Introduction
In the last decades, in order to gain more insight into electron transfer processes, extensive studies have been carried out on the optical behavior of chromophoric molecular systems consisting of electron donor and acceptor groups connected via different bridges [
1,
2]. Because of its long fluorescence lifetime (up to 450 ns) [
3], high fluorescence quantum yield [
4] and its ability to act as a donor [
5-
9] as well as acceptor [
9-
12], pyrene has often been chosen as an ideal charge transfer partner. In some cases, the pyrene moiety was attached to chelating “polypyridyl” systems, which are known to coordinate d
7 to d
10 metal-ions, and through this specific metal ion complexation the excited state properties can be tuned. As conjugated spacers between the pyrene and the metal centers, phenylene or ethynylene-phenylene have been reported. Among the innocent metal ions able to coordinate bipyridine units, Zn(II) has received attention because of its ability to enhance the emission of the ligands and tune some of the properties. For example, in the poly- and oligo-phenylene vinylene (OPV) derivatives covalently linked to a bipyridine system reported by Wasielewski
et al. [
13] an enhancement of the electronic delocalization on the polymer backbone and a red shift of the emission was observed upon addition of particular ions [e.g. Zn(II)]. The emissive excited state was suggested to have charge transfer character. More recently, systems have been used in order to show the occurrence of emissive charge-transfer states in their zinc (II) complexes, such as bipyridine linked to a donor group [
14] or to a pyrene unit via an oligo-phenylene bridge [
15,
16], conjugated pyrene-thiophene-terpyridine [
17], or OPV terminated by a terpyridine unit. [
18] Also in free ligands such as bipyridine bound to pyrene [
19] and terpyridine linked in its 4′-position to a dimethylaniline group [
20] charge transfer emission was observed.
The special interest in the terpyridine derivative systems lies in e.g. the strong blue emission of their zinc complexes. Furthermore, the zinc can be used to assemble different units and build up long rod-like linear structures which can be considered coordination polymers. [
21,
22] Due to the dynamic nature of the systems and to the high emission quantum yield in the blue region, zinc bis(phenylterpyridine) derivatives [
23,
24] have attracted the attention in the material-science field. Two recent reviews on polypyridyl systems in conjunction with aromatic units [
25] and with respect to molecular wires [
26] are available.
In order to design systems where the emission can be tuned and to have a full understanding of the free ligand and its behavior once coordinated to zinc, we have investigated the properties of a pyrene substituted terpyridine, its zinc complex and the bis-protonated form of the ligand. The synthesis, electrochemical behavior and the photophysics of these species composed of a terpyridine (Tpy) ligand which is linked to a phenyl substituted with two pyrene units in meta positions (
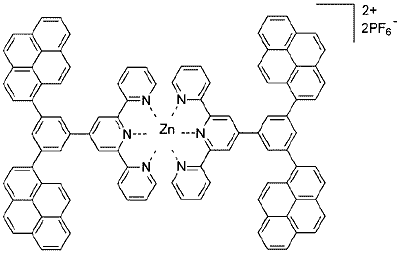
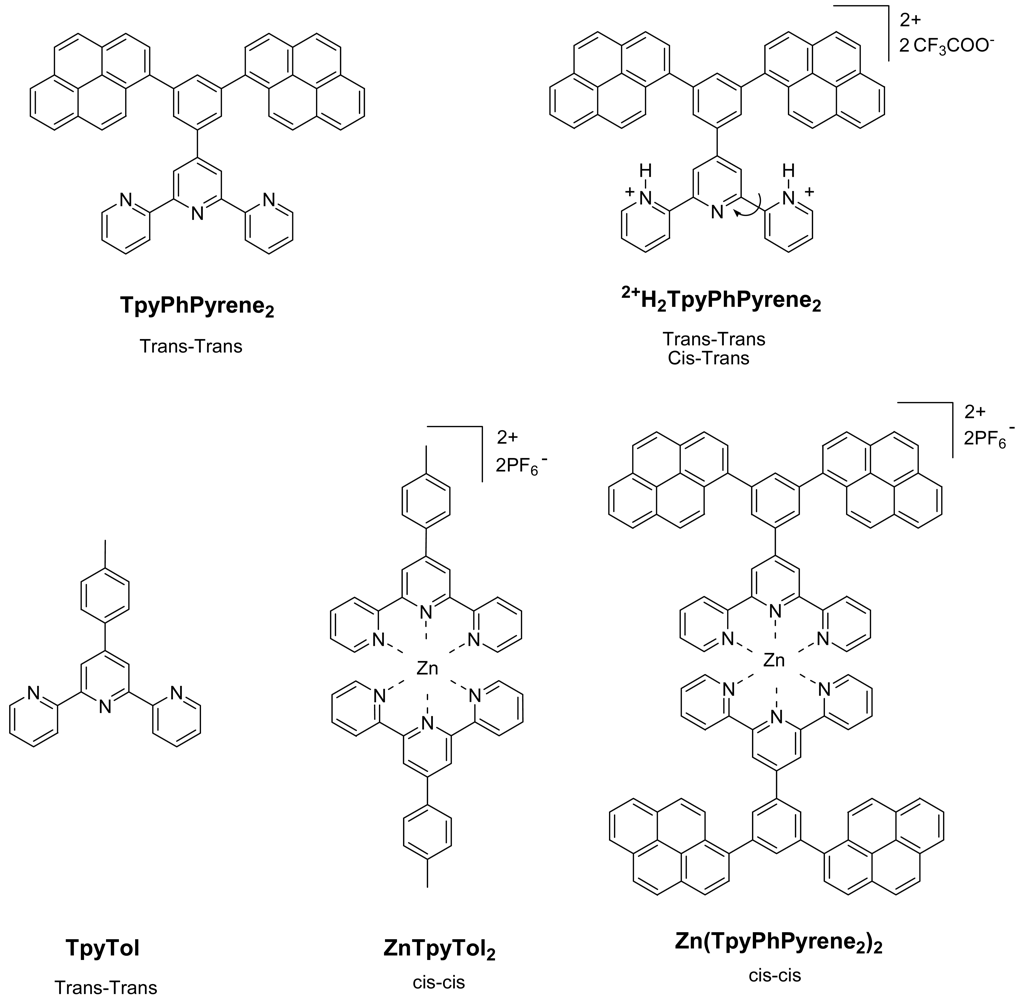
Figure 1) are described.
The term dendrimer, a contraction of the Greek words dendros for “tree” and meros for “part”, can be applied to branched, tree like molecules. Zn(TpyPhPyrene2)2 has four pyrene branches and a Zn(Tpy)2 core and as such can be considered a first generation dendrimer. Expansion of the dendrimer into higher generations can in principle be attained by functionalization of each pyrene with, e.g. a 3,5-bis(pyrene-1-yl)-phenyl unit at the 8-position. Zn(TpyPhPyrene2)2 is a branched molecule and a simple (first generation) model for such highly branched fluorescent dendrimers, but it is also a dendritic polarity probe. As will be shown, Zn(TpyPhPyrene2)2 is very sensitive towards polar solvent molecules and as such can be described as a fluorescent dendritic sensor for the polarity of its environment.
Results and Discussion
Synthesis
The di-halogenated terpyridine (
TpyPhBr2) was prepared by using a slight modification of a literature method [
27] (see Experimental section). The free ligand
TpyPhPyrene2 was synthesized by coupling
TpyPhBr2 with 1-pyreneboronic acid following the Suzuki cross coupling procedure (
Scheme 1) [
28,
29].
The zinc complex was prepared by refluxing
TpyPhPyrene2 with ZnCl
2 in methanol followed by precipitation of the hexafluorophosphate salt by addition of ammonium hexafluorophosphate, as reported before (see
Scheme 1) [
30].
The bis-protonated molecule was prepared in situ prior to measurement by adding 100 μL of a 1 M trifluoroacetic acid solution (in the solvent required) to a 2 mL solution of TpyPhPyrene2 (Abs ≈ 0.2).
The ligand TpyPhPyrene2 and the zinc complex Zn(TpyPhPyrene2)2 were isolated and fully characterized by 1H-NMR, 13C-NMR and High Resolution Electrospray Ionization mass spectrometry.
