Critical Evaluation of Acetylcholine Determination in Rat Brain Microdialysates using Ion-Pair Liquid Chromatography with Amperometric Detection

Abstract
:1. Introduction
2. Results and Discussion
2.1. Method optimization
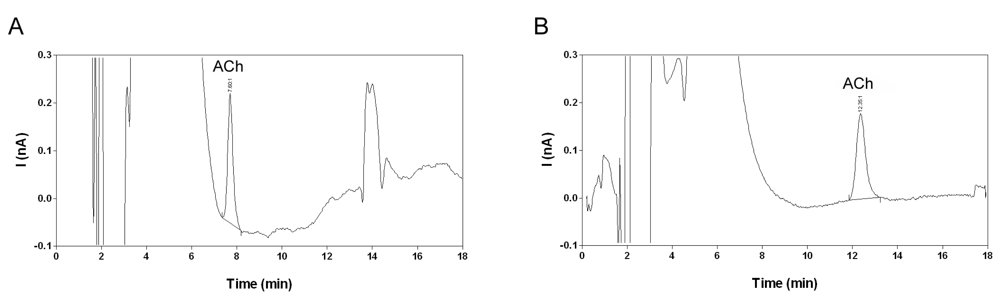
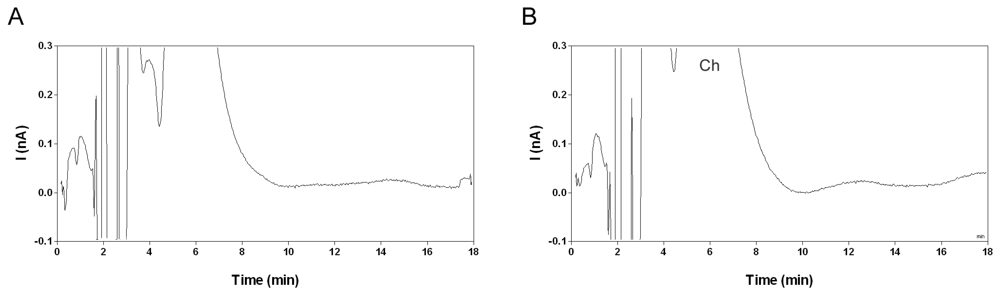
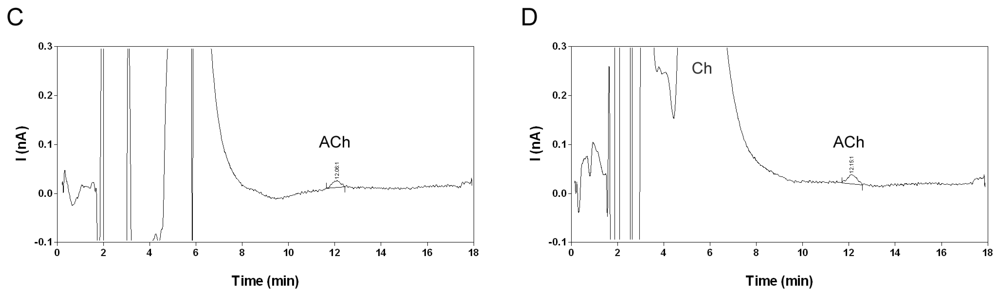
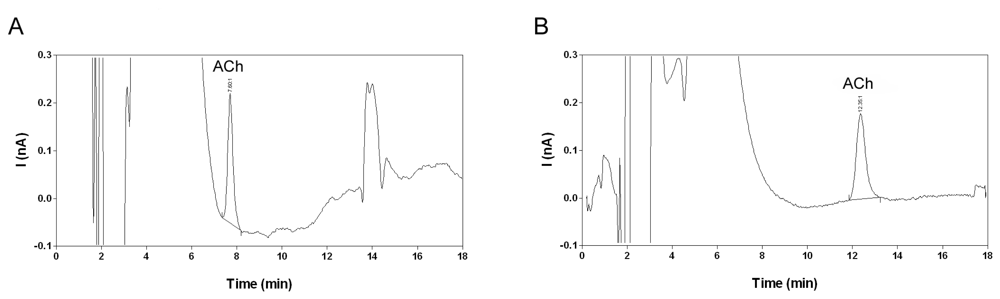
2.2. Selectivity and limit of quantification
2.3. Linearity and matrix effects
2.4. Precision and accuracy
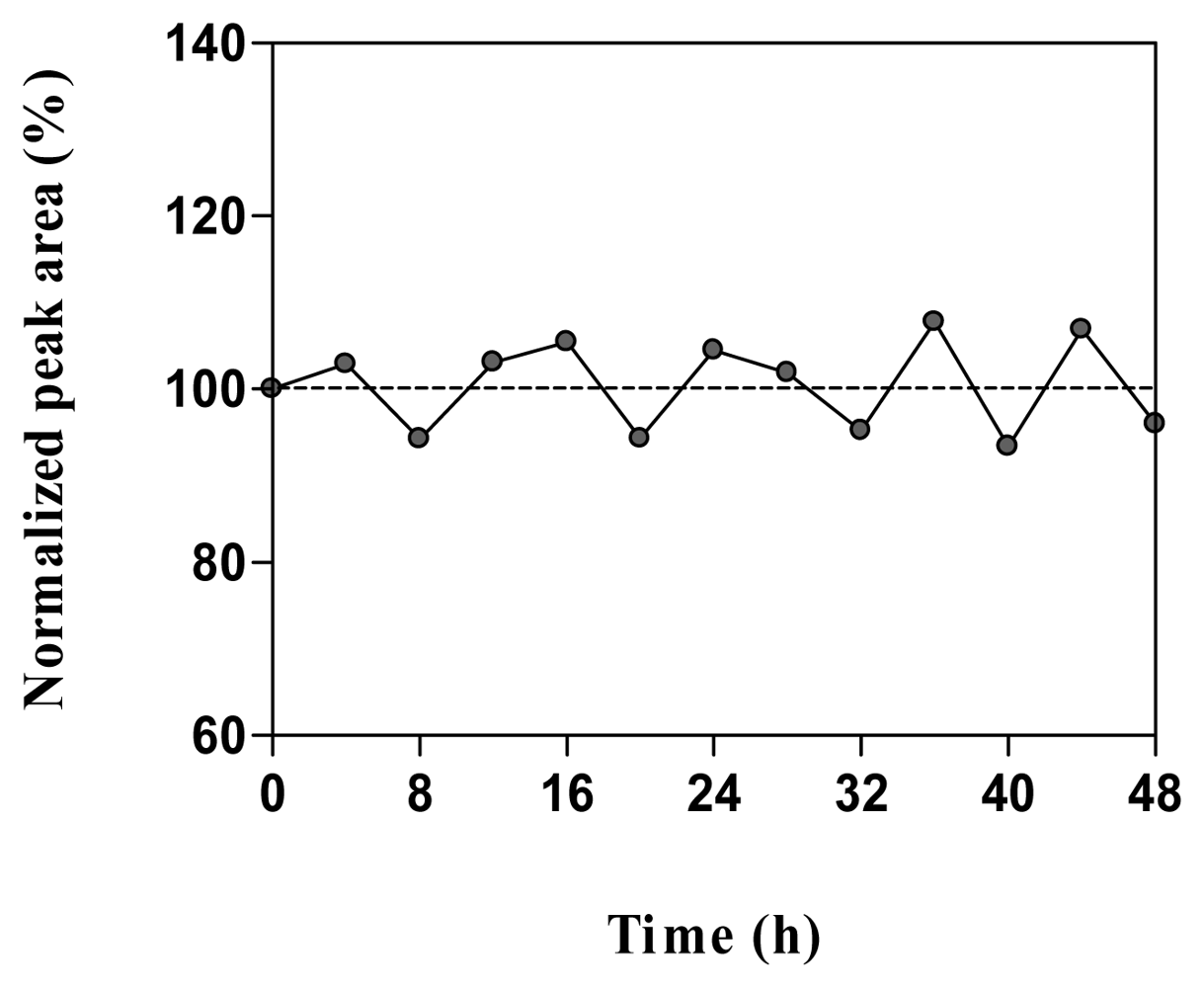
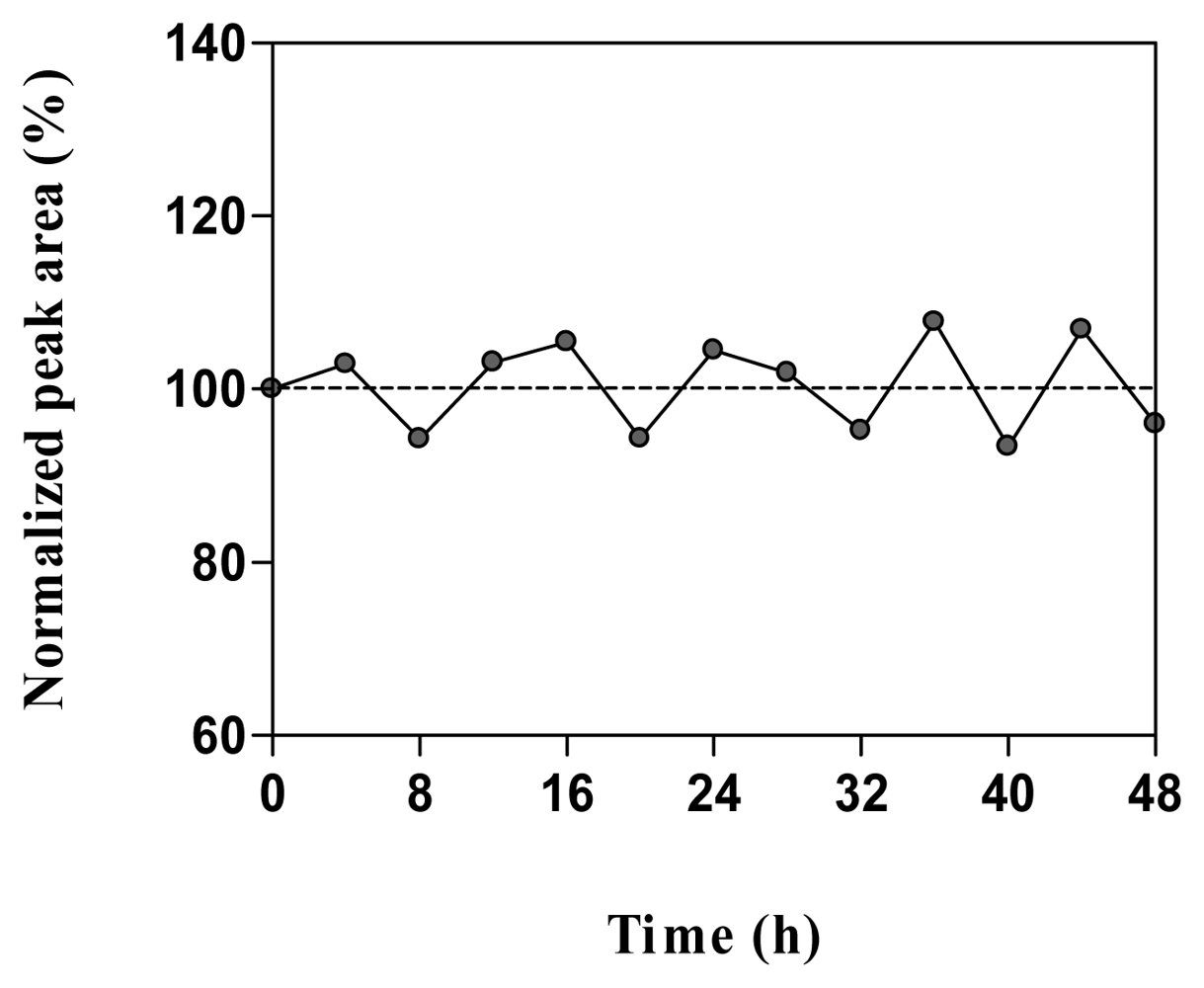
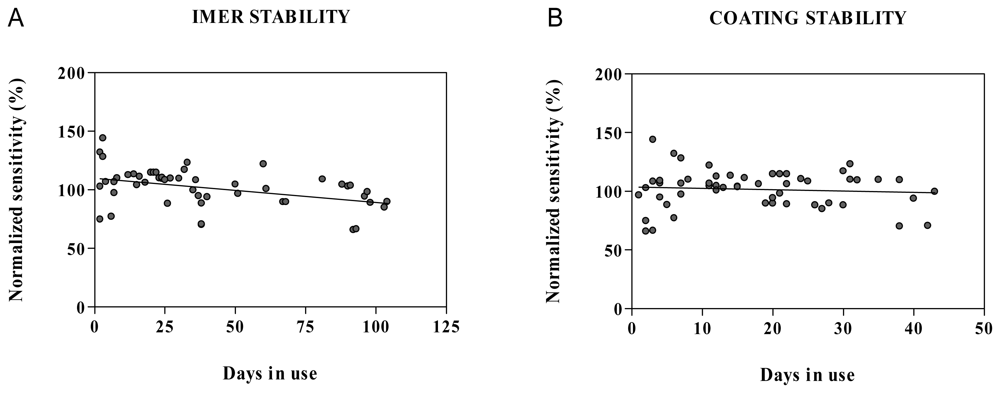
2.5. Stability in water and microdialysis matrix
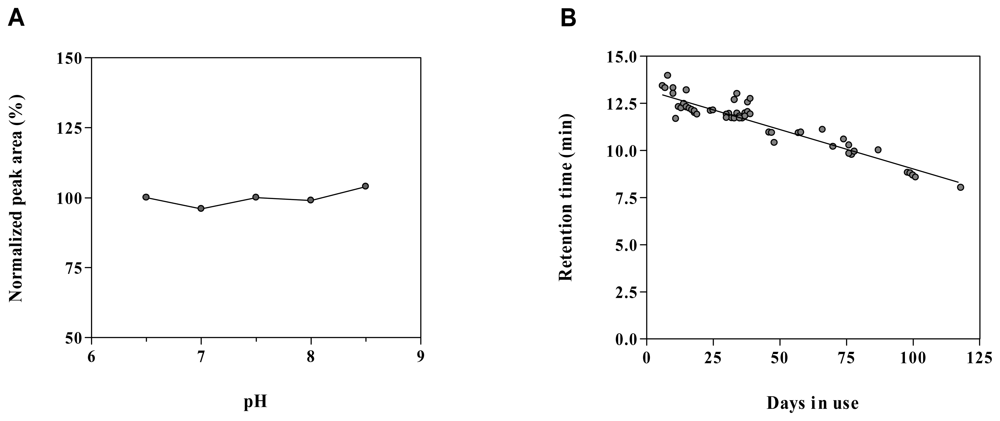
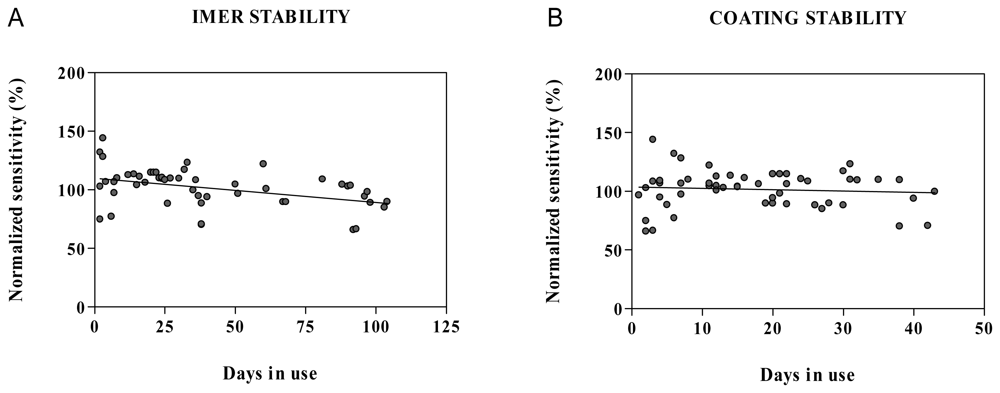
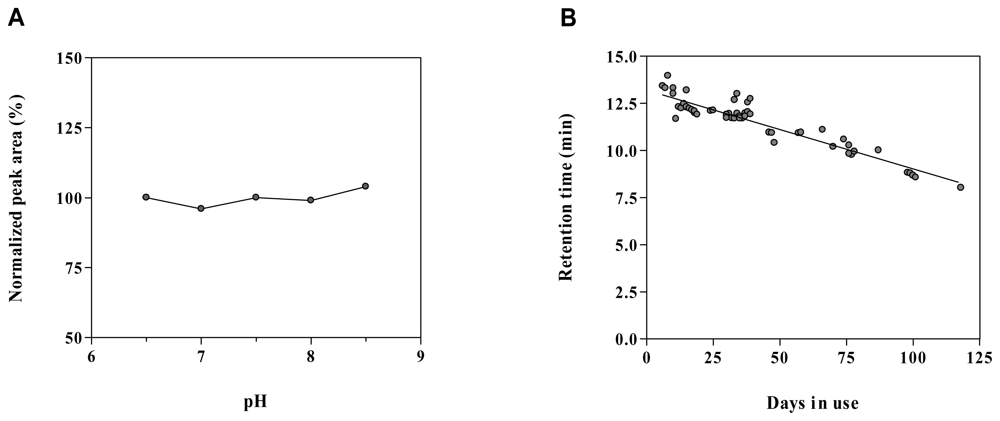
2.6. Method robustness and system suitability
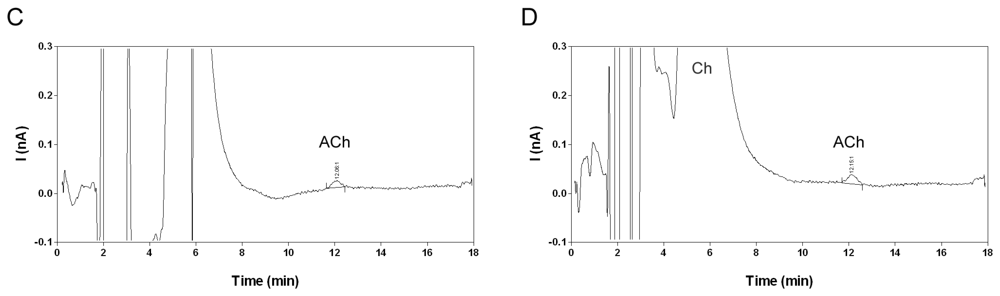
2.7. Analysis of microdialysis samples
3. Experimental Section
3.1. Chemicals
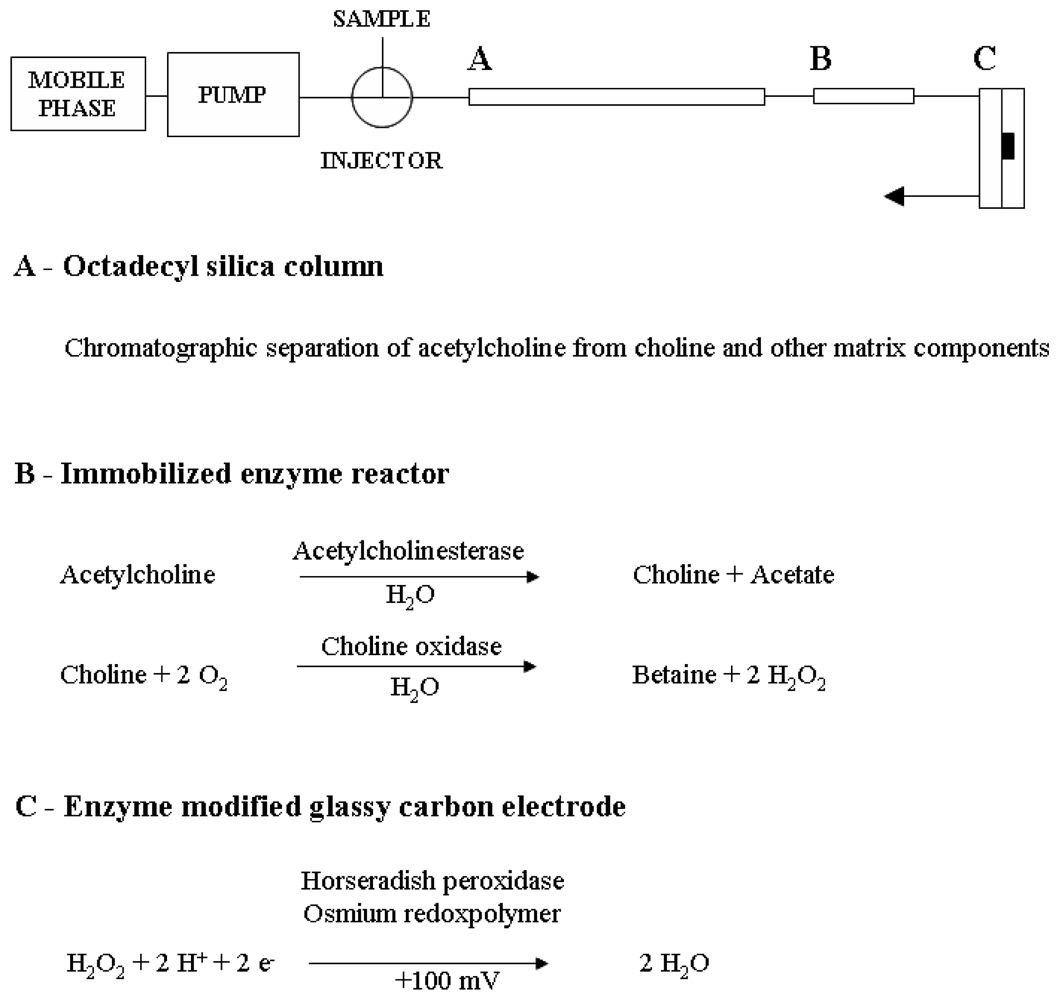
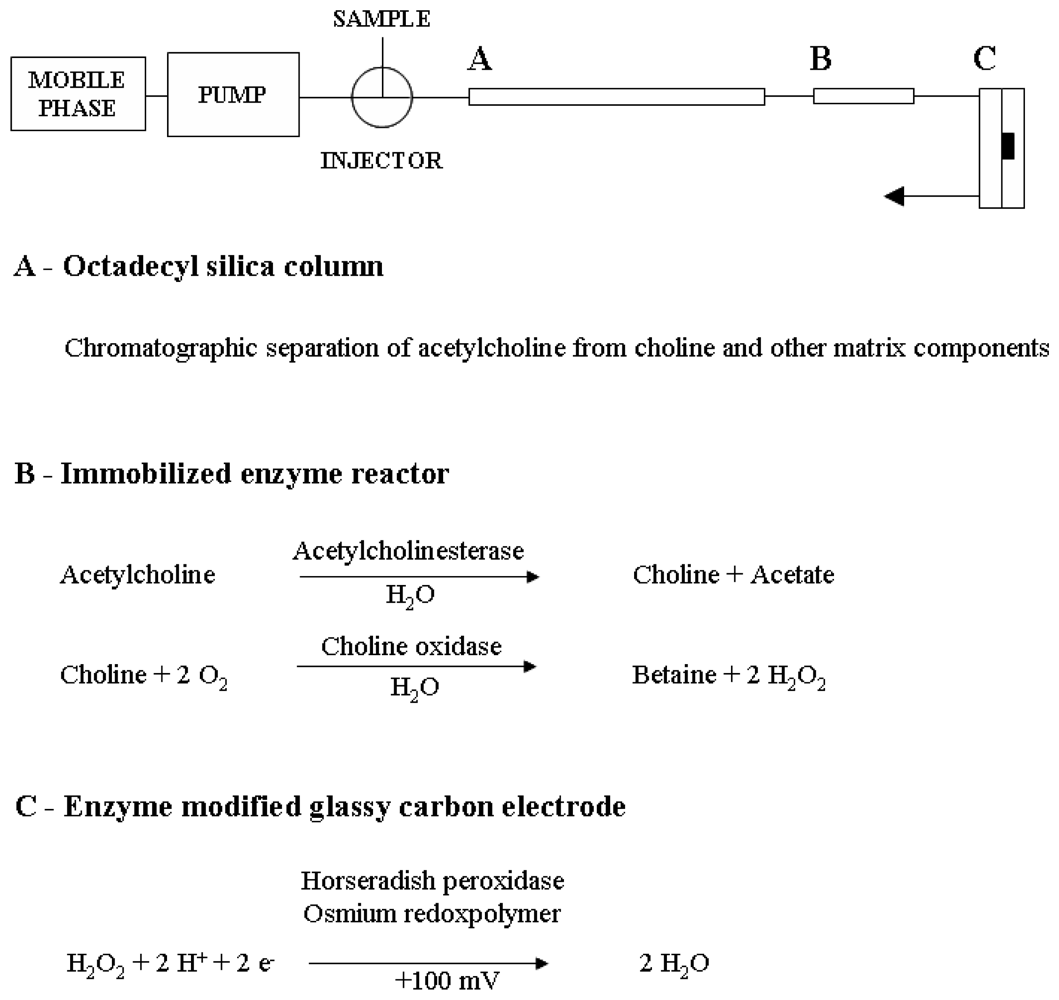
3.2. Liquid chromatography equipment and conditions
3.3. Enzyme reactors and enzyme-modified amperometric detection
3.4. Microdialysis equipment and sampling
3.5. Preparation of stock solutions, calibration standards and quality control samples
3.6. Statistical analysis
4. Conclusions
Acknowledgments
References and Notes
- Hasselmo, M.E.; Bower, J.M. Acetylcholine and memory. Trends Neurosci. 1993, 6, 218–222. [Google Scholar]
