Immunotherapeutic Concepts to Target Acute Myeloid Leukemia: Focusing on the Role of Monoclonal Antibodies, Hypomethylating Agents and the Leukemic Microenvironment

Abstract
:1. Introduction
2. Monoclonal Antibodies (mAbs)
2.1. CD33
2.2. CD123
2.3. CD133
2.4. CD64
2.5. C-Type Lectin-Like Molecule 1
2.6. Other Targets for Antibody-Directed Therapy
2.7. Targeting AML Stem Cells
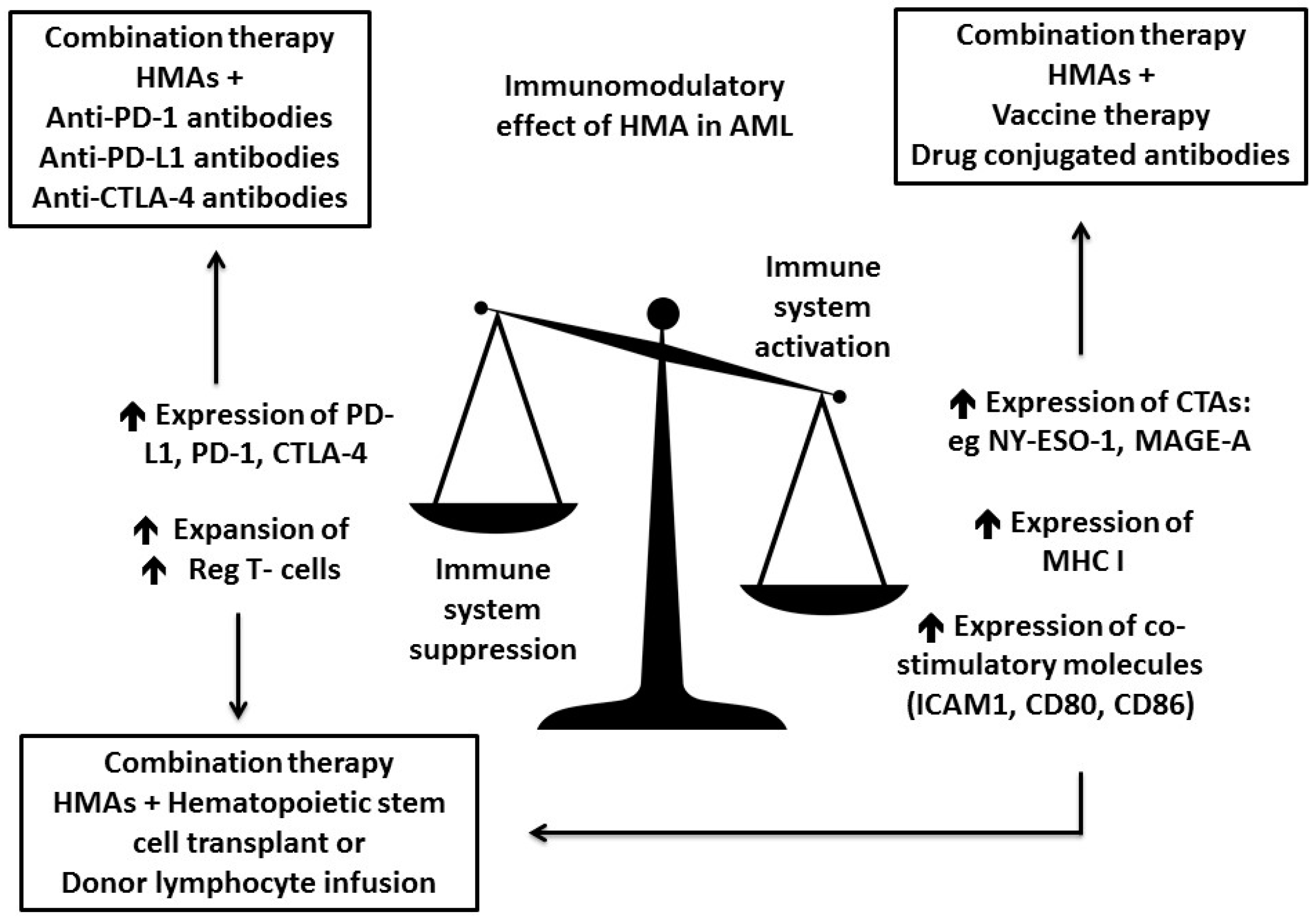
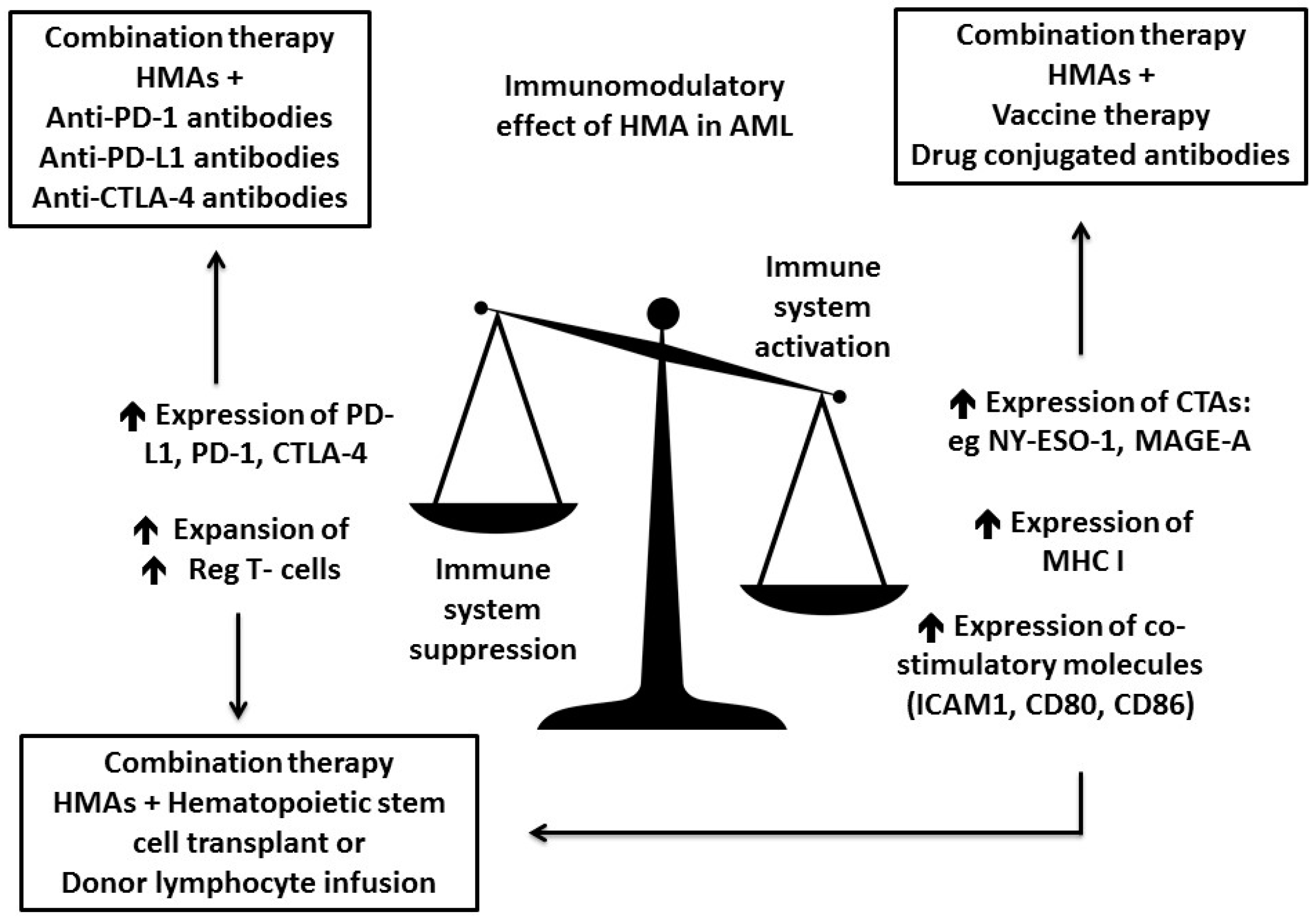
3. Increase Antigenicity of AML Cells by Hypomethylating Agents (HMAs)
3.1. HMA Enhance Antigen Presentation
3.2. HMA Enhance Checkpoint Inhibition
3.3. HMA Might Enhance GVL Effect While Reducing GVHD
4. Modulation of the Leukemic Immune Microenvironment
4.1. Small Molecule Immunomodulatory Drugs (IMiDs)
4.2. Immunosuppressive Factors Expressed and Secreted by the Tumor or Tumor Microenvironment
4.3. Myeloid-Derived Suppressor Cells
4.4. Tumor Associated Macrophages
4.5. Tumor Associated Neutrophils
4.6. Regulatory T Cells
4.7. Tumor Expressing Inhibitory Molecules, Cytotoxic CD8+ T Cells Exhaustion and Checkpoint Inhibitors
5. Conclusions
Conflicts of Interest
References
- Estey, E. Why is progress in acute myeloid leukemia so slow? Semin. Hematol. 2015, 52, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Dombret, H.; Gardin, C. An update of current treatments for adult acute myeloid leukemia. Blood 2016, 127, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Schmitt, A.; Rojewski, M.T.; Chen, J.; Giannopoulos, K.; Fei, F.; Yu, Y.; Götz, M.; Heyduk, M.; Ritter, G.; et al. RHAMM-R3 peptide vaccination in patients with acute myeloid leukemia, myelodysplastic syndrome, and multiple myeloma elicits immunologic and clinical responses. Blood 2008, 111, 1357–1365. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, K.; Yong, A.S.; Mielke, S.; Savani, B.N.; Jafarpour, B.; Eniafe, R.; Quan Le, R.; Musse, L.; Boss, C.; Childs, R.; et al. Lymphodepletion is permissive to the development of spontaneous T-cell responses to the self-antigen PR1 early after allogeneic stem cell transplantation and in patients with acute myeloid leukemia undergoing WT1 peptide vaccination following chemotherapy. Cancer Immunol. Immunother. 2012, 61, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Saini, K.S.; Azim, H.A., Jr.; Cocorocchio, E.; Vanazzi, A.; Saini, M.L.; Raviele, P.R.; Pruneri, G.; Peccatori, F.A. Rituximab in Hodgkin lymphoma: Is the target always a hit? Cancer Treat. Rev. 2011, 37, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.; Stein, A.; Gokbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Jilani, I.; Estey, E.; Huh, Y.; Joe, Y.; Manshouri, T.; Yared, M.; Giles, F.; Kantarjian, H.; Cortes, J.; Thomas, D.; et al. Differences in CD33 intensity between various myeloid neoplasms. Am. J. Clin. Pathol. 2002, 118, 560–566. [Google Scholar] [CrossRef] [PubMed]
- O’Hear, C.; Rubnitz, J.E. Recent research and future prospects for gemtuzumab ozogamicin: Could it make a comeback? Expert Rev. Hematol. 2014, 7, 427–429. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.L.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: Results of the MRC AML15 trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Russell, N.H.; Hills, R.K.; Kell, J.; Freeman, S.; Kjeldsen, L.; Hunter, A.E.; Yin, J.; Craddock, C.F.; Dufva, I.H.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy improves survival in older patients with acute myeloid leukemia. J. Clin. Oncol. 2012, 30, 3924–3931. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.K.; Hills, R.K.; Hunter, A.E.; Milligan, D.; Kell, W.J.; Wheatley, K.; Yin, J.; McMullin, M.F.; Dignum, H.; Bowen, D.; et al. The addition of gemtuzumab ozogamicin to low-dose Ara-C improves remission rate but does not significantly prolong survival in older patients with acute myeloid leukaemia: Results from the LRF AML14 and NCRI AML16 pick-a-winner comparison. Leukemia 2013, 27, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014, 15, 986–996. [Google Scholar] [CrossRef]
