
Conformational Flexibility Differentiates Naturally Occurring Bet v 1 Isoforms


Abstract
:
1. Introduction
2. Results
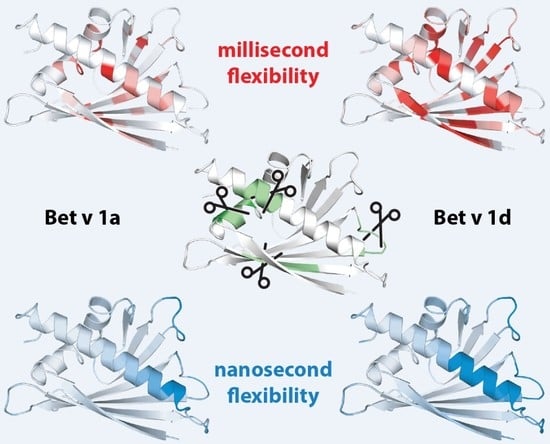
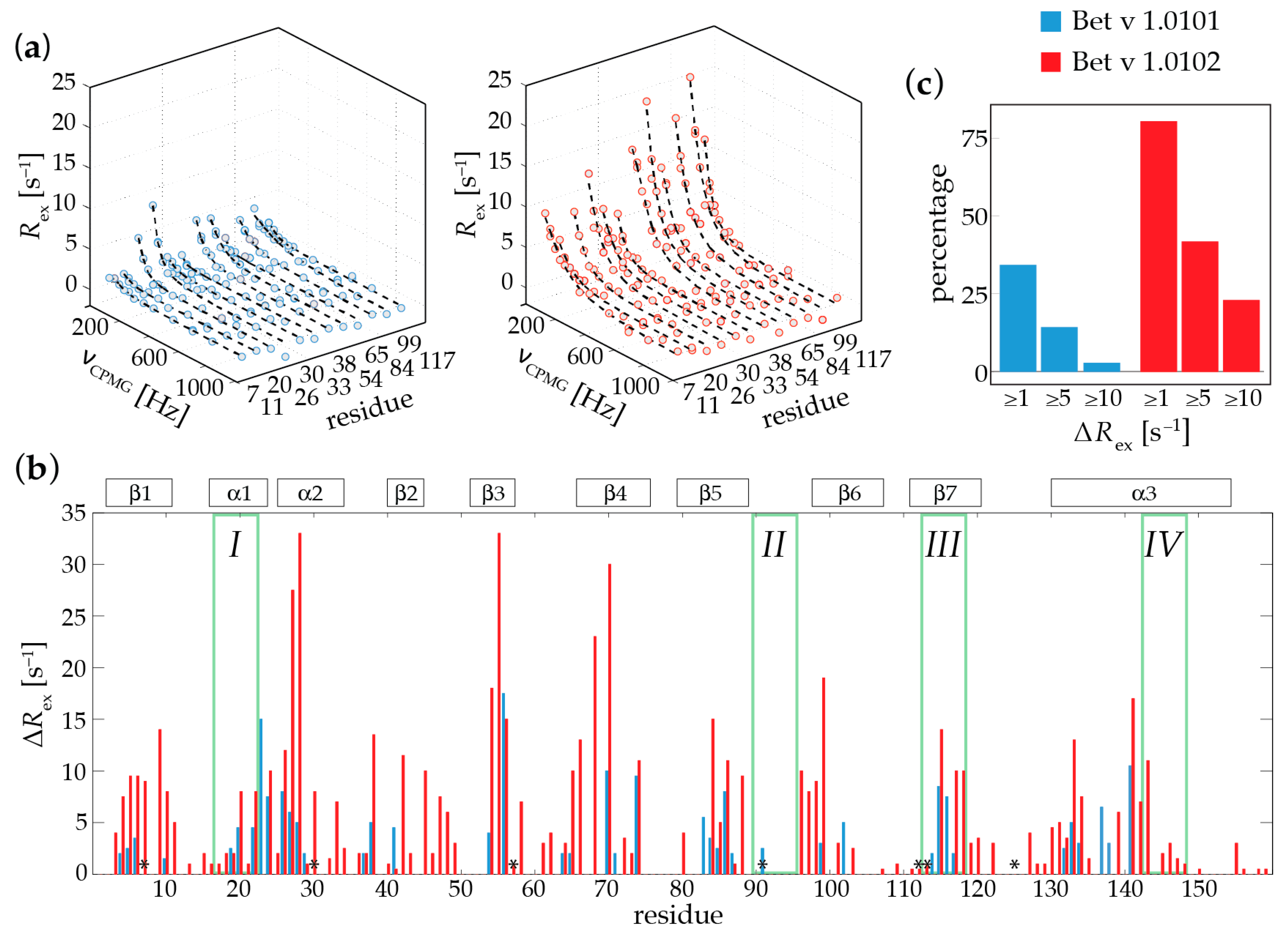
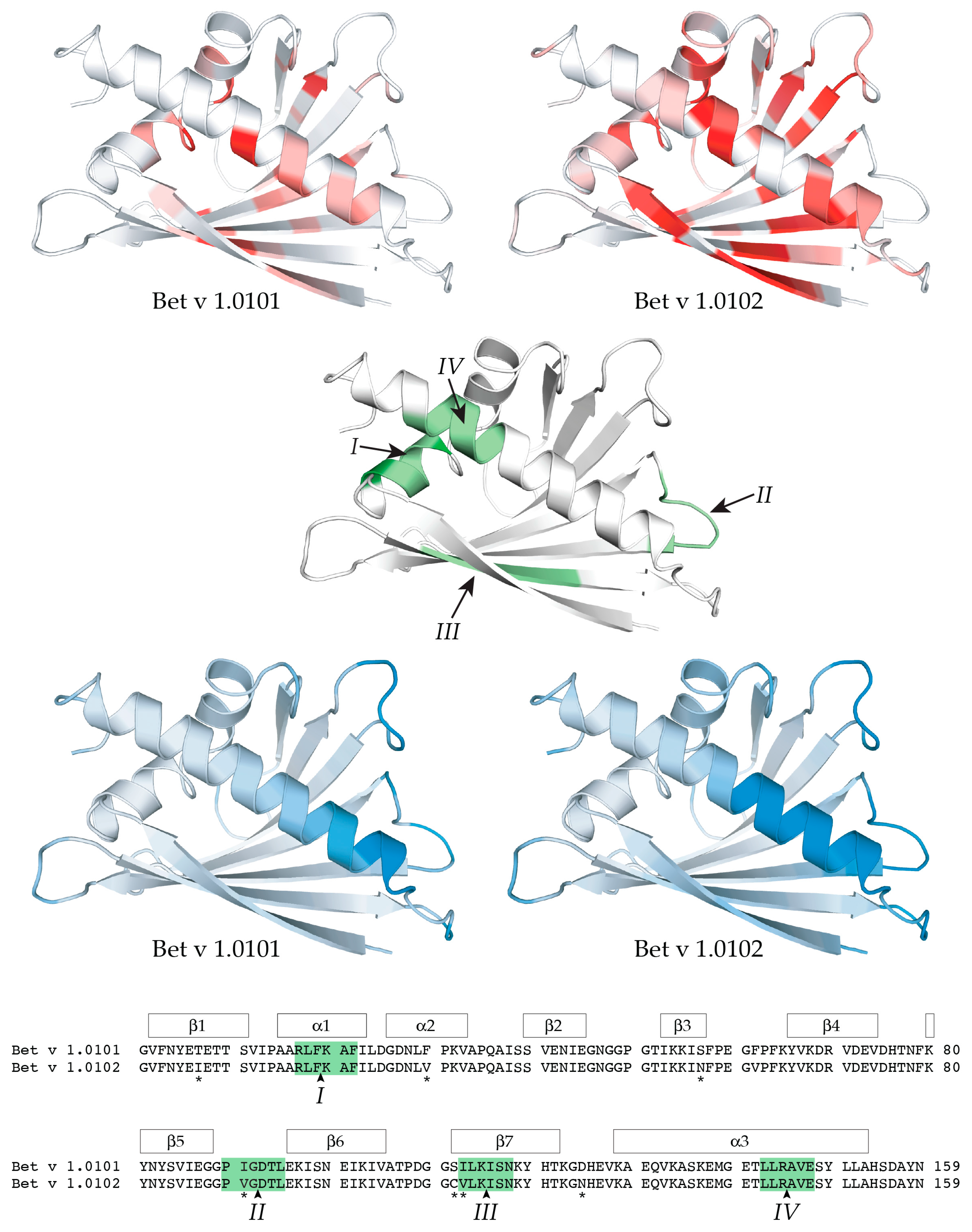
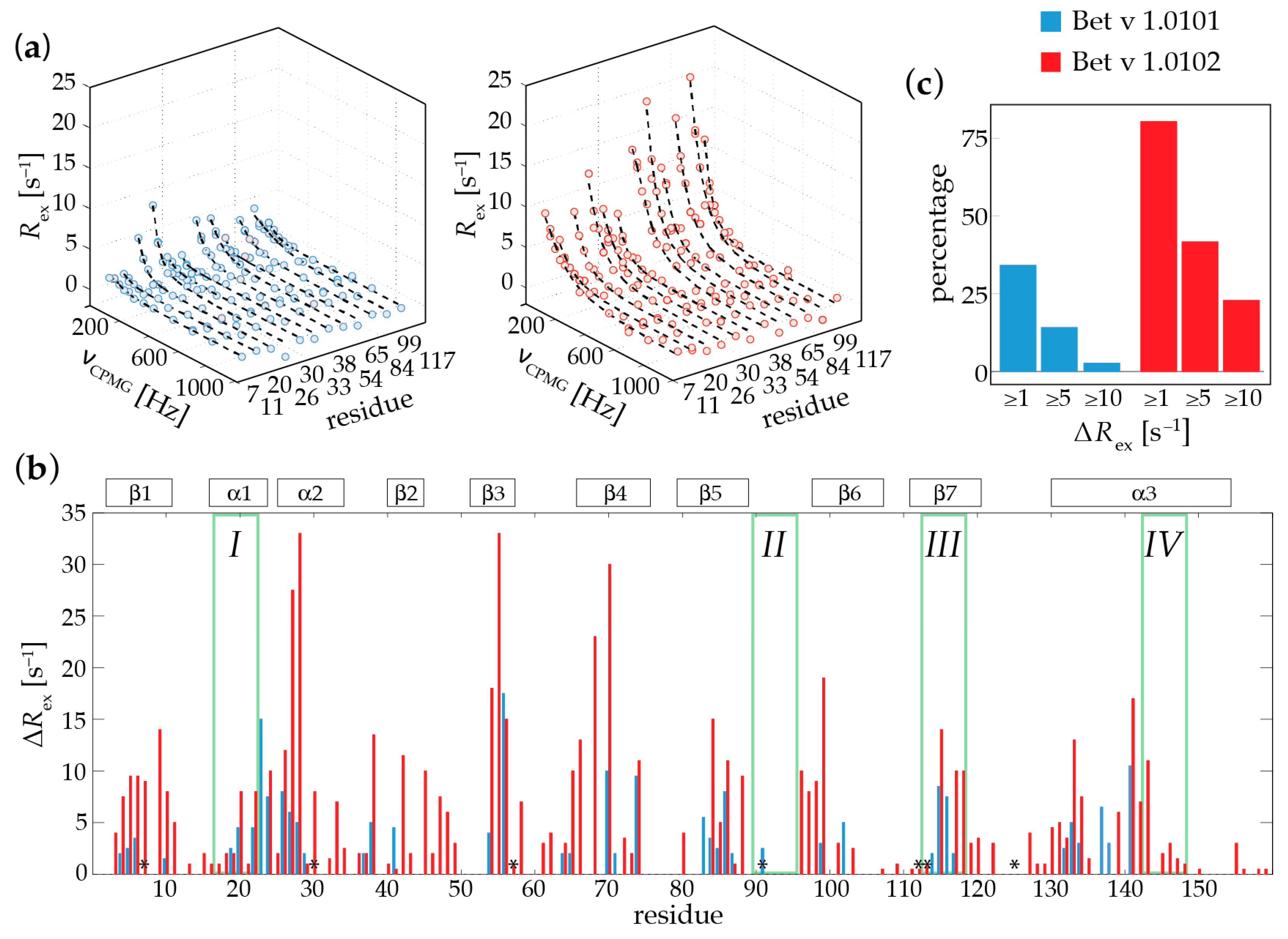
2.1. Microsecond-Millisecond Conformational Flexibility
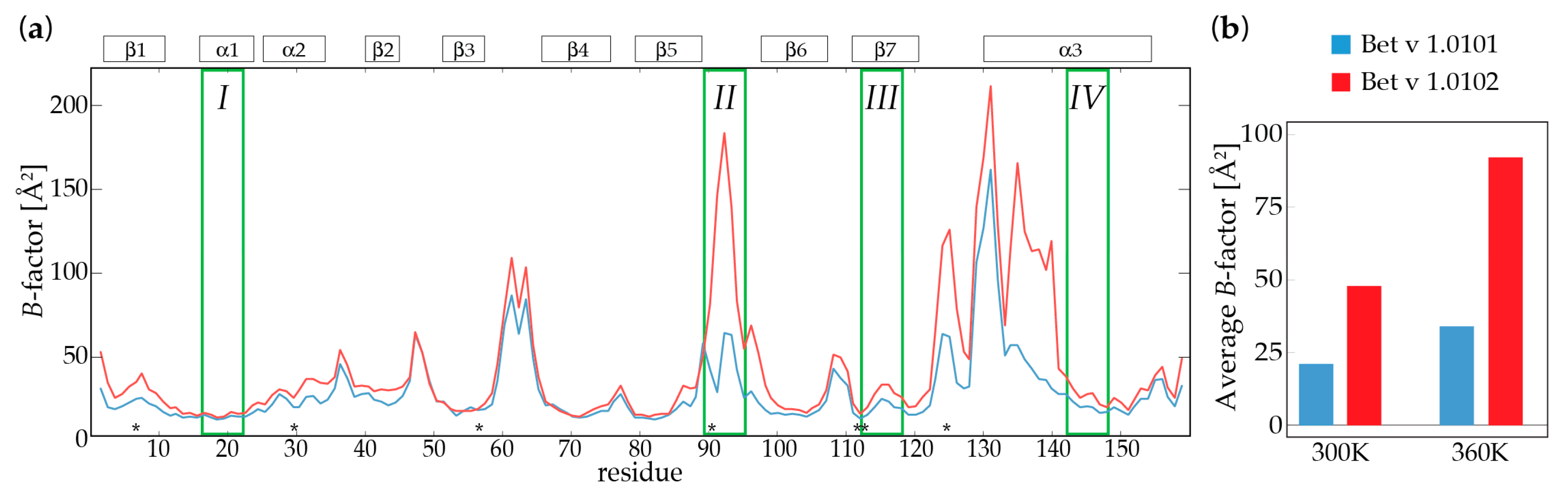
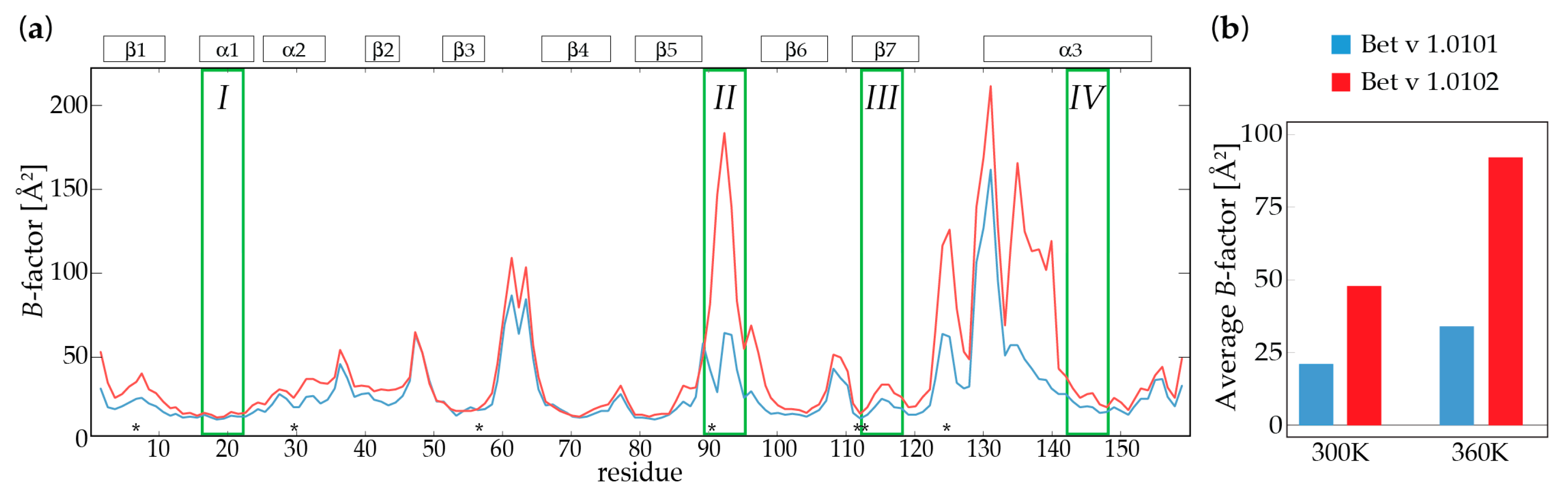
2.2. Molecular Dynamics Simulations
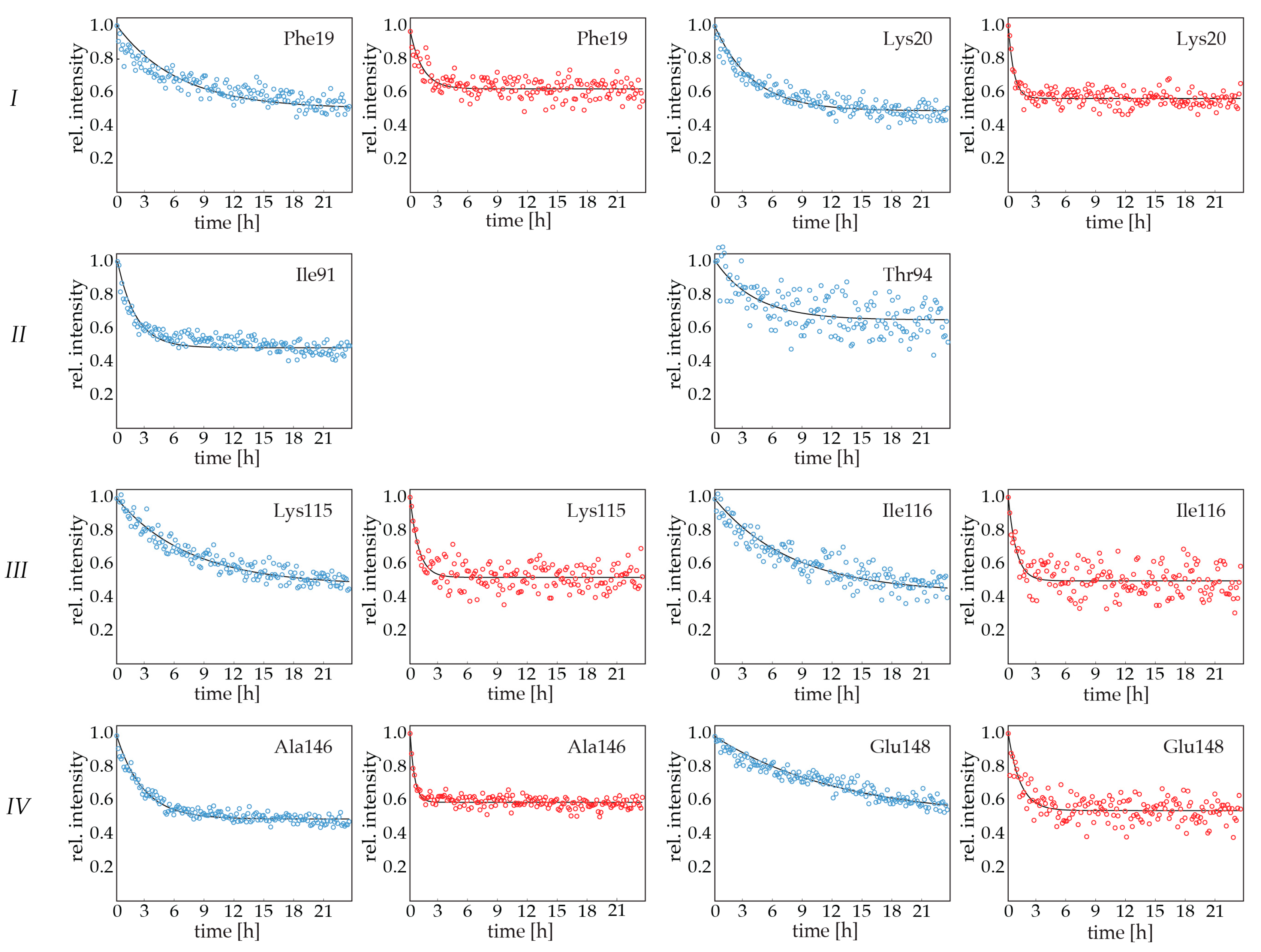
2.3. Backbone Amide Hydrogen-Deuterium Exchange
3. Discussion
4. Materials and Methods
4.1. NMR Sample Preparation
4.2. NMR Experiments and Data Analysis
4.3. Molecular Dynamics Simulations
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Matricardi, P.M.; Kleine-Tebbe, J.; Hoffmann, H.J.; Valenta, R.; Hilger, C.; Hofmaier, S.; Aalberse, R.C.; Agache, I.; Asero, R.; Ballmer-Weber, B.; et