- Pisani, A.; Bernardi, G.; Ding, J.; Surmeier, D.J. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 2007, 10, 545–553. [Google Scholar]
- Perry, E.; Walker, M.; Grace, J.; Perry, R. Acetylcholine in mind: a neurotransmitter correlate of consciousness? Trends Neurosci. 1999, 6, 273–280. [Google Scholar]
- Graves, L.; Pack, A.; Abel, T. Sleep and memory: a molecular perspective. Trends Neurosci. 2001, 4, 237–243. [Google Scholar]
- Sarter, M.; Parikh, V. Choline transporters, cholinergic transmission and cognition. Nat. Rev. Neurosci. 2005, 1, 48–56. [Google Scholar]
- Wess, J.; Eglen, R.M.; Gautam, D. Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat. Rev. Drug Discov. 2007, 9, 721–733. [Google Scholar]
- Stahl, M.S. Essential Psychopharmacology: neuroscientific basis and practical application, 2nd Edition. ed; Cambridge University Press: New York, 2000; pp. 467–471. [Google Scholar]
- Tsai, T.H. Separation methods used in the determination of choline and acetylcholine. J. Chromatogr. B. 2000, 747, 111–122. [Google Scholar]
- Ungerstedt, U.; Hallstrom, A. In vivo microdialysis--a new approach to the analysis of neurotransmitters in the brain. Life Sci. 1987, 41, 861–864. [Google Scholar]
- Brooks, J.M.; Sarter, M.; Bruno, J.P. D2-like receptors in nucleus accumbens negatively modulate acetylcholine release in prefrontal cortex. Neuropharmacology 2007, 53, 455–463. [Google Scholar]