- Friedrich, M.; Henn, A.; Raum, T.; Bajtus, M.; Matthes, K.; Hendrich, L.; Wahl, J.; Hoffmann, P.; Kischel, R.; Kvesic, M.; et al. Preclinical characterization of AMG 330, a CD3/CD33-bispecific T-cell-engaging antibody with potential for treatment of acute myelogenous leukemia. Mol. Cancer Ther. 2014, 13, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Testa, U.; Pelosi, E.; Frankel, A. CD 123 is a membrane biomarker and a therapeutic target in hematologic malignancies. Biomark. Res. 2014, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Busfield, S.J.; Biondo, M.; Wong, M.; Ramshaw, H.S.; Lee, E.M.; Ghosh, S.; Braley, H.; Panousis, C.; Roberts, A.W.; He, S.Z.; et al. Targeting of acute myeloid leukemia in vitro and in vivo with an anti-CD123 mAb engineered for optimal ADCC. Leukemia 2014, 28, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.D.; Roboz, G.J.; Walter, R.B.; Altman, J.K.; Ferguson, A.; Curcio, T.J.; Orlowski, K.F.; Garrett, L.; Busfield, S.J.; Barnden, M.; et al. First-in man, phase 1 study of CSL362 (anti-IL3Rα / anti-CD123 monoclonal antibody) in patients with CD123+ acute myeloid leukemia (AML) in CR at high risk for early relapse. Blood 2014, 124, 120. [Google Scholar]
- Lane, A.A.; Sweet, K.L.; Wang, E.S.; Donnellan, W.B.; Walter, R.B.; Stein, A.S.; Rizzieri, D.A.; Carraway, H.E.; Mantzaris, L.; Prebet, T.; et al. Results from ongoing phase 2 trial of SL-401 as consolidation therapy in patients with acute myeloid leukemia (AML) in remission with high relapse risk including minimal residual disease (MRD). Blood 2016, 128, 215. [Google Scholar]
- Chichili, G.R.; Huang, L.; Li, H.; Burke, S.; He, L.; Tang, Q.; Jin, L.; Gorlatov, S.; Ciccarone, V.; Chen, F.; et al. A CD3 × CD123 bispecific DART for redirecting host T cells to myelogenous leukemia: Preclinical activity and safety in nonhuman primates. Sci. Transl. Med. 2015, 7, 289ra82. [Google Scholar] [PubMed]
- Li, J.; Zhong, X.Y.; Li, Z.Y.; Cai, J.F.; Zou, L.; Li, J.M.; Yang, T.; Liu, W. CD133 expression in osteosarcoma and derivation of CD133+ cells. Mol. Med. Rep. 2013, 7, 577–584. [Google Scholar] [PubMed]
- Vercauteren, S.M.; Sutherland, H.J. CD133 (AC133) expression on AML cells and progenitors. Cytotherapy 2001, 3, 449–459. [Google Scholar] [PubMed]
- Ferrandina, G.; Petrillo, M.; Bonanno, G.; Scambia, G. Targeting CD133 antigen in cancer. Expert Opin. Ther. Targets 2009, 13, 823–837. [Google Scholar] [PubMed]
- Rothfelder, K.; Koerner, S.; Andre, M.; Leibold, J.; Kousis, P.; Buehring, H.J.; Haen, S.P.; Kuebler, A.; Kanz, L.; Grosse-Hovest, L.; et al. Induction of NK cell reactivity against myeloid leukemia by a novel Fc-optimized CD133 antibody. Blood 2015, 126, 3793. [Google Scholar]
- Ball, E.D.; McDermott, J.; Griffin, J.D.; Davey, F.R.; Davis, R.; Bloomfield, C.D. Expression of the three myeloid cell-associated immunoglobulin G Fc receptors defined by murine monoclonal antibodies on normal bone marrow and acute leukemia cells. Blood 1989, 73, 1951–1956. [Google Scholar] [PubMed]
- Krasinskas, A.M.; Wasik, M.A.; Kamoun, M.; Schretzenmair, R.; Moore, J.; Salhany, K.E. The usefulness of CD64, other monocyte-associated antigens, and CD45 gating in the subclassification of acute myeloid leukemias with monocytic differentiation. Am. J. Clin. Pathol. 1998, 110, 797–805. [Google Scholar] [PubMed]
- Stahnke, B.; Thepen, T.; Stocker, M.; Rosinke, R.; Jost, E.; Fischer, R.; Tur, M.K.; Barth, S. Granzyme B-H22(scFv), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol. Cancer Ther. 2008, 7, 2924–2932. [Google Scholar] [PubMed]
- Bakker, A.B.; van den Oudenrijn, S.; Bakker, A.Q.; Feller, N.; van Meijer, M.; Bia, J.A.; Jongeneelen, M.A.C.; Visser, T.J.; Bijl, N.; Geuijen, C.A.W.; et al. C-type lectin-like molecule-1: A novel myeloid cell surface marker associated with acute myeloid leukemia. Cancer Res. 2004, 64, 8443–8450. [Google Scholar] [PubMed]
- Zhao, X.; Singh, S.; Pardoux, C.; Zhao, J.; His, E.D.; Abo, A.; Korver, W. Targeting C-type lectin-like molecule-1 for antibody-mediated immunotherapy in acute myeloid leukemia. Haematologica 2010, 95, 71–78. [Google Scholar] [PubMed]