Electrochemistry
The study of the electrochemical properties of the ligand
TpyPhPyrene2 was performed in deaerated THF (vs SCE). Two irreversible cathodic peaks at -1.57 V (small hump) and -1.91 V (involving at least two electrons) are observed by cyclic voltammetry. On the basis of the comparison to literature data [
31,
32], we attribute the lowest reduction process to the terpyridine reduction in which the central pyridine is reduced first around -1.57 V and the pyrenes reducing at higher voltage (-1.9 V). The oxidation process that occurs at E
p,a= +1.36 V is also irreversible. This is consistent with the oxidation of the pyrene, which occurs at this potential in acetonitrile [
33]. In this case, the lack of reversibility in the redox behavior is suggested to arise because of the formation of a dimer by π-π interaction of one radical cation of pyrene with a second neutral pyrene molecule [
34].
For Zn(TpyPhPyrene2)2, no metal centered redox processes are expected for the d10 zinc ion and none were observed. In this case, the same irreversible redox processes were observed without a significant shift.
As
TpyPhPyrene2 contains an electron donor (pyrene) and an electron acceptor (Tpy), it can display charge transfer characteristics. From the electrochemical data, it is possible to estimate the thermodynamic energy change for electron transfer (Gibbs free energy change: ΔGeT) in such a system by using a Weller-type analysis [
35]. The E
00 can be estimated using the maximum emission of the donor at 77 K in butyronitrile matrix (3.28 eV, see luminescence part). Neglecting the work term, it is found that the ΔG
eT is negative in the solvent in which the electrochemistry was performed, THF, (E
ox - E
red - E
00 = 1.36 + 1.56 - 3.28 = -0.36 V). As the redox processes are irreversible, the value obtained is a rough estimate. This negative value shows that the process is thermodynamically allowed. The increase of the solvent polarity, relative to THF, will result in a more negative ΔG
eT.
UV/Visible absorption
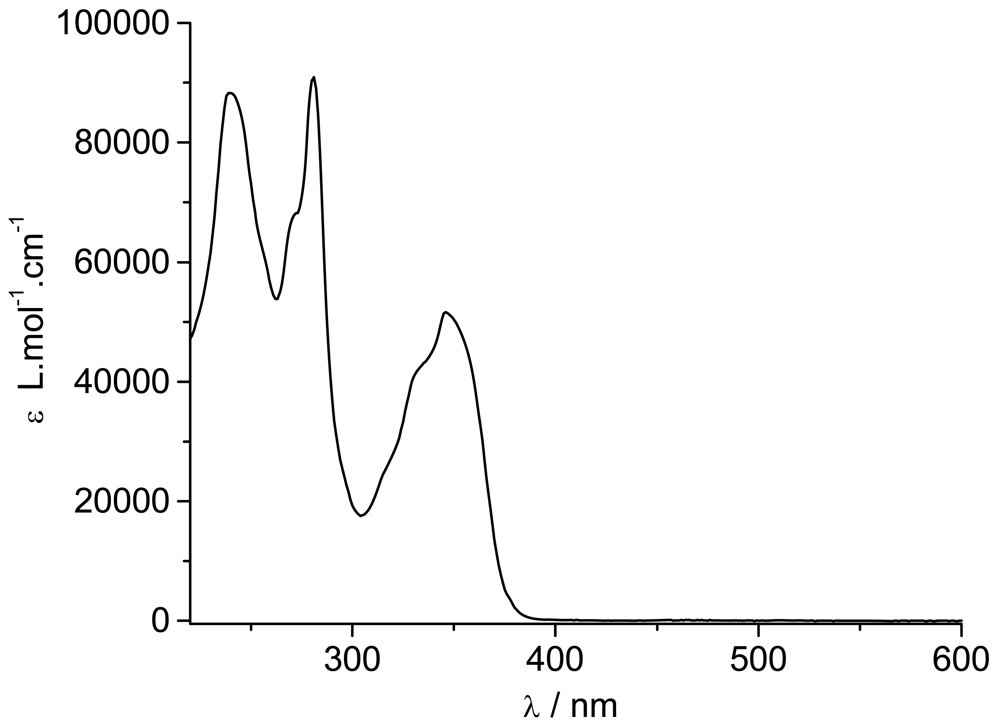
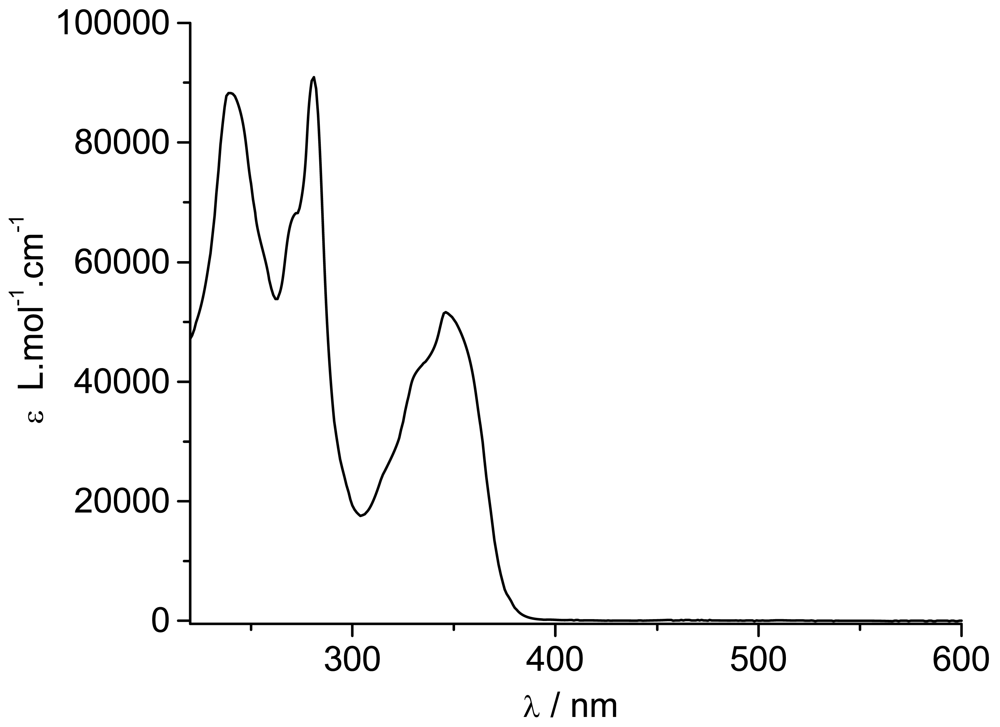
The ground state absorption spectrum of
TpyPhPyrene2 is shown in
Figure 2. It shows three absorption bands centered at 240 nm, 280 nm and 347 nm with two shoulders at 270 nm and 332 nm. The spectra obtained in low polarity solvents (e.g. toluene, tetrahydrofuran, dichloromethane) and in high polarity (e.g. acetonitrile, dimethylformamide) environment are identical, within experimental error.
By comparing the spectrum with those of 1-phenylpyrene [
10,
36] and of substituted phenyl-terpyridine [
20], all bands can be identified. The 240 nm band, the shoulder at 270 nm and the 347 nm bands belong to the allowed π-π* transitions of the phenylpyrene moiety while the band at 280 nm is attributed to π-π* transitions of the Tpy unit. The second shoulder at 332 nm seems to be due to a contribution from both parts. Apart from the Tpy bands, the spectrum consists of the characteristic absorption bands of substituted pyrene where the typical vibronic structure of pyrene is broadened by the interaction between the π-electron systems of the pyrene (or alkylpyrene [
37,
38]) and the phenyl units [
19].
The molar absorption coefficients are quite high for each band (see
Table 1) revealing allowed π-π* transitions (
Table 1). If a weak charge transfer band should be observed, we are expecting it in the UV region (around 340 nm
) as also reported elsewhere [
23,
39].
