scholar]
- Bolick, D.T.; Srinivasan, S.; Kim, K.W.; Hatley, M.E.; Clemens, J.J.; Whetzel, A.; Ferger, N.; Macdonald, T.L.; Davis, M.D.; Tsao, P.S.; Lynch, K.R.; Hedrick, C.C. Sphingosine-1-phosphate prevents tumor necrosis factor-alpha-mediated monocyte adhesion to aortic endothelium in mice. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 976–981. [Google Scholar]
- Theilmeler, G.; Schmidt, C.; Hermann, J.; Keul, P.; Schäfers, M.; Herrgott, I.; Mersmann, J.; Larmann, J.; Hermann, S.; Stypmann, J.; et al. High-density lipoproteins and their constituent, sphingosine-1-phosphate, deirectly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophosphlipid receptor. Circulation 2006, 114, 1403–1409. [Google Scholar]
- Whetzel, A.M.; Bolick, D.T.; Srinivasan, S.; Macdonald, T.L.; Morris, M.A.; Ley, K.; Hedrick, C.C. Sphingosine-1 phosphate prevents monocyte/endothelial interactions in tyoe 1 diabetic NOD mice through activation of the S1P1 receptor. Circ. Res. 2006, 99, 731–739. [Google Scholar]
- Aoki, S.; Yatomi, Y.; Shimosawa, T.; Yamashita, H.; Kitayama, J.; Tsuno, N.H.; Takahashi, K.; Ozaki, T. The suppressive effct of sphingosine 1-phosphate on monocyte-endothelium adhesion may be mediated by the rearrangement of the endothelial integrins alpha(5)beta(1) and alpha(v)beta(3). J. Thromb. Haemost. 2007, 5, 1292–1301. [Google Scholar]
- Lin, C.I.; Chen, C.N.; Chen, J.H.; Lee, H. Lysophospholipids increase IL-8 and MCP-1 expression in human umbilical cord vein endothelial cells through an IL-1-dependent mechanism. J. Cell Biochem. 2006, 99, 1216–1232. [Google Scholar]
- Lin, C.I.; Chen, C.N.; Lin, P.W.; Lee, H. Sphingosine 1-phosphate regulates inflammation-related genes in human endothelial cells through S1P1 and S1P3. Biochem. Biophys. Res. Commun. 2007, 355, 895–901. [Google Scholar]
- Klul, M.J.; Hla, T. Role of the sphingosine 1-phosphate receptor EDG-1 in vascular smooth muscle cell proliferation and migration. Circ. Res. 2001, 89, 496–502. [Google Scholar]
- Usui, S.; Sugimoto, N.; Takuwa, N.; Sakagami, S.; Takata, S.; Kaneko, S.; Takuwa, Y. Blood lipid mediator sphingosine 1-phosphate potently stimulates platelet-derived growth factor-A and -B chain expression through S1P1-Gi-Ras-MAPK-dependent induction of Kruppel-like factor 5. J. Biol. Chem. 2004, 279, 12300–12311. [Google Scholar]
- Takashima, S.; Sugimoto, N.; Takuwa, N.; Okamoto, Y.; Yoshioka, K.; Takamura, M.; Takata, S.; Kaneko, S.; Takuwa, Y. G12/13 and Gq mediate S1P2-induced inhibition of Rac and migration in vascular smooth muscle in a manner dependent on Rho but not Rho kinase. Cardiovasc. Res. 2008, 79, 689–697. [Google Scholar]
- Shimizu, T.; Nakazawa, T.; Cho, A.; Dastvan, F.; Shilling, D.; Daum, G.; Reidy, M.A. Sphingosine 1-phosphate receptor 2 negatively regulates neointimal formation in mouse arteries. Circ. Res. 2007, 101, 995–1000. [Google Scholar]
- Wamhoff, B.R.; Lynch, K.R.; Macdonald, T.L.; Owens, G.K. Sphingosine-1-phosphate receptor subtypes differentially regulate smooth muscle phenotype. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1454–1461. [Google Scholar]
- Medlin, M.D.; Staus, D.P.; Dubash, A.D.; Taylor, J.M.; Mack, C.P. Sphingsoine 1-phosphate receptor 2 signals through leukemia associated RhoGEF (LARG), to promote smooth muscle cell differentiation. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1779–1786. [Google Scholar]
- Olivera, A.; Eisner, C.; Kitamura, Y.; Dillahunt, S.; Allende, L.; Tuymetova, G.; Watford, W.; Meylan, F.; Diesner, S.C.; Li, L.; Schnermann, J.; Proia, R.L.; Rivera, J. Sphingosine kinase 1 and sphingosine-1-phosphate receptor 2 are vital to recovery from anaphylactic shock in mice. J. Clin. Invest. 2010, 120, 1429–1440. [Google Scholar]