- Bajaj, J.; Konuma, T.; Lytle, N.K.; Kwon, H.Y.; Ablack, J.N.; Cantor, J.M.; Rizzieri, D.; Chuah, C.; Oehler, V.G.; Broome, E.H.; et al. CD98-mediated adhesive signaling enables the establishment and propagation of acute myelogenous leukemia. Cancer Cell 2016, 30, 792–805. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-resistant human acute myeloid leukemia cells are not enriched for leukemic stem cells but require oxidative metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Ikegawa, S.; Doki, N.; Kurosawa, S.; Yamaguchi, T.; Sakaguchi, M.; Harada, K.; Yamamoto, K.; Hino, Y.; Shingai, N.; Senoo, Y.; et al. CD25 expression on residual leukemic blasts at the time of allogeneic hematopoietic stem cell transplant predicts relapse in patients with acute myeloid leukemia without complete remission. Leuk Lymphoma 2016, 57, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Gonen, M.; Sun, Z.; Figueroa, M.E.; Patel, J.P.; Abdel-Wahab, O.; Racevskis, J.; Ketterling, R.P.; Fernandez, H.; Rowe, J.M.; Tallman, M.S.; et al. CD25 expression status improves prognostic risk classification in AML independent of established biomarkers: ECOG phase 3 trial, E1900. Blood 2012, 120, 2297–2306. [Google Scholar] [CrossRef] [PubMed]
- Madhumathi, J.; Sridevi, S.; Verma, R.S. CD25 targeted therapy of chemotherapy resistant leukemic stem cells using DR5 specific TRAIL peptide. Stem Cell Res. 2017, 19, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Keyhani, A.; Huh, Y.O.; Jendiroba, D.; Pagliaro, L.; Cortez, J.; Pierce, S.; Pearlman, M.; Estey, E.; Kantarjian, H.; Freireich, E.J. Increased CD38 expression is associated with favorable prognosis in adult acute leukemia. Leuk Res. 2000, 24, 153–159. [Google Scholar] [CrossRef]
- Dos Santos, C.; Xiaochuan, S.; Chenghui, Z.; Ndikuyeze, G.H.; Glover, J.; Secreto, T.; Doshi, P.; Sasser, K.; Danet-Desnoyers, G. Anti-leukemic activity of daratumumab in acute myeloid leukemia cells and patient-derived xenografts. Blood 2014, 124, 2312. [Google Scholar]
- Pollyea, D.A.; Gutman, J.A.; Gore, L.; Smith, C.A.; Jordan, C.T. Targeting acute myeloid leukemia stem cells: A review and principles for the development of clinical trials. Haematologica 2014, 99, 1277–1284. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Jorgensen, J.L.; Brooks, C.; Shi, C.; Zhang, Q.; Nogueras Gonzalez, G.M.; Cavazos, A.; Pan, R.; Mu, H.; Wang, S.A.; et al. Anti-leukemia efficacy and mechanisms of action of SL-101, a novel anti-CD123 antibody-conjugate, in acute myeloid leukemia. Clin. Cancer Res. 2015, 15, S14. [Google Scholar] [CrossRef]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M.; et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef] [PubMed]
- Zeidan, A.M.; Stahl, M.; Komrokji, R. Emerging biological therapies for the treatment of myelodysplastic syndromes. Expert Opin. Emerg. Drugs 2016, 21, 283–300. [Google Scholar] [CrossRef] [PubMed]
- Podoltsev, N.A.; Stahl, M.; Zeidan, A.M.; Gore, S.D. Selecting initial treatment of acute myeloid leukaemia in older adults. Blood Rev. 2016, 31, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Prebet, T.; Gore, S.D.; Esterni, B.; Gardin, C.; Itzykson, R.; Thepot, S.; Dreyfus, F.; Rauzy, O.B.; Recher, C.; Adès, L.; et al. Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure. J. Clin. Oncol. 2011, 29, 3322–3327. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.P.N.; de Veaux, M.; Perreault, S.; Itzykson, R.; Ritchie, E.K.; Sekeres, M.A.; Fathi, A.T.; Komrokji, R.S.; Bhatt, V.R.; Al-Kali, A.; et al. The use of hypomethylating agents (HMAs) in patients with relapsed and refractory acute myeloid leukemia (RR-AML): Clinical outcomes and their predictors in a large international patient cohort. Blood 2016, 128, 1063. [Google Scholar]
- Stahl, M.; Gore, S.D.; Vey, N.; Prebet, T. Lost in translation? Ten years of development of histone deacetylase inhibitors in acute myeloid leukemia and myelodysplastic syndromes. Expert Opin. Investig. Drugs 2016, 25, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Zeidan, A.M. Hypomethylating agents in combination with histone deacetylase inhibitors in higher risk myelodysplastic syndromes: Is there a light at the end of the tunnel? Cancer 2017, 123, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Kohrman, N.; Gore, S.D.; Kim, T.K.; Zeidan, A.M.; Prebet, T. Epigenetics in cancer: A hematological perspective. PLoS Genet. 2016, 12, e1006193. [Google Scholar] [CrossRef] [PubMed]