The UV/Visible spectrum of the zinc complex
Zn(
TpyPhPyrene2)
2 is almost super-imposable over that of the free ligand
TpyPhPyrene2 (
Figure 3). Only the lowest energy transition is slightly blue shifted upon addition of the zinc(II) (from 347 nm to 342 nm). This shift, accompanied by an increase of the molar absorption coefficient, can be attributed to zinc complexes where a new band, growing around 330 nm, is reported. This band is attributed to the cis-cis form for other zinc(II) terpyridine complexes where the nitrogen complexation induces the ligand to convert from the trans-trans to cis-cis form [
40,
41]. Moreover, the increase in the molar absorption coefficient of the 342 nm band is accompanied by a slight decrease of the 281 nm band (from 88,400 L mol
-1 cm
-1 to 78,000 L mol
-1 cm
-1;
Table 1). This increase of the molar absorption coefficient can be attributed to an Intramolecular Ligand Charge Transfer (
1ILCT) transition, which can appear as a shoulder at 340 nm [
23,
39]. In order to identify the ILCT absorption band, a differential spectrum was obtained by subtracting the UV/visible absorption spectrum of
TpyPhPyrene2 from the one of
Zn(
TpyPhpyrene2)
2 (divided by 2) both in THF solution (see
Figure 4).
A band at 340 nm can be observed, which we assign to an
1ILCT transition. The appearance of such a band (with a relatively high extinction coefficient) suggests that upon complexation with the zinc ion, the electron withdrawing character of the Tpy moiety increases and therefore, the donor acceptor interaction becomes stronger. The
1ILCT transition appears to borrow intensity from the pyrene-Tpy based transitions which results in the enhancement of the charge transfer absorption band that is overlapping with the pyrene bands [
42-
46]. The structure in the CT absorption band can be due to the presence of two conformations or to a slight spectral shift as compared to the reference systems.
Upon addition of trifluoroacetic acid to the solution of
TpyPhPyrene2, a bis-protonation is expected [
23]. The absorption spectrum is almost identical to that of the zinc complex (
Figure 3). The main difference is a negligible blue shift of the lowest energy band compared to the free ligand (346 nm for
2+H2TpyPhPyrene2; 347 nm for
TpyPhPyrene2) which shows that upon protonation, conformational changes are not the same as upon zinc complex formation. We believe, in fact, that only one of the pyridine rings changes conformation from trans to cis in order to “coordinate” one proton with the central pyridine [
47]. The molar absorption coefficient of the 347 nm band of
2+H2TpyPhPyrene2, (
Table 1) is higher than that of
TpyPhPyrene2 and
Zn(
TpyPhPyrene2)
2, which indicates that the
1ILCT absorption can be observed here as well and that this transition is even more favorable than for
Zn(
TpyPhPyrene2)
2.
Luminescence study
The fluorescence emission spectra of
TpyPhPyrene2 and
Zn(
TpyPhPyrene2)
2 were found to be independent on the excitation wavelength for all solvents studied. Interestingly, the shape of the emission bands and the emission maxima depend strongly on the solvent used (
Figure 5). These phenomena are attributed to a solvatochromic shift of the emission maximum as often observed for charge transfer transitions [
19,
48-
50]. The shape of the emission bands is independent of the sample concentration (in the 10
-4 to 10
-8 M range), thus ruling out intermolecular excimer formation.
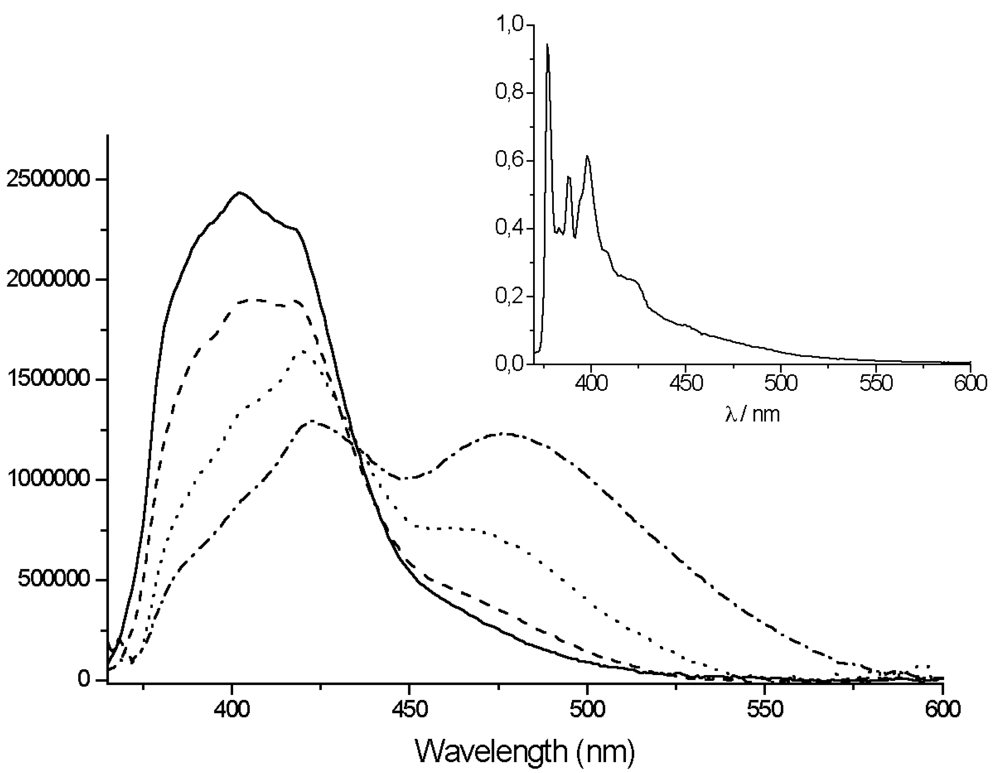
The emission spectrum of the free ligand in THF (
Figure 5) shows a broad but somewhat structured emission band centered at 400 nm that is attributed to the local excited (LE) state of the pyrene. However, in nitrile solvents the spectra show a dual emission with a second band arising around 470 to 480 nm (with increasing polarity).
The appearance of the low energy emission band for the TpyPhPyrene2 ligand in highly polar solvents implies the existence of a low lying excited state polar in nature and stabilized by the solvent that is due to a charge separation between the electron donor (pyrene) and the electron acceptor (terpyridine) moieties of the molecule.
Upon closer examination of
Figure 5, it can be seen that the so called local excited state emission also changes shape with polarity. Whereas the spectra in THF and valeronitrile have a typical shape that can be attributed to “substituted pyrene”, the spectra in propionitrile and acetonitrile clearly have a different shape. This is in contrast with a π–π* transition localized on the pyrene (Ham effect: increase of 0-0 transition with polarity). In fact, these spectral features are very similar to tolyl-terpyridine (
TpyTol) emission in polar solvent. It thus appears that several close lying excited states are present and a reordering of states occurs upon solvent polarity change. A comparison of the emission spectrum of
TpyPhPyrene2 with that of
TpyTol is given in
Figure 6.
Clearly, a simple picture with only one LE state is not fully correct and a manifold of two or more states must be considered as fluorescent levels. Of course, it is not possible to fully rule out the possibility that the 425 nm band is just an enhancement of the vibronic structure of the pyrene (or 1,3-dipyrenyl-benzene) that gains intensity vs the other vibration levels due to the change in solvent.
The excited state lifetimes were determined in a number of solvents (see
table 2). The LE state displays a lifetime between 17 and 20 ns in most non-polar solvents. In polar media, a bi-exponential decay of the emission can be observed due to the formation of a lower lying
1ILCT state (
Figure 5) that has a shorter lifetime (approximately 6 ns,
Table 2) and a lower energy maximum with increasing the polarity of the solvent.
The luminescence quantum yield of the overall emission (LE + ILCT) for
TpyPhPyrene2 varies between 12 % and 29 % (see
Table 2). The population of the CT state can be modulated by changing the polarity in a series of nitrile solvents (
Table 3). The increase of the amplitude of the 6 ns component with increasing the polarity reveals a more efficient transfer to the
1ILCT. In the same table, we have collected the different excited state lifetimes for the two emissions (LE, τ
1 and ILCT, τ
2) as well as the different weights (Amplitude τ). We have analyzed the behavior in a series of nitrile solvent where only the polarity increases going from butyronitrile to acetonitrile. All the spectra have been repeated in deaerated solutions but no influence of oxygen was observed.