- Cerbai, F.; Giovannini, M.G.; Melani, C.; Enz, A.; Pepeu, G. N1phenethyl-norcymserine, a selective butyrylcholinesterase inhibitor, increases acetylcholine release in rat cerebral cortex: A comparison with donepezil and rivastigmine. Eur. J. Pharmacol. 2007, 572, 142–150. [Google Scholar]
- Huang, T.; Yang, L.; Gitzen, J.; Kissinger, P.T.; Vreeke, M.; Heller, A. Detection of basal acetylcholine in rat brain microdialysate. J. Chromatogr. B. 1995, 670, 323–327. [Google Scholar]
- Kato, T.; Liu, J.K.; Yamamoto, K.; Osborne, P.G.; Niwa, O. Detection of basal acetylcholine release in the microdialysis of rat frontal cortex by high-performance liquid chromatography using a horseradish peroxidase-osmium redox polymer electrode with pre-enzyme reactor. J. Chromatogr. B. 1996, 682, 162–6. [Google Scholar]
- Millan, M.J.D.; Cara, B.; Dekeyne, A.; Panayi, F.; De Groote, L.; Sicard, D.; Cistarelli, L.; Billiras, R.; Gobert, A. Selective blockade of dopamine D(3) versus D(2) receptors enhances frontocortical cholinergic transmission and social memory in rats: a parallel neurochemical and behavioural analysis. J. Neurochem. 2007, 100, 1047–1061. [Google Scholar]
- Yamamoto, K.; Sato, K.; Chikuma, T.; Kato, T. A highly sensitive and stable detection of acetylcholine by HPLC-osmium-horseradish peroxidase redox polymer electrode coated on a gold radial flow ring disk. Anal. Chim. Acta. 2004, 521, 209–214. [Google Scholar]