- Heninger, E.; Krueger, T.E.; Lang, J.M. Augmenting antitumor immune responses with epigenetic modifying agents. Front. Immunol. 2015, 6, 29. [Google Scholar] [PubMed]
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Akers, S.N.; Odunsi, K.; Karpf, A.R. Regulation of cancer germline antigen gene expression: Implications for cancer immunotherapy. Future Oncol. 2010, 6, 717–732. [Google Scholar] [CrossRef] [PubMed]
- Atanackovic, D.; Luetkens, T.; Kloth, B.; Fuchs, G.; Cao, Y.; Hildebrandt, Y.; Meyer, S.; Bartels, K.; Reinhard, H.; Lajmi, N.; et al. Cancer-testis antigen expression and its epigenetic modulation in acute myeloid leukemia. Am. J. Hematol. 2011, 86, 918–922. [Google Scholar] [CrossRef] [PubMed]
- Chambost, H.; van Baren, N.; Brasseur, F.; Olive, D. MAGE-A genes are not expressed in human leukemias. Leukemia 2001, 15, 1769–1771. [Google Scholar] [CrossRef] [PubMed]
- Ortmann, C.A.; Eisele, L.; Nuckel, H.; Klein-Hitpass, L.; Fuhrer, A.; Duhrsen, U.; Zeschnigk, M. Aberrant hypomethylation of the cancer-testis antigen PRAME correlates with PRAME expression in acute myeloid leukemia. Ann. Hematol. 2008, 87, 809–818. [Google Scholar] [CrossRef] [PubMed]
- Goodyear, O.; Agathanggelou, A.; Novitzky-Basso, I.; Siddique, S.; McSkeane, T.; Ryan, G.; Vyas, P.; Cavenagh, J.; Stankovic, T.; Moss, P.; et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood 2010, 116, 1908–1918. [Google Scholar] [CrossRef] [PubMed]
- Almstedt, M.; Blagitko-Dorfs, N.; Duque-Afonso, J.; Karbach, J.; Pfeifer, D.; Jager, E.; Lübbert, M. The DNA demethylating agent 5-aza-2′-deoxycytidine induces expression of NY-ESO-1 and other cancer/testis antigens in myeloid leukemia cells. Leuk. Res. 2010, 34, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Paluch, B.E.; Matsuzaki, J.; James, S.R.; Collamat-Lai, G.; Blagitko-Dorfs, N.; Ford, L.A.; Naqash, R.; Lübbert, M.; Karpf, A.R.; et al. Induction of cancer testis antigen expression in circulating acute myeloid leukemia blasts following hypomethylating agent monotherapy. Oncotarget 2016, 7, 12840–12856. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.; Paluch, B.E.; Matsuzaki, J.; James, S.R.; Collamat-Lai, G.; Karbach, J.; Nemeth, M.J.; Taverna, P.; Karpf, A.R.; Griffiths, E.A. Immunomodulatory action of SGI-110, a hypomethylating agent, in acute myeloid leukemia cells and xenografts. Leuk. Res. 2014, 38, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.X.; Mei, Z.Y.; Zhou, J.H.; Yao, Y.S.; Li, Y.H.; Xu, Y.H.; Li, J.X.; Gao, X.N.; Zhou, M.H.; Jiang, M.M.; et al. Low dose decitabine treatment induces CD80 expression in cancer cells and stimulates tumor specific cytotoxic T lymphocyte responses. PLoS ONE 2013, 8, e62924. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, P.M.J.; Paluch, B.E.; Brumberger, Z.; Kaufman, S.; Karpf, A.R.; Odunsi, K.; Miller, A.; Kocent, J.; Wang, E.S.; Nemeth, M.J.; et al. NY-ESO-1 vaccination in combination with decitabine for patients with MDS induces CD4+ and CD8+ T-cell responses. Blood 2015, 126, 2873. [Google Scholar]
- Fathi, A.T.; Erba, H.P.; Lancet, J.E.; Stein, E.M.; Ravandi, F.; Faderl, S.; Walter, R.B.; Advani, A.; DeAngelo, D.J.; Kovacsovics, T.J.; et al. Vadastuximab talirine plus hypomethylating agents: A well-tolerated regimen with high remission rate in frontline older patients with acute myeloid leukemia (AML). Blood 2016, 128, 591. [Google Scholar]
- Zhang, L.; Gajewski, T.F.; Kline, J. PD-1/PD-L1 interactions inhibit antitumor immune responses in a murine acute myeloid leukemia model. Blood 2009, 114, 1545–1552. [Google Scholar] [CrossRef] [PubMed]
- Kronig, H.; Kremmler, L.; Haller, B.; Englert, C.; Peschel, C.; Andreesen, R.; Blank, C.U. Interferon-induced programmed death-ligand 1 (PD-L1/B7-H1) expression increases on human acute myeloid leukemia blast cells during treatment. Eur. J. Haematol. 2014, 92, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Basu, S.; Garcia-Manero, G.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Hendrickson, S.; Pierce, S.; Ning, J.; Konopleva, M.; et al. Phase IB/II study of nivolumab in combination with azacytidine (AZA) in patients (pts) with relapsed acute myeloid leukemia (AML). Blood 2016, 128, 763. [Google Scholar]