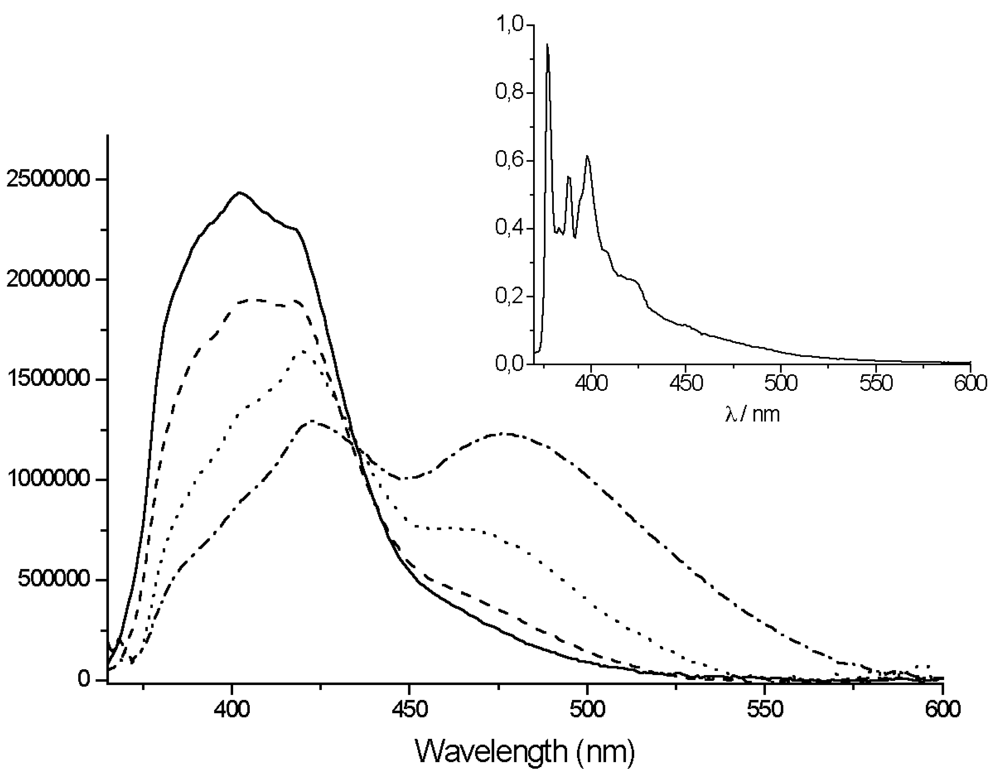
For
Zn(
TpyPhPyrene2)
2, two bands can be observed in the emission spectra (
Figure 7): a sharp local emission band and a broad structureless charge transfer emission band. The high energy band that is slightly structured can be attributed either to the pyrene or to the Tpy perturbed by the zinc ion.
The lowest energy band observed at 518 nm (in THF) to 616 nm (in acetonitrile) is strongly solvatochromic and is red shifted in all solvents, as compared to the
TpyPhPyrene2 emission (
Table 4). Such a solvent dependence is again incompatible with localized π-π* emission from either the Tpy or the pyrene moiety, but it is consistent with emission from a
1ILCT state. This state emits at lower energy when the zinc is coordinated to
TpyPhPyrene2 due to the ionic charges introduced by the zinc which makes the Tpy a stronger electron withdrawing group. A strong decrease of the quantum yield of emission was observed as compared to that of the free ligand (cf.
Tables 2 and
4). The quantum yields measured for
Zn(TpyPhPyrene2)2 show a slight variation: from more than 5% in a nonpolar solvent to less than 0.3% in polar media. The emission intensity drops strongly when passing from dichloromethane (Δƒ = 0.218) to acetone (Δƒ = 0.284).
The emission centered around 400 nm shows a mono-exponential decay on the order of 1 to 2 nanoseconds in every solvent (much shorter than that of the free ligand). The emission lifetime on the red side of the spectrum, which is attributed to the 1ILCT shows a lifetime between 3 and 14 ns. It is interesting to note that for Zn(TpyPhPyrene2)2, the 1ILCT lifetime is decreasing when going to more polar solvents which is in agreement with the decrease in the energy gap between the excited state and the ground state (energy gap law).
The bis-protonated terpyridine 2+H2TpyPhPyrene2 in chlorinated solvents shows the same emission bands as the zinc complex. For this system, the LE state emission can be estimated to be reduced more than 500 times and the 1ILCT emission is more red-shifted. Clearly, protonation or coordination of the nitrogen atoms of the TpyPhPyrene2 strongly increases the non-radiative processes in this system.
In rigid matrices at low temperature (77 K), the fluorescence emission spectra of all compounds show emission bands with vibronic structure and no structureless CT-emission band can be observed (see inset
Figures 5 and