- Zhu, Y.; Wong, P.S.; Cregor, M.; Gitzen, J.F.; Coury, L.A.; Kissinger, P.T. In vivo microdialysis and reverse phase ion pair liquid chromatography/tandem mass spectrometry for the determination and identification of acetylcholine and related compounds in rat brain. Rapid Commun. Mass Spectrom. 2000, 14, 1695–700. [Google Scholar]
- Hows, M.E.; Organ, A.J.; Murray, S.; Dawson, L.A.; Foxton, R.; Heidbreder, C; et al. High-performance liquid chromatography/tandem mass spectrometry assay for the rapid high sensitivity measurement of basal acetylcholine from microdialysates. J. Neurosci. Meth. 2002, 121, 33–39. [Google Scholar]
- Keski-Rahkonen, P.; Lehtonen, M.; Ihalainen, J.; Sarajarvi, T.; Auriola, S. Quantitative determination of acetylcholine in microdialysis samples using liquid chromatography/atmospheric pressure spray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 2933–2943. [Google Scholar]
- Uutela, P.; Reinila, R.; Piepponen, P.; Ketola, R.A.; Kostiainen, R. Analysis of acetylcholine and choline in microdialysis samples by liquid chromatography/tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 2950–2956. [Google Scholar]
- Potter, P.E.; Meek, J.L.; Neff, N.H. Acetylcholine and choline in neuronal tissue measured by HPLC with electrochemical detection. J. Neurochem. 1983, 41, 188–194. [Google Scholar]