- Czibere, A.; Bruns, I.; Kroger, N.; Platzbecker, U.; Lind, J.; Zohren, F.; Fenk, R.; Germing, U.; Schröder, T.; Gräf, T.; et al. 5-Azacytidine for the treatment of patients with acute myeloid leukemia or myelodysplastic syndrome who relapse after allo-SCT: A retrospective analysis. Bone Marrow Transplant. 2010, 45, 872. [Google Scholar] [CrossRef] [PubMed]
- Lal, G.; Zhang, N.; van der Touw, W.; Ding, Y.; Ju, W.; Bottinger, E.P.; Reid, S.P.; Levy, D.E.; Bromberg, J.S. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J. Immunol. 2009, 182, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Polansky, J.K.; Kretschmer, K.; Freyer, J.; Floess, S.; Garbe, A.; Baron, U.; Olek, S.; Hamann, A.; von Boehmer, H.; Huehn, J. DNA methylation controls Foxp3 gene expression. Eur. J. Immunol. 2008, 38, 1654–1663. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Ritchey, J.; Prior, J.L.; Holt, M.; Shannon, W.D.; Deych, E.; Piwnica-Worms, D.R.; DiPersio, J.F. In vivo administration of hypomethylating agents mitigate graft-versus-host disease without sacrificing graft-versus-leukemia. Blood 2010, 116, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Abarca, L.I.; Gutierrez-Cosio, S.; Santamaria, C.; Caballero-Velazquez, T.; Blanco, B.; Herrero-Sanchez, C.; García, J.L.; Carrancio, S.; Hernández-Campo, P.; González, F.J.; et al. Immunomodulatory effect of 5-azacytidine (5-azaC): Potential role in the transplantation setting. Blood 2010, 115, 107–121. [Google Scholar] [CrossRef] [PubMed]
- Goodyear, O.C.; Dennis, M.; Jilani, N.Y.; Loke, J.; Siddique, S.; Ryan, G.; Nunnick, J.; Khanum, R.; Raghavan, M.; Cook, M.; et al. Azacitidine augments expansion of regulatory T cells after allogeneic stem cell transplantation in patients with acute myeloid leukemia (AML). Blood 2012, 119, 3361–3369. [Google Scholar] [CrossRef] [PubMed]
- Maffei, R.; Fiorcari, S.; Bulgarelli, J.; Rizzotto, L.; Martinelli, S.; Rigolin, G.M.; Debbia, G.; Castelli, I.; Bonacorsi, G.; Santachiara, R.; et al. Endothelium-mediated survival of leukemic cells and angiogenesis-related factors are affected by lenalidomide treatment in chronic lymphocytic leukemia. Exp. Hematol. 2014, 42, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Kantarjian, H.; Estrov, Z.; Faderl, S.; Ravandi, F.; Rey, K.; Cortes, J.; Borthakur, G. A phase II study of lenalidomide alone in relapsed/refractory acute myeloid leukemia or high-risk myelodysplastic syndromes with chromosome 5 abnormalities. Clin. Lymphoma Myeloma Leuk. 2012, 12, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Sekeres, M.A.; Gundacker, H.; Lancet, J.; Advani, A.; Petersdorf, S.; Liesveld, J.; Mulford, D.; Norwood, T.; Willman, C.L.; Appelbaum, F.R.; et al. A phase 2 study of lenalidomide monotherapy in patients with deletion 5q acute myeloid leukemia: Southwest oncology group study S0605. Blood 2011, 118, 523–528. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.A.; Uy, G.L.; Trinkaus, K.; Nelson, A.D.; Demland, J.; Abboud, C.N.; Cashen, A.F.; Stockerl-Goldstein, K.E.; Westervelt, P.; DiPersio, J.F.; et al. A phase 2 study of high-dose lenalidomide as initial therapy for older patients with acute myeloid leukemia. Blood 2011, 117, 1828–1833. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H. Blocking IDO activity to enhance anti-tumor immunity. Front. Biosci. 2012, 4, 734–745. [Google Scholar] [CrossRef]
- Mahoney, K.M.; Rennert, P.D.; Freeman, G.J. Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 2015, 14, 561–584. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Ben, Q.; Tu, S.; Dong, W.; Qi, X.; Wu, Y. Serum interleukin-33 levels in patients with gastric cancer. Dig. Dis. Sci. 2011, 56, 3596–3601. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.A.; Fu, Y.; Zhang, D.N.; Zhang, J. Serum IL-33 as a diagnostic and prognostic marker in non- small cell lung cancer. Asian Pac. J. Cancer Prev. 2013, 14, 2563–2566. [Google Scholar] [CrossRef] [PubMed]