- Keul, P.; Lucke, S.; von Wnuck Lipinski, K.; Bode, C.; Gräler, M.; Heusch, G.; Levkau, B. Sphingosine-1-phosphate receptor 3 promotes recruitment of monocyte/macrophages in inflammation and atherosclerosis. Circ. Res. 2010. [Google Scholar] [CrossRef]
- Doung, C.Q.; Bared, S.M.; Abu-Khader, A.; Buechler, C.; Schmitz, A.; Schmitz, G. Expression of the lysophospholipid receptor family and investigation of lysophospholid-mediated responses in human macrophages. Biochim. Biophys. Acta 2004, 1682, 112–119. [Google Scholar]
- Fueller, M.; Wang, D.A.; Tigyi, G.; Siess, W. Activation of human monocytic cells by lysophosphatidic acid and sphingosine-1-phosphate. Cell Signal. 2003, 15, 367–375. [Google Scholar]
- Ryu, J.; Kim, H.J.; Chang, E.J.; Huang, H.; Banno, Y.; Kim, H.H. Sphingosine 1-phosphate as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. EMBO J. 2006, 25, 5840–5851. [Google Scholar]
- Hughes, J.E.; Srinivasan, S.; Lynch, K.R.; Proia, R.L.; Ferdek, P.; Hedrick, C.C. Sphingosine-1-phosphate induces an anti-inflammatory phenotype in macrophages. Circ. Res. 2008, 102, 950–958. [Google Scholar]
- Michaud, J.; Im, D.S.; Hla, T. Inhibitory role of sphingosine 1-phosphate receptor 2 in macrophage recruitment during inflammation. J. Immunol. 2010, 184, 1475–1483. [Google Scholar]
- Zhi, L.; Leung, B.P.; Melendez, A.J. Sphingosine kinase 1 regulates pro-inflammatory responses triggered by TNFalpha in primary human monocytes. J. Cell Physiol. 2006, 208, 109–115. [Google Scholar]
- Duenas, A.I.; Aceves, M.; Fernandez-Pisonero, L.; Gomez, C.; Orduna, A.; Crespo, M.S.; Garcia-Rodriguez, C. Selective attenuation of Toll-like receptor2 signaling may explain the atheroprotective effect of sphingosine 1-phosphate. Cardiovasc. Res. 2008, 79, 537–544. [Google Scholar]
- Geng, Y.J.; Libby, P. Progression of atheroma. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1370–1380. [Google Scholar]
- Weigert, A.; Johann, A.M.; von Knethen, A.; Schmidt, H.; Geisslinger, G.; Brune, B. Apoptotic cells promote macrophage survival by releasing the antiapoptotic mediator sphingosine-1-phosphate. Blood 2006, 108, 1635–1642. [Google Scholar]
- Weis, N.; Weigert, A.; von Knethen, A.; Brune, B. Heme oxygenase-1 contributes to an alternative macrophage activation profile induced by apoptotic cell supernatants. Mol. Biol. Cell 2009, 20, 1280–1288. [Google Scholar]
- Kharel, Y.; Lee, S.; Snyder, A.H.; Sheasley-O'neill, S.L.; Morris, M.A.; Setiady, Y.; Zhu, R.; Zigler, M.A.; Burcin, T.L.; Ley, K.; Tung, K.S.; Engelhard, V.H.; Macdonald, T.L.; Pearson-White, S.; Lynch, K.R. Sphingosine kinase 2 is required for modulation of lymphocyte traffic by FTY720. J. Biol. Chem. 2005, 280, 36865–36872. [Google Scholar]
- Chiba, K. FTY720, a new class of immunomodulator, inhibits lymphocyte egress from secondary lymphoid tissue and thymus by agonistic activity at sphingosine 1-phosphate receptors. Pharmacol. Ther. 2005, 108, 308–319. [Google Scholar]
- Mandala, S.; Hajdu, R.; Bergstrom, J.; Quackenbush, E.; Xie, J.; Milligan, J.; Thornton, R.; Shei, G.J.; Card, D.; Keohane, C.; Rosenbach, M.; Hale, J.; Lynch, C.L.; Rupprecht, K.; Parsons, W.; Rosen, H. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science 2002, 296, 346–349. [Google Scholar]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allede, M.L.; Pria, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 2004, 427, 355–360. [Google Scholar]
- Arkas, O.; Kury, P.; Kieseier, B.; Hartung, H.P. Fingolimod is a potential novel therapy for multiple sclerosis. Nat. Rev. Neurol. 2010, 6, 373–382. [Google Scholar]
- Klingenberg, R.; Nofer, J.R.; Rudling, M.; Bea, F.; Blessing, E.; Preusch, M.; Grone, H.J.; Katus, H.A.; Hansson, G.K.; Dengler, T.J. Sphingosine-1-phosphate analogue FTY720 causes lymphocyte redistribution and hypercholesterolemia in apoE-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2392–2399. [Google Scholar]