7) indicating that the intramolecular electron transfer does not occur at low temperature, which corroborates the room temperature assignments. The long excited state lifetimes at 77 K (89 ns for
TpyPhPyrene2 and 21 ns for
Zn(TpyPhPyrene2)2) also indicate that the structured fluorescence is a pyrene-based emission since the corresponding Tpy emission decays in less than 1 ns. At this temperature, even for the zinc complex, no emission coming from the Tpy triplet state can be observed as for other Tpy derivatives. This suggests that the triplet state of the Tpy is quenched by a very fast triplet-triplet energy transfer from the Tpy state to the lowest
3Py* state located around 640 nm [
51] which is apparently non-emissive at 77 K in this case.
Transient Absorption spectroscopy
To gain a better insight into the processes in and the nature of the excited states of
TpyPhPyrene2 and of
Zn(TpyPhPyrene2)2 at room temperature, time resolved absorption spectroscopy was employed. Spectra on the nanosecond time scale are depicted in
Figures 8 to
10.
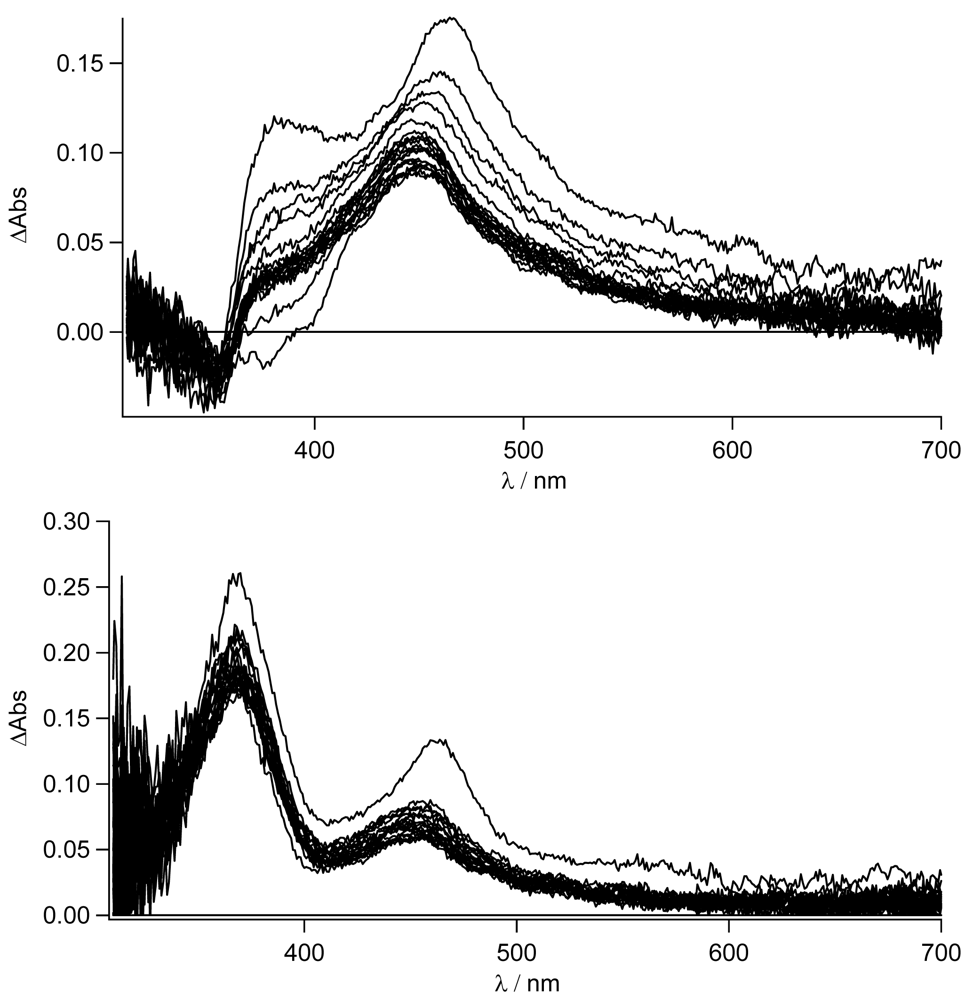
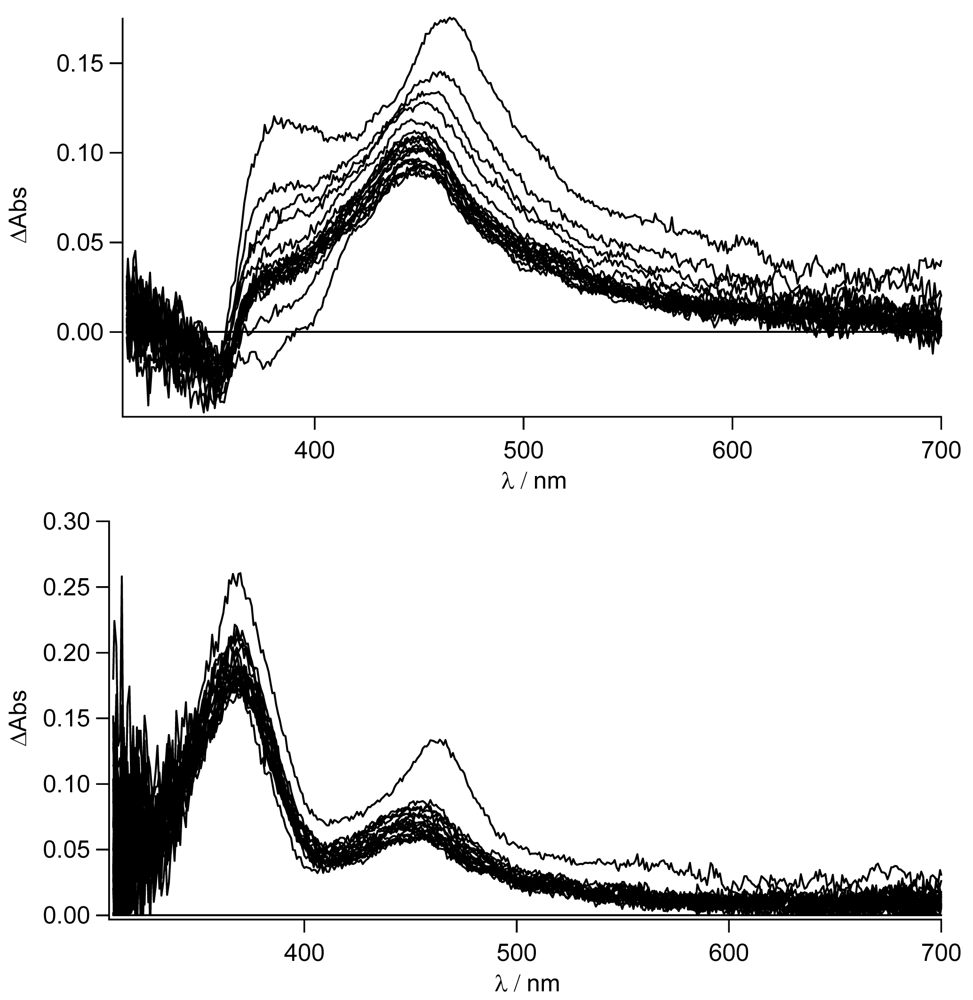
In
Figure 8, the transient absorption spectra of
TpyPhPyrene2 and of
Zn(TpyPhPyrene2)2 in CH
3CN are depicted. Both spectra are characterized by two positive absorption bands at 463 nm and at 380 nm which can be attributed to the radical cation of the pyrene (463 nm) and the radical anion of the terpyridine (380 nm). For both compounds, the charge separated state decays bi-exponential on a 6 and ∼12 ns timescale and is converted into a blue shifted absorption band with a long lifetime. The bi-exponential decay indicates excited state equilibration. The long-lived species has a maximum absorption at 450 nm and can be assigned to a triplet-triplet absorption (see
Figure 8).
It is interesting to notice that, as for the emission spectroscopy, in CH
3CN
TpyPhPyrene2 has a longer lived CT state (τ
em = 6-8 ns, see
Table 3) as compared to that of the zinc complex (τ
em = 3 ns, see
Table 4). In the transient spectra, a similar kinetic behavior is observed. The decay of the triplet state can be followed better in
Figure 9, which shows similar spectra as in
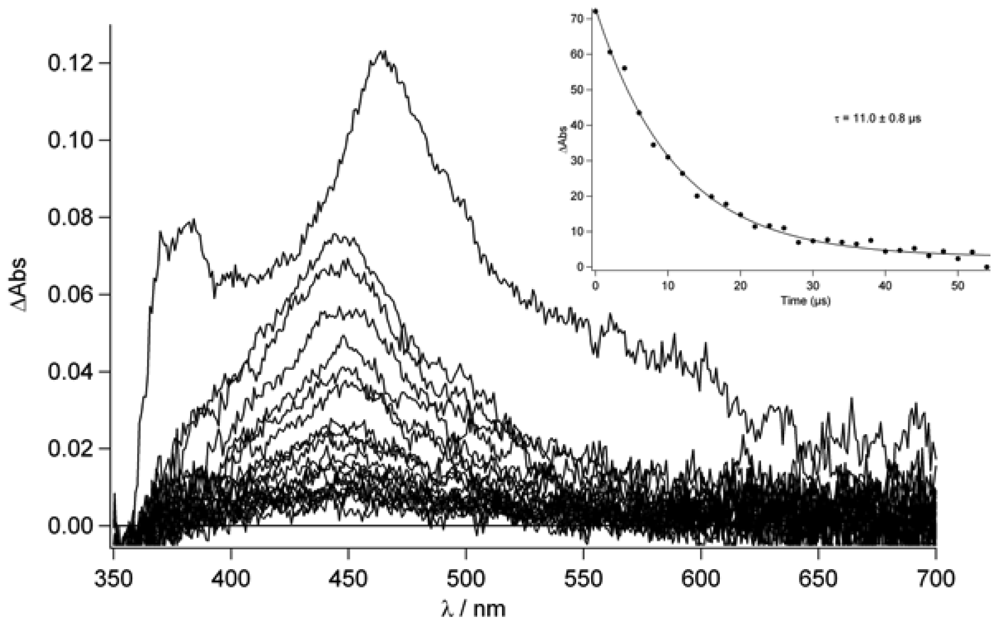
Figure 8 in deaerated conditions and with 2 μs incremental time delay. As it can be seen, the lifetime of this excited state is ca. 11.0 μs (see inset
Figure 9) while, in aerated condition, a lifetime of 240 ns is found.