- Gramatzki, D.; Frei, K.; Cathomas, G.; Moch, H.; Weller, M.; Mertz, K.D. Interleukin-33 in human gliomas: Expression and prognostic significance. Oncol. Lett. 2016, 12, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Levescot, A.; Flamant, S.; Basbous, S.; Jacomet, F.; Feraud, O.; Anne Bourgeois, E.; Bonnet, M.L.; Giraud, C.; Roy, L.; Barra, A.; et al. BCR-ABL-induced deregulation of the IL-33/ST2 pathway in CD34+ progenitors from chronic myeloid leukemia patients. Cancer Res. 2014, 74, 2669–2676. [Google Scholar] [CrossRef] [PubMed]
- Mager, L.F.; Riether, C.; Schurch, C.M.; Banz, Y.; Wasmer, M.H.; Stuber, R.; Theocharides, A.P.; Li, X.; Xia, Y.; Saito, H.; et al. IL-33 signaling contributes to the pathogenesis of myeloproliferative neoplasms. J. Clin. Investig. 2015, 125, 2579–2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasmer, M.H.; Krebs, P. The role of IL-33-dependent inflammation in the tumor microenvironment. Front. Immunol. 2016, 7, 682. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, L.; Wang, H.; Xiong, S.; Li, Y.; Tao, Q.; Xiao, W.; Qin, H.; Wang, Y.; Zhai, Z. Tumor-induced CD14+HLA-DR−/low myeloid-derived suppressor cells correlate with tumor progression and outcome of therapy in multiple myeloma patients. Cancer Immunol. Immunother. 2015, 64, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.F.; Kuan, F.C.; Yen, T.C.; Lu, M.S.; Lin, P.Y.; Chung, Y.H.; Chen, W.C.; Lee, K.D. IL-6-stimulated CD11b+ CD14+ HLA-DR-myeloid-derived suppressor cells, are associated with progression and poor prognosis in squamous cell carcinoma of the esophagus. Oncotarget 2014, 5, 8716–8728. [Google Scholar] [CrossRef] [PubMed]
- Laborde, R.R.; Lin, Y.; Gustafson, M.P.; Bulur, P.A.; Dietz, A.B. Cancer vaccines in the world of immune suppressive monocytes (CD14(+)HLA-DR(lo/neg) Cells): The gateway to improved responses. Front. Immunol. 2014, 5, 147. [Google Scholar] [CrossRef] [PubMed]
- Movahedi, K.; Guilliams, M.; van den Bossche, J.; Van den Bergh, R.; Gysemans, C.; Beschin, A.; De Baetselier, P.; van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Eksioglu, E.A.; Zhou, J.; Zhang, L.; Djeu, J.; Fortenbery, N.; Epling-Burnette, P.; Van Bijnen, S.; Dolstra, H.; Cannon, J.; et al. Induction of myelodysplasia by myeloid-derived suppressor cells. J. Clin. Investig. 2013, 123, 4595–4611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyzer, A.R.; Stroopinsky, D.; Rajabi, H.; Washington, A.; Tagde, A.; Coll, M.; Fung, J.; Bryant, M.P.; Cole, L.; Palmer, K.; et al. MUC1 mediated induction of myeloid-derived suppressor cells in patients with acute myeloid leukemia. Blood 2017, 129, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Wesolowski, R.; Markowitz, J.; Carson, W.E., 3rd. Myeloid derived suppressor cells—A new therapeutic target in the treatment of cancer. J. Immunother. Cancer 2013, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef] [PubMed]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef] [PubMed]
- Al-Matary, Y.S.; Botezatu, L.; Opalka, B.; Hones, J.M.; Lams, R.F.; Thivakaran, A.; Schütte, J.; Köster, R.; Lennartz, K.; Schroeder, T.; et al. Acute myeloid leukemia cells polarize macrophages towards a leukemia supporting state in a growth factor independence 1 dependent manner. Haematologica 2016, 101, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [PubMed]
- Pekarek, L.A.; Starr, B.A.; Toledano, A.Y.; Schreiber, H. Inhibition of tumor growth by elimination of granulocytes. J. Exp. Med. 1995, 181, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Shojaei, F.; Singh, M.; Thompson, J.D.; Ferrara, N. Role of Bv8 in neutrophil-dependent angiogenesis in a transgenic model of cancer progression. Proc. Natl. Acad. Sci. USA 2008, 105, 2640–2645. [Google Scholar] [CrossRef] [PubMed]
- Youn, J.I.; Gabrilovich, D.I. The biology of myeloid-derived suppressor cells: The blessing and the curse of morphological and functional heterogeneity. Eur. J. Immunol. 2010, 40, 2969–2975. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Workman, C.J.; Vignali, D.A. Targeting regulatory T cells in tumors. FEBS J. 2016, 283, 2731–2748. [Google Scholar] [CrossRef] [PubMed]