- Hansson, G.K.; Libby, P. The immune response in atherosclerosis: a double-edged sword. Nat. Rev. Immunol. 2006, 6, 508–519. [Google Scholar]
- Du, W.; Takuwa, N.; Yoshioka, K.; Okamoto, Y.; Gonda, K.; Sugihara, K.; Fukamizu, A.; Asano, M.; Takuwa, Y. S1P(2), the G protein-coupled receptor for sphingosine-1-phosphate, negatively regulates tumor angiogenesis and tumor growth in vivo in mice. Cancer Res. 2010, 70, 772–781. [Google Scholar]
- Wolfrum, S.; Dendorfer, A.; Rikitake, Y.; Stalker, T.J.; Gong, Y.; Scalia, R.; Dominiak, P.; Liao, J.K. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler. Thromb. Vasc. Biol 2004, 24, 1842–1847. [Google Scholar]
- Benitah, S.A.; Valeron, P.F.; Lacal, J.C. ROCK and nuclear factor-kappaB-dependent activation of cyclooxygenase-2 by Rho GTPases: effects on tumor growth and therapeutic consequences. Mol. Biol.Cell 2003, 14, 3041–3054. [Google Scholar]
- Bot, I.; de Jager, S.C.; Zernecke, A.; Lindstedt, K.A.; van Berkel, T.J.; Weber, C.; Biessen, E.A. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation 2007, 115, 2516–2525. [Google Scholar]
- Gu, L.; Okada, Y.; Clinton, S.K.; Gerard, C.; Sukhova, G.K.; Libby, P.; Rollins, B.J. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell. 1998, 2, 275–281. [Google Scholar]
- Zhu, S.N.; Chen, M.; sJongstra-Bilen, J.; Cybulsky, M.I. GM-CSF regulates initiaml cell proliferation in nascent atherosclerotic lesions. J. Exp. Med. 2009, 206, 2141–2149. [Google Scholar]
- Tabas, I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2255–2264. [Google Scholar]
- Ozawa, K.; Hirata, K.; Yamamoto, K. 2003, Pyrazolopyridine derivatives and medicinal use thereof. World Intellectual Property Organization Patent WO/2003/051876 A1. filed December 13, 2002, and issued June 26, 2003. [Google Scholar]
- Rader, D.J.; Daugherty, A. Translating molecular discoveries into new therapies for atherosclerosis. Nature 2008, 451, 904–913. [Google Scholar]
- Laufs, U.; La Fata, V.; Plutzky, J.; Liao, J.K. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation 1998, 97, 1129–1135. [Google Scholar]
- Libby, P.; Aikawa, M. Stabilization of atherosclerotic plaques: new mechanisms and clinical targets. Nat. Med. 2002, 8, 1257–1262. [Google Scholar]
- Aiello, R.J.; Bourassa, P.A.; Lindsey, S.; Weng, W.; Freeman, A.; Showell, H. J. Leukotriene B4 receptor antagonism reduces monocytic foam cells in mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 443–449. [Google Scholar]
- Veillard, N.R.; Kwak, B.; Pelli, G.; Mulhaupt, F.; James, R.W.; Proudfoot, A.E.; Mach, F. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ Res 2004, 94, 253–261. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Okamoto, Y.; Wang, F.; Yoshioka, K.; Takuwa, N.; Takuwa, Y. Sphingosine-1-Phosphate-Specific G Protein-Coupled Receptors as Novel Therapeutic Targets for Atherosclerosis. Pharmaceuticals 2011, 4, 117-137. https://doi.org/10.3390/ph4010117
Okamoto Y, Wang F, Yoshioka K, Takuwa N, Takuwa Y. Sphingosine-1-Phosphate-Specific G Protein-Coupled Receptors as Novel Therapeutic Targets for Atherosclerosis. Pharmaceuticals. 2011; 4(1):117-137. https://doi.org/10.3390/ph4010117
Chicago/Turabian StyleOkamoto, Yasuo, Fei Wang, Kazuaki Yoshioka, Noriko Takuwa, and Yoh Takuwa. 2011. "Sphingosine-1-Phosphate-Specific G Protein-Coupled Receptors as Novel Therapeutic Targets for Atherosclerosis" Pharmaceuticals 4, no. 1: 117-137. https://doi.org/10.3390/ph4010117
APA StyleOkamoto, Y., Wang, F., Yoshioka, K., Takuwa, N., & Takuwa, Y. (2011). Sphingosine-1-Phosphate-Specific G Protein-Coupled Receptors as Novel Therapeutic Targets for Atherosclerosis. Pharmaceuticals, 4(1), 117-137. https://doi.org/10.3390/ph4010117