In order to establish if the triplet state belongs to the pyrene moiety or to the Tpy unit, the transient absorption spectrum of
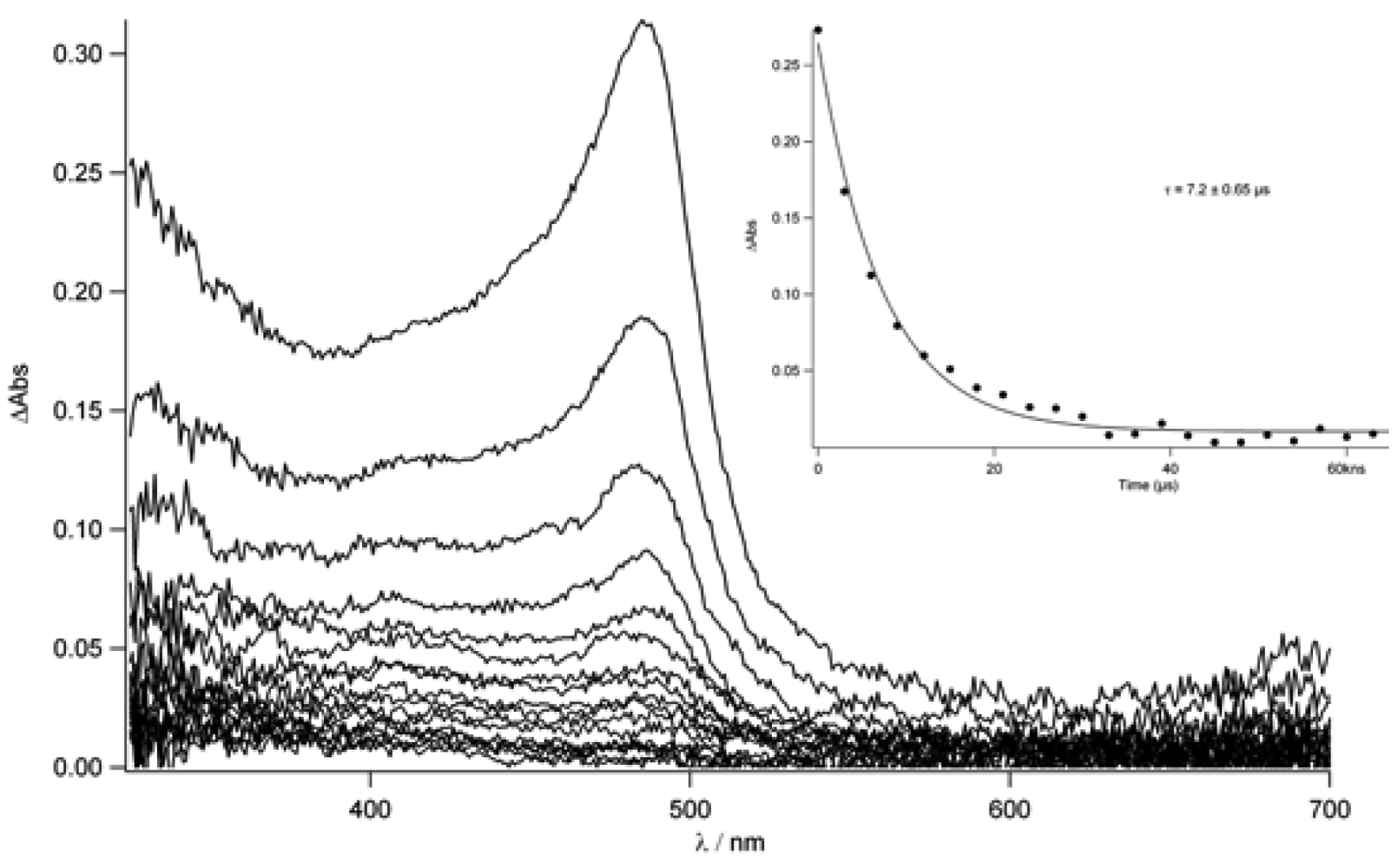
TpyTol in acetonitrile was recorded (
Figure 10). In fact, this spectrum shows a sharp peak around 490 nm which decays within 530 ns in aerated conditions while, after degassing, a longer lifetime was found (7.2 μs). This absorption can clearly be attributed to the terpyridine triplet state (T
1→T
n).
On the basis of this experimental evidence and on the literature reports [
52,
53], we assigned the long lived transition observed in
Figure 9 to the T
1→T
n absorption of the pyrene moiety. In our systems, the T
1→T
n is localized in the 430-460 nm region. It thus seems most likely that phenyl substitution of pyrene shifts its triplet-triplet absorption to slightly longer wavelengths than that reported for unsubstituted pyrene (410-430 nm).
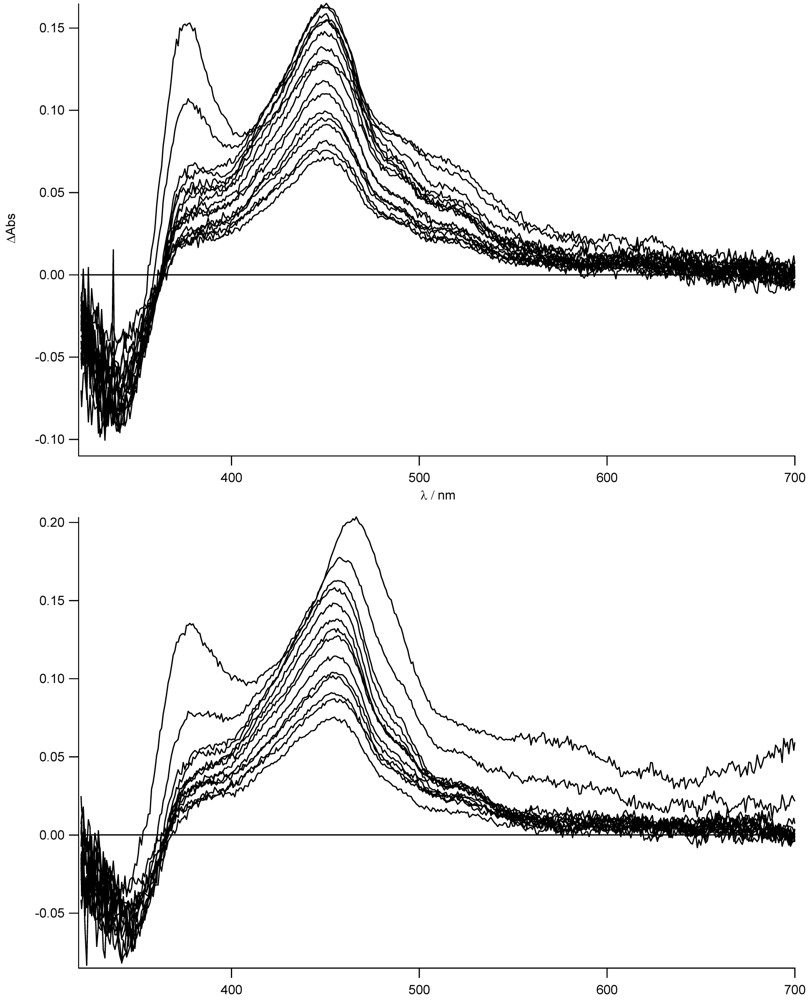
To support the formation of the charge separated state and to prove that this effect is strongly dependent on the solvent and more important on the presence of the zinc, we have measured the transient absorption spectra in THF for
TpyPhPyrene2 and
Zn(TpyPhPyrene2)2. The results are shown in
Figure 11.
As can be easily seen for the free ligand in the nonpolar solvent, no evidence (lack of the 463 nm band) of the CT state is observed. The local singlet excited state, populated in the first nanoseconds, is visible (see also later) and transforms into the long lived triplet state. On the other hand, for the zinc complex in THF, the stronger charge transfer character of its excited state is mirrored in the formation of the charge separated state at 463 nm as was already reported in acetonitrile (see
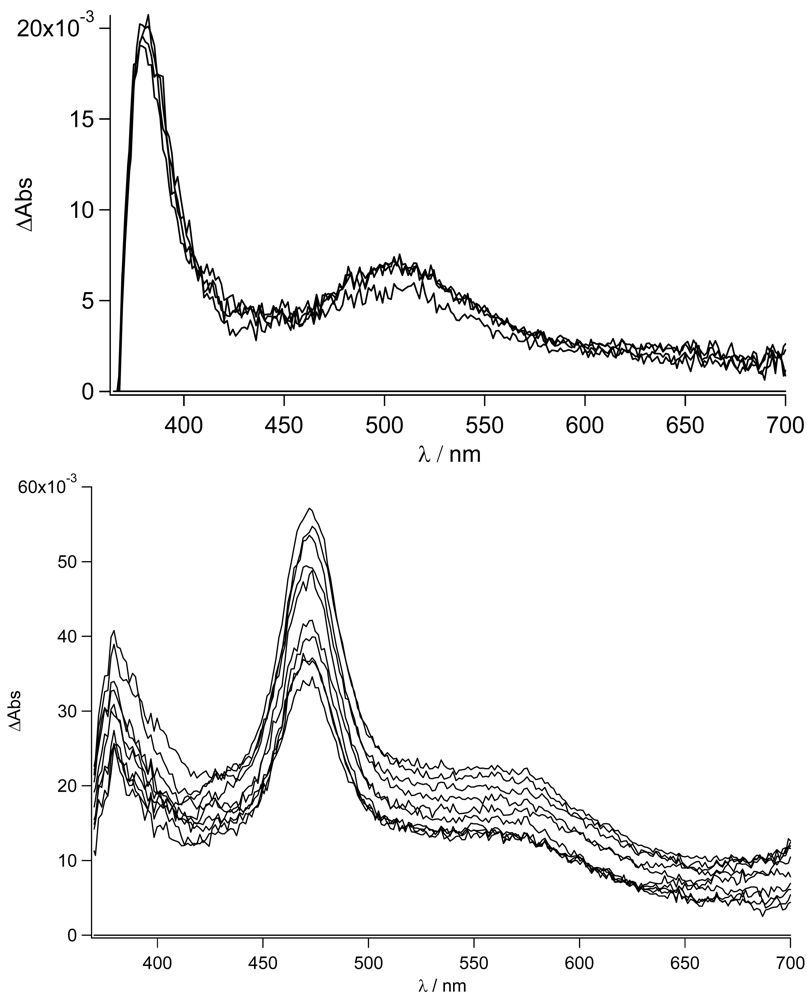
Figure 8). In this case, no indication of the population of the LE state is observed. The decay to the triplet state of the pyrene fits with the longer lived CT lifetime measured by time resolved emission spectroscopy (τ = 14 ns). The sub-nanosecond transient absorption spectra are shown in
Figure 12 in THF and acetonitrile, respectively, for
TpyPhPyrene2.