- Turnis, M.E.; Sawant, D.V.; Szymczak-Workman, A.L.; Andrews, L.P.; Delgoffe, G.M.; Yano, H.; Beres, A.J.; Vogel, P.; Workman, C.J.; Vignali, D.A.A. Interleukin-35 limits anti-tumor immunity. Immunity 2016, 44, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Cai, S.F.; Fehniger, T.A.; Song, J.; Collins, L.I.; Piwnica-Worms, D.R.; Ley, T.J. Granzyme B and perforin are important for regulatory T cell-mediated suppression of tumor clearance. Immunity 2007, 27, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Bucher, C.; Munger, M.E.; Highfill, S.L.; Tolar, J.; Munn, D.H.; Levine, B.L.; Riddle, M.; June, C.H.; Vallera, D.A.; et al. Depletion of endogenous tumor-associated regulatory T cells improves the efficacy of adoptive cytotoxic T-cell immunotherapy in murine acute myeloid leukemia. Blood 2009, 114, 3793–3802. [Google Scholar] [CrossRef] [PubMed]
- Ustun, C.; Miller, J.S.; Munn, D.H.; Weisdorf, D.J.; Blazar, B.R. Regulatory T cells in acute myelogenous leukemia: Is it time for immunomodulation? Blood 2011, 118, 5084–5095. [Google Scholar] [CrossRef] [PubMed]
- Teague, R.M.; Kline, J. Immune evasion in acute myeloid leukemia: Current concepts and future directions. J. Immunother. Cancer 2013, 1, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.V.; Chemnitz, J.M.; Frauwirth, K.A.; Lanfranco, A.R.; Braunstein, I.; Kobayashi, S.V.; Linsley, P.S.; Thompson, C.B.; Riley, J.L. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005, 25, 9543–9553. [Google Scholar] [CrossRef] [PubMed]
- Armand, P. Immune checkpoint blockade in hematologic malignancies. Blood 2015, 125, 3393–3400. [Google Scholar] [CrossRef] [PubMed]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Casulo, C.; Yahalom, J.; Schoder, H.; Barr, P.M.; Caron, P.; Chiu, A.; Constine, L.S.; Drullinsky, P.; Friedberg, J.W.; et al. Brentuximab vedotin and AVD followed by involved-site radiotherapy in early stage, unfavorable risk Hodgkin lymphoma. Blood 2016, 128, 1458–1464. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: Preliminary results of a phase Ib study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A.; Yamashita, T.; Tamura, H.; Zhao, W.; Tsuji, T.; Shimizu, M.; Shinya, E.; Takahashi, H.; Tamada, K.; Chen, L.; et al. Interferon-γ and tumor necrosis factor-α induce an immunoinhibitory molecule, B7-H1, via nuclear factor-κB activation in blasts in myelodysplastic syndromes. Blood 2010, 116, 1124–1131. [Google Scholar] [CrossRef] [PubMed]
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.B.; McSweeney, P.; et al. Ipilimumab for patients with relapse after allogeneic transplantation. N. Engl. J. Med. 2016, 375, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Michot, J.M.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: A comprehensive review. Eur. J. Cancer 2016, 54, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Gubin, M.M.; Schreiber, R.D.; Smyth, M.J. New insights into cancer immunoediting and its three component phases—Elimination, equilibrium and escape. Curr. Opin. Immunol. 2014, 27, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Basu, S.; Garcia-Manero, G.; Cortes, J.E.; Ravandi, F.; Ning, J.; Xiao, L.; Juliana, L.; Kornblau, S.M.; Konopleva, M.; et al. Defining the immune checkpoint landscape in patients (pts) with acute myeloid leukemia (AML). Blood 2016, 128, 2900. [Google Scholar]
- Garcia-Manero, G.; Daver, N.G.; Montalban-Bravo, G.; Jabbour, E.J.; DiNardo, C.D.; Kornblau, S.M.; Bose, P.; Alvarado, Y.; Ohanian, M.; Borthakur, G.; et al. A phase II study evaluating the combination of nivolumab (Nivo) or ipilimumab (Ipi) with azacitidine in pts with previously treated or untreated myelodysplastic syndromes (MDS). Blood 2016, 128, 344. [Google Scholar]
- Mellman, I.; Coukos, G.; Dranoff, G. Cancer immunotherapy comes of age. Nature 2011, 480, 480–489. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Target | Drug | Trial Phase | Patient Population | Single Agent/Combination | Ref./Identifier | Status |
|---|---|---|---|---|---|---|
| CD33 | IMGN779 | I | Adult patients with relapsed/refractory CD33+ AML | Single agent | NCT02674763 | Recruiting |