- Tsai, T.R.; Cham, T.M.; Chen, K.C.; Chen, C.F.; Tsai, T.H. Determination of acetylcholine by online microdialysis coupled with pre- and post-microbore column enzyme reactors with electrochemical detection. J Chromatogr. B. 1996, 678, 151–155. [Google Scholar]
- Kehr, J.; Dechent, P.; Kato, T.; Ogren, S.O. Simultaneous determination of acetylcholine, choline and physostigmine in microdialysis samples from rat hippocampus by microbore liquid chromatography/electrochemistry on peroxidase redox polymer coated electrodes. J. Neurosci. Meth. 1998, 83, 143–150. [Google Scholar]
- Sotoyama, H; Zhu, Y.; Gitzen, J.; Xie, F.; Kissinger, P. Feasibility of Ion-Pair Reversed-Phase Liquid Chromatography/Electrochemistry Detection for Determination of Acetylcholine in Microdialysates Collected Without Acetylcholinesterase Inhibitors. Curr. Separations 2002, 20, 11–16. [Google Scholar]
- Carter, A.J.; Kehr, J. Microbore high-performance liquid chromatographic method for measuring acetylcholine in microdialysis samples: optimizing performance of platinum electrodes. J. Chromatogr. B. 1997, 692, 207–212. [Google Scholar]
- Ichikawa, J.; Dai, J.; Meltzer, H.Y. Acetylcholinesterase Inhibitors Are Neither Necessary nor Desirable for Microdialysis Studies of Brain Acetylcholine. Curr. Separations 2000, 19, 37–44. [Google Scholar]