al. EAACI Molecular allergology user’s guide. Pediatr. Allergy Immunol. 2016, 27 (Suppl. S23), 1–250. [Google Scholar] [CrossRef] [PubMed]
- Jarolim, E.; Rumpold, H.; Endler, A.T.; Ebner, H.; Breitenbach, M.; Scheiner, O.; Kraft, D. IgE and IgG antibodies of patients with allergy to birch pollen as tools to define the allergen profile of Betula verrucosa. Allergy 1989, 44, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Moverare, R.; Westritschnig, K.; Svensson, M.; Hayek, B.; Bende, M.; Pauli, G.; Sorva, R.; Haahtela, T.; Valenta, R.; Elfman, L. Different IgE reactivity profiles in birch pollen-sensitive patients from six European populations revealed by recombinant allergens: An imprint of local sensitization. Int. Arch. Allergy Immunol. 2002, 128, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Hauser, M.; Asam, C.; Himly, M.; Palazzo, P.; Voltolini, S.; Montanari, C.; Briza, P.; Bernardi, M.L.; Mari, A.; Ferreira, F.; et al. Bet v 1-like pollen allergens of multiple Fagales species can sensitize atopic individuals. Clin. Exp. Allergy 2011, 41, 1804–1814. [Google Scholar] [CrossRef] [PubMed]
- Swoboda, I.; Jilek, A.; Ferreira, F.; Engel, E.; Hoffmann-Sommergruber, K.; Scheiner, O.; Kraft, D.; Breiteneder, H.; Pittenauer, E.; Schmid, E.; et al. Isoforms of Bet v 1, the major birch pollen allergen, analyzed by liquid chromatography, mass spectrometry, and cDNA cloning. J. Biol. Chem. 1995, 270, 2607–2613. [Google Scholar] [CrossRef] [PubMed]