As expected from the nanosecond transient absorption spectra and in accordance with the emission spectra that were reported for
TpyPhPyrene2 (see
Figure 5), the transient absorption spectra obtained in the two solvents are quite different. In the nonpolar medium THF, the only visible transitions are those due to S
1→S
n of the Tpy, at high energy (390 nm), and those belonging to the pyrene units (in the 470-530 nm region) [
54,
55]. In the polar solvent (acetonitrile), the spectra present the singlet absorption of the Tpy (at 390 nm) but, at lower energy, two distinct features are present which cannot be attributed to the pyrene singlet absorption (
Figure 12 bottom). In fact, as we have already discussed, a charge transfer in acetonitrile is thermodynamically allowed leading to the formation of the radical cation of the pyrene [
56,
57] and the radical anion of the Tpy clearly visible at 463 nm and 550 nm, respectively. This CT state decays on a nanosecond time scale as mentioned before. The formation of the CT state from the singlet excited state could not be detected, indicating direct charge transfer state formation by excitation in the CT absorption band (optical electron transfer). In our time regime (up to 1ns), no other states are populated and due to the long lifetime (τ = 17 ns), no clear decay is observed. It is interesting to notice that the band at 470 nm does not show a clearly monoexponential decay. This may be due to the formation of the pyrene triplet state that, unfortunately, overlaps with the CT absorption band.
For the zinc complex and the bis-protonated species, the electron transfer is expected to be more exergonic and, indeed, the transient absorption spectra show the typical features of the formation of the CT state (see
Figure 13).
Conclusions
The properties of 3,5-bis(pyrene-1-yl)-4′-phenyl-2,2′:6′,2″-terpyridine, its bis-protonated form and its zinc complex were studied. The electrochemistry of
TpyPhPyrene2 in combination with a Weller-type analysis shows that photoinduced electron transfer process is thermodynamically allowed in polar solvents. The steady state and time resolved spectroscopy indicate the presence of a photoinduced electron transfer process for
Zn(TpyPhPyrene2)2) in all solvents, and for
TpyPhPyrene2 only in polar solvents. A general deactivation pathway is proposed (
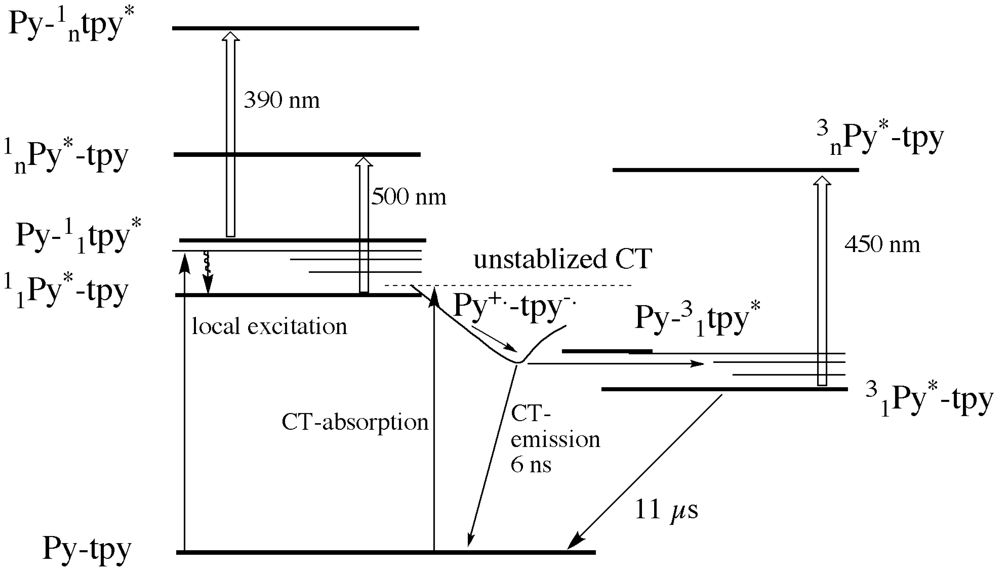
Scheme 2). The systems displays a charge transfer absorption band, and charge transfer emission in many solvents.
As can be seen in
Scheme 2, the un-relaxed charge transfer state to which direct absorption can occur is very close in energy to both the local excited state of the pyrene and of the terpyridine. The excited state absorption of the latter two excited states can be observed for the ligand in non-polar solvents. In polar solvent and for the zinc complex, we observe a population of a short lived charge transfer state that shows emission and that decays mainly to a triplet state localized on the pyrene unit. The transient absorption spectroscopy confirmed the charge separation phenomenon revealing typical bands of the radical cation of the pyrene and radical anion of the Tpy.
The modulation of the emission of metal complexes by substitution with chromophoric units is a delicate interplay of different excited states where the strongly emissive properties (of e.g. zinc-bis-terpyridyl) can be lost through charge transfer states with small quantum yields of emission and to dark local triplet states. Strongly emissive charge transfer states like observed for the free ligand
TpyPhPyrene2 are still relatively rare and difficult to design, but white light emission [
58] (for e.g. OLED lightning purposes) is attainable.
The substitution of phenyl-terpyridines with electron donating pyrene groups can lead to the development of new charge transfer systems that have special emissive properties that can be modulated by complexation.
Experimental
General
All chemicals were purchased from Acros or Aldrich and were used as received. All solvents for synthesis were analytic grade. For the spectroscopy, spectroscopic grade solvents were used. 1H-NMR spectra were obtained with a Varian Gemini-300 spectrometer. Chemical shifts (δ) are given in ppm, using the deuterated solvent as internal standard.
Synthesis of TpyPhBr2
To a solution of 3,5-bromobenzaldehyde (1 eq) in methanol (50 mL) and aqueous sodium hydroxide (1 mol/L, 1 eq) was added 2-acetylpyridine (1.05 eq) and the mixture was stirred for 3 hours. The precipitate was filtered off on a glass filter and recrystallised from ethanol. The precipitate (3,5-dibromoazachalchone) was dissolved in dichloromethane (100 mL) and was then washed with water and dried over MgSO4. The solvent was evaporated under vacuum and the solid was used without further purification.
In a round bottom flask, potassium tertbutoxide (2 eq) was solubilized in dry THF (100 mL) Then, acetylpyridine (1 eq) was added. The reaction was stirred for 4 hours (becoming slowly pink) at room temperature under nitrogen. 3,5- Dibromoazachalchone (1 eq) in dry THF (25 mL) was added. The reaction was stirred under nitrogen at room temperature for 4 hours (becoming quickly red). Then, THF was evaporated. Ammonium acetate (10 Eq) in ethanol (50 mL) was added and the reaction was stirred at 70 °C with the flask aerated during 36 hours. Then, the solvent (black solution) was removed. The solid was dispersed in water and filtered. This solid was intensively washed with water then solubilized in CHCl3 and dried over MgSO4. The product was recrystallized from ethanol (3 times). Yield: 43% (M= 467.17 g/mol); 1H-NMR (300 MHz, CDCl3): δ (ppm) = 8.76 ppm (d, 3J= 4.8 Hz, 2 H), 8.70 ppm (s, 2 H), 8.66ppm (s, 2 H), 7.97 ppm (d, 4J= 1.5 Hz, 2 H), 7.91 ppm (t, 3J= 8.1 Hz, 2 H), 7.76 ppm (d, 4J= 1.5 Hz, 1 H), 7.36 ppm (t, 3J= 4.8 Hz, 2 H).