- Herzog, C.D.; Nowak, K.A.; Sarter, M.; Bruno, J.P. Microdialysis without acetylcholinesterase inhibition reveals an age-related attenuation in stimulated cortical acetylcholine release. Neurobiol. Aging 2003, 24, 861–863. [Google Scholar]
- Izurieta-Sanchez, P.; Jonkers, N.; Sarre, S.; Ebinger, G.; Michotte, Y. Neostigmine influences the L-dopa-induced extracellular dopamine levels in the striatum. Brain Res. 2000, 856, 250–253. [Google Scholar]
- Eva, C.; Hadjiconstantinou, M.; Neff, N.H.; Meek, J.L. Acetylcholine measurement by high-performance liquid chromatography using an enzyme-loaded postcolumn reactor. Anal. Biochem. 1984, 143, 320–324. [Google Scholar]
- Damsma, G.; Westerink, B.H.; de Vries, J.B.; Van den Berg, C.J.; Horn, A.S. Measurement of acetylcholine release in freely moving rats by means of automated intracerebral dialysis. J. Neurochem. 1987, 48, 1523–1528. [Google Scholar]
- Damsma, G.; Westerink, B.H.; Imperato, A.; Rollema, H.; de Vries, J.B.; Horn, A.S. Automated brain dialysis of acetylcholine in freely moving rats: detection of basal acetylcholine. Life Sci. 1987, 41, 873–876. [Google Scholar]
- Guerrieri, A.; Palmisano, F. An acetylcholinesterase/choline oxidase-based amperometric biosensors as a liquid chromatography detector for acetylcholine and choline determination in brain tissue homogenates. Anal. Chem. 2001, 73, 2875–2882. [Google Scholar]
- Tindall, G.W.; Perry, R.L. Explanation for the enhanced dissolution of silica column packing in high pH phosphate and carbonate buffers. J. Chromatogr. A. 2003, 988, 309–312. [Google Scholar]