| CD33 | Gemtuzumab ozogamicin | II | Patients up to 70 years with AML induction/re-induction failure, AML in CR1 with poor cytogenetics, AML in 2nd CR with MRD, AML in 3rd CR, AML in refractory relapse but ≤25% BM blasts, MDS with >6% BM blasts at diagnosis, secondary MDS with ≤5% BM blasts at diagnosis
Note: disease must express >/=10% CD33+ for patients with AML | Combination with busulfan and cyclophosphamide | NCT02221310 | Recruiting |
| CD33 | AMV564 | I | Adult patients with relapsed/refractory AML | Single agent | NCT03144245 | Recruiting |
| CD33 | SGN-CD33A | III | Adult patients with newly diagnosed, previously untreated intermediate or adverse risk de novo or secondary AML | Combination with azacitidine or decitabine | NCT02785900 | Recruiting |
| CD123 | SGN-CD123A | I | Adult patients up to 74 years with relapsed/refractory CD123-detectable AML following at least 2 but no more than 3 prior regimens; patients may be eligible after only 1 previous regimen if in a high risk category | Single agent | NCT02848248 | Recruiting |
| CD123 | XmAb14045 | I | Adult patients with primary or secondary AML , B-cell Acute lymphocytic leukemia (ALL), blastic plasmacytoid dendritic cell neoplasm (BPDCN), Chronic myeloid leukemia (CML) in blast phase, resistant or intolerant to tyrosine kinase inhibitors; patients with relapsed or refractory disease with no available standard therapy | Single agent | NCT02730312 | Recruiting |
| CD123 | JNJ-56022473 (CSL362) | II/III | Elderly patients, 65 years or older with de novo or secondary AML | Combination with decitabine | NCT02472145 | Recruiting |
| CD123 | MGD006 | I | Adult patients with primary or secondary AML or MDS with an International prognostic scoring system (IPSS) category of intermediate 2 or high risk | Single agent | NCT02152956 | Recruiting |
| CD123 | SL-401 | I/II | Adult patients with AML in first or second CR or CRi | Single agent | NCT02270463 | Recruiting |
| PD-L1 | Durvalumab (MEDI4736) | II | Adult patients with MDS or elderly patients (≥65 years) with newly diagnosed de novo AML or secondary AML | Combination with azacitidine | NCT02775903 | Recruiting |
| PD-L1 | Atezolizumab | I | Adult patients with relapsed refractory AML; elderly patients with treatment naiive AML who are unfit for induction chemotherapy | Combination with guadecitabine | NCT02892318 | Recruiting |
| PD-1 | Nivolumab | II | Adult patients with relapsed/ refractory AML | Combination with azacitidine; Combination with ipilimumab and azacitidine | NCT02397720 | Recruiting |
| PD-1 | Pembrolizumab | II | Adult patients with relapsed/ refractory AML | Combination with azacitidine | NCT02845297 | Recruiting |
| CTLA-4 | Ipilimumab | I | Adult patients with relapsed/refractory AML or MDS;
Elderly patients (≥75 years) with treatment naïve de novo or secondary AML | Combination with decitabine | NCT02890329 | Recruiting |
| IDO | Indoximod | I/II | Adult patients with newly diagnosed AML | Combination with “7+3” | NCT02835729 | Recruiting |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gbolahan, O.B.; Zeidan, A.M.; Stahl, M.; Abu Zaid, M.; Farag, S.; Paczesny, S.; Konig, H. Immunotherapeutic Concepts to Target Acute Myeloid Leukemia: Focusing on the Role of Monoclonal Antibodies, Hypomethylating Agents and the Leukemic Microenvironment. Int. J. Mol. Sci. 2017, 18, 1660. https://doi.org/10.3390/ijms18081660
Gbolahan OB, Zeidan AM, Stahl M, Abu Zaid M, Farag S, Paczesny S, Konig H. Immunotherapeutic Concepts to Target Acute Myeloid Leukemia: Focusing on the Role of Monoclonal Antibodies, Hypomethylating Agents and the Leukemic Microenvironment. International Journal of Molecular Sciences. 2017; 18(8):1660. https://doi.org/10.3390/ijms18081660
Chicago/Turabian StyleGbolahan, Olumide Babajide, Amer M. Zeidan, Maximilian Stahl, Mohammad Abu Zaid, Sherif Farag, Sophie Paczesny, and Heiko Konig. 2017. "Immunotherapeutic Concepts to Target Acute Myeloid Leukemia: Focusing on the Role of Monoclonal Antibodies, Hypomethylating Agents and the Leukemic Microenvironment" International Journal of Molecular Sciences 18, no. 8: 1660. https://doi.org/10.3390/ijms18081660