- Schenk, M.F.; Gilissen, L.J.; Esselink, G.D.; Smulders, M.J. Seven different genes encode a diverse mixture of isoforms of Bet v 1, the major birch pollen allergen. BMC Genom. 2006, 7, 168. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.; Michalska, K.; Sikorski, M.; Jaskolski, M. Structural and functional aspects of PR-10 proteins. FEBS J. 2013, 280, 1169–1199. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.; Hirtenlehner, K.; Jilek, A.; Godnik-Cvar, J.; Breiteneder, H.; Grimm, R.; Hoffmann-Sommergruber, K.; Scheiner, O.; Kraft, D.; Breitenbach, M.; et al. Dissection of immunoglobulin E and T lymphocyte reactivity of isoforms of the major birch pollen allergen Bet v 1: Potential use of hypoallergenic isoforms for immunotherapy. J. Exp. Med. 1996, 183, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Radauer, C.; Bublin, M.; Hoffmann-Sommergruber, K.; Kopp, T.; Greisenegger, E.K.; Vogel, L.; Vieths, S.; Scheiner, O.; Breiteneder, H. Naturally occurring hypoallergenic Bet v 1 isoforms fail to induce IgE responses in individuals with birch pollen allergy. J. Allergy Clin. Immunol. 2008, 121, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Smole, U.; Balazs, N.; Hoffmann-Sommergruber, K.; Radauer, C.; Hafner, C.; Wallner, M.; Ferreira, F.; Grossinger, R.; de Jong, E.C.; Wagner, S.; et al. Differential T-cell responses and allergen uptake after exposure of dendritic cells to the birch pollen allergens Bet v 1.0101, Bet v 1.0401 and Bet v 1.1001. Immunobiology 2010, 215, 903–909. [Google Scholar] [CrossRef] [PubMed]
- Watts, C. Antigen processing in the endocytic compartment. Curr. Opin. Immunol. 2001, 13, 26–31. [Google Scholar] [CrossRef]