- Larsson, N.; Ruzgas, T.; Gorton, L.; Kokaia, M.; Kissinger, P.; Csöregi, E. Design and development of an amperometric biosensor for acetylcholine determination in brain microdialysates. Electrochim. Acta. 1998, 43, 3541–3554. [Google Scholar]
- Ventura, D.A; Nickelly, J.G. Pulse dampening system for high pressure liquid chromatography. Anal. Chem. 1978, 50, 1017–1018. [Google Scholar]
- Claessens, H.A.l; van Straten, M.A. Review on the chemical and thermal stability of stationary phases for reversed-phase liquid chromatography. J. Chromatogr. A. 2004, 1060, 23–41. [Google Scholar]
- Gunaratna, P.C.; Wilson, G.S. Optimization of multienzyme flow reactors for determination of acetylcholine. Anal. Chem. 1990, 62, 402–407. [Google Scholar]
- Guidance for Industry, Bioanalytical Method Validation. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research, Center for Veterinary Medicine, 2000. http://www.fda.gov/CDER/GUIDANCE/4252fnl.pdf.
- Sletten, D.M.; Nickander, K.K.; Low, P.A. Stability of acetylcholine chloride solution in autonomic testing. J. Neurol. Sci. 2005, 234, 1–3. [Google Scholar]
- Day, J.C.; Kornecook, T.J.; Quirion, R. Application of in vivo microdialysis to the study of cholinergic systems. Methods 2001, 23, 21–39. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Added Concentration (nM) | Mean ± SD measured concentration (nM) | Repeatability (RSD %) | Inter-day precision (RSD %) | Accuracy (%) | ||