Synthesis of TpyPhPyrene2
In a 100 mL Schlenk flask, TpyPhBr2 (100 mg, 0.214 mmol), pyrene-1-boronic acid (132 mg, 0.535 mmol) and cesium carbonate (418 mg, 1.284 mmol) were mixed in DMF (30 mL) and the solution was degassed. A catalytic amount of Pd(PPh3)4 was added (25 mg, 0.021 mmol). The reaction was heated during 60 hours at 100°C under nitrogen. The solution was filtered and the DMF was removed with toluene (azeotrope). The solid was washed with pentane and filtered off. The solid was solubilized in chloroform dried with MgSO4, filtered and evaporated under vaccum. The solid was purified by chromatography (alumina oxide) using a mixture dichloromethane/ethyl acetate (7:1). Yield: 35%, m = 54 mg (M = 709.86 g/mol); ESI-MS: 710.20; 1H-NMR (300 MHz, CD2Cl2) (see below): δ (ppm) = 8.98 (s, 2 H), 8.70 (d, 3J= 7.8 Hz, 2 H), 8.66 (d, 3J= 5.4 Hz, 2 H), 8.45 (d, 3J= 9.0 Hz, 2 H), 8.35-8.00 (m, 19 H), 7.89 (dt, 3J= 7.2 Hz, 4J= 1.8 Hz, 2 H), 7.36 (t, 3J= 4.8 Hz, 2 H); 13C-NMR (125.7 MHz, CD3CN): δ (ppm) = 156.2, 156.1, 151.9, 151.3, 142.4, 140.6, 137.8, 137.1, 132.4, 131.7, 131.1, 128.8, 128.0, 127.7, 126.6, 126.5, 126.4, 125.4, 125.2, 125.1, 124.9, 124.0, 123.8, 123.4, 123.1, 122.0, 119, 8, 119.2; UV/vis [λmax (nm) (ε (× 104 M-1.cm-1)), in acetonitrile]: 238 (8.8), 281 (8.6), 347 (5.0); Luminescence [λmax (nm), in dichloromethane, (77K)]: 399, (377, 388, 398).
Synthesis of Zn(TpyPhPyrene2)2 2 PF6
In a 50 mL Schlenk flask, TpyPhPyrene2 (50 mg, 0.070 mmol) and ZnCl2 (5 mg, 0.037 mmol) were suspended in methanol (15 mL) and refluxed for 4 hours. Then, the solution was cooled down, filtered and the isolated material dissolved in a solution of THF (20 mL). A water solution of ammonium hexafluorophosphate was added precipitating the corresponding zinc hexafluorophosphate salt. The precipitate was filtered, washed extensively with water and ether yielding a yellow solid. Yield: 76 %, m = 47 mg (M = 1775.03 g/mol); ESI-MS: 742.30 (- 2 PF6); 1H-NMR (300 MHz, CD2Cl2): δ (ppm) = 9.00 (s, 2 H), 8.53 (m, 4 H), 8.38 (dd, 3J= 8.1 Hz, 2 H), 8.30-8.00 (m, 19 H), 7.89 (d, 3J= 4.8 Hz, 2 H), 7.45 (dd, 3J= 5.4 Hz, 2 H); UV/vis [λmax (nm) (ε (× 104 M-1.cm-1)), in acetonitrile]: 240 (17.2), 281 (15.6), 342 (11.1); Luminescence [λmax (nm), in dichloromethane, (77K)]: 540, (377, 388, 398).
Cyclic voltammetry
Cyclic voltammetry was performed in a three-electrode cell equipped with a platinum milli-electrode and a platinum wire counter-electrode. A silver wire served as quasi-reference electrode and its potential was checked against the ferricinium/ ferrocene couple (Fc+/Fc) before and after each experiment. The electrolytic media involved THF (HPLC grade), and 0.1 M of tetrabutylammonium hexafluorophosphate (TBAHP - puriss quality). All experiments were performed in a glove box containing dry, oxygen-free (< 1ppm) argon, at room temperature. Electrochemical experiments were carried out with an EGG PAR 273A potentiostat.
Spectra
Electronic absorption spectra were recorded with a Lambda 19 NIR model from Perkin-Elmer. Fluorescence spectra were recorded in non-deoxygenated solvents (spectroscopic grade) at 20°C with a QM-4/QuantaMaster™ fluorometer from PTI® equipped with rapid mono-channel detection and continuous excitation source. Quantum yields were determined using Quinine bisulfate as a standard reference (Φf = 0.54 at 20°C in 1 N H2SO4). For time resolved spectroscopy, samples were dissolved in spectroscopic solvents and filtered (0.4 μm PVDF HPLC-filters) to remove particles and potential aggregates. The samples had an absorbance of ca. 0.7 – 0.9 (1 cm) for nanosecond transient measurements and of ca. 0.3 – 0.7 (2 mm) for femtosecond transient at the excitation wavelength. The UV/vis absorption spectra of the samples were measured before and after the laser experiments and were found to be virtually identical, thus ruling out any possible degradation or chemical change of the samples. All photophysical data reported here have a 5 to 10 % error limit, unless indicated otherwise. The experiments were performed at room temperature.
Sub-nanosecond transient absorption experiments were performed with a Spectra-Physics Hurricane Titanium:Sapphire regenerative amplifier system. The full spectrum setup was based on an optical parametric amplifier (Spectra-Physics OPA 800C) as the pump. The residual fundamental light, from the pump OPA, was used for white light generation, which was detected with a CCD spectrograph (Ocean Optics). The polarization of the pump light was controlled by a Berek Polarization Compensator (New Focus). The Berek-Polarizer was always included in the setup to provide the Magic-Angle conditions. The probe light was double-passed over a delay line (Physik Instrumente, M-531DD) that provides an experimental time window of 3.6 ns with a maximal resolution of 0.6 fs/step. The OPA was used to generate excitation pulses at 530 nm. The laser output was typically 3.5-5 μJ pulse-1 (130 fs FWHM) with a repetition rate of 1 kHz. The samples were placed into cells of 2 mm path length (Hellma) and were stirred with a downward projected PTFE shaft, using a direct drive spectro-stir (SPECTRO-CELL). This stir system was also used for the white light generation in a 2 mm water cell.
Time-resolved fluorescence measurements were performed on a picosecond single photon counting (SPC) setup. The frequency doubled (300-340 nm, 1 ps, 3.8 MHz) output of a cavity dumped DCM dye laser (Coherent model 700) pumped by a mode-locked Ar-ion laser (Coherent 486 AS Mode Locker, Coherent Innova 200 laser) was used as the excitation source. A (Hamamatsu R3809) micro channel plate photomultiplier was used as detector. The instrument response (∼17 ps FWHM) was recorded using the Raman scattering of a doubly de-ionized water sample. Time windows (4000 channels) of 5 ns (1.25 ps/channel) – 50 ns (12.5 ps/channel) could be used, enabling the measurement of lifetimes of 5 ps - 40 ns. The recorded traces were deconvoluted with the system response and fitted to an exponential function using the Igor Pro program.
In nanosecond pump-probe experiments, for excitation a (Coherent) Infinity Nd: YAG-XPO laser was used. The laser illuminated a slit of 10 × 2 mm. Perpendicular to this, the probe light provided by an EG&G (FX504) low pressure Xenon lamp, which irradiated the sample through a 1 mm pinhole. The overlap of the two beams falls within the first two millimeter of the cell, after the slit. The probe light from both the signal and the reference channels is then collected in optical fibers which are connected to an Acton SpectraPro-150 spectrograph which is coupled to a Princeton Instruments ICCD-576-G/RB-EM gated intensified CCD camera. Using a 5 ns gate this camera simultaneously records the spectrally dispersed light from both optical fibers on separate stripes of the CCD. De-aeration was performed by bubbling with Argon for 20 to 30 minutes.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}