|---|---|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | ||||
| 0.30 | 0.30 ± 0.04 | 0.32 ± 0.05 | 0.32 ± 0.05 | 15.7 | 8.6 | 103.6 |
| 1.00 | 1.02 ± 0.07 | 0.98 ± 0.11 | 1.03 ± 0.05 | 7.4 | 5.1 | 100.9 |
| 3.00 | 2.95 ± 0.06 | 3.03 ± 0.27 | 2.88 ± 0.07 | 4.4 | 5.1 | 98.4 |
| 10.0 | 9.60 ± 0.37 | 9.79 ± 0.44 | 9.96 ± 0.50 | 4.4 | 4.0 | 97.8 |
© 2008 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
De Bundel, D.; Sarre, S.; Van Eeckhaut, A.; Smolders, I.; Michotte, Y. Critical Evaluation of Acetylcholine Determination in Rat Brain Microdialysates using Ion-Pair Liquid Chromatography with Amperometric Detection. Sensors 2008, 8, 5171-5185. https://doi.org/10.3390/s8085171
De Bundel D, Sarre S, Van Eeckhaut A, Smolders I, Michotte Y. Critical Evaluation of Acetylcholine Determination in Rat Brain Microdialysates using Ion-Pair Liquid Chromatography with Amperometric Detection. Sensors. 2008; 8(8):5171-5185. https://doi.org/10.3390/s8085171
Chicago/Turabian StyleDe Bundel, Dimitri, Sophie Sarre, Ann Van Eeckhaut, Ilse Smolders, and Yvette Michotte. 2008. "Critical Evaluation of Acetylcholine Determination in Rat Brain Microdialysates using Ion-Pair Liquid Chromatography with Amperometric Detection" Sensors 8, no. 8: 5171-5185. https://doi.org/10.3390/s8085171