- Egger, M.; Jurets, A.; Wallner, M.; Briza, P.; Ruzek, S.; Hainzl, S.; Pichler, U.; Kitzmuller, C.; Bohle, B.; Huber, C.G.; et al. Assessing protein immunogenicity with a dendritic cell line-derived endolysosomal degradome. PLoS ONE 2011, 6, e17278. [Google Scholar] [CrossRef] [PubMed]
- Freier, R.; Dall, E.; Brandstetter, H. Protease recognition sites in Bet v 1a are cryptic, explaining its slow processing relevant to its allergenicity. Sci. Rep. 2015, 5, 12707. [Google Scholar] [CrossRef] [PubMed]
- Mittag, T.; Kay, L.E.; Forman-Kay, J.D. Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit. 2010, 23, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Karplus, M.; Kuriyan, J. Molecular dynamics and protein function. Proc. Natl. Acad. Sci. USA 2005, 102, 6679–6685. [Google Scholar] [CrossRef] [PubMed]
- Mittermaier, A.K.; Kay, L.E. Observing biological dynamics at atomic resolution using NMR. Trends Biochem. Sci. 2009, 34, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Gajhede, M.; Osmark, P.; Poulsen, F.M.; Ipsen, H.; Larsen, J.N.; van Neerven, R.J.J.; Schou, C.; Lowenstein, H.; Spangfort, M.D. X-ray and NMR structure of Bet v 1, the origin of birch pollen allergy. Nat. Struct. Biol. 1996, 3, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Kofler, S.; Asam, C.; Eckhard, U.; Wallner, M.; Ferreira, F.; Brandstetter, H. Crystallographically mapped ligand binding differs in high and low IgE binding isoforms of birch pollen allergen Bet v 1. J. Mol. Biol. 2012, 422, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Krishna, M.M.; Hoang, L.; Lin, Y.; Englander, S.W. Hydrogen exchange methods to study protein folding. Methods 2004, 34, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Skinner, J.J.; Lim, W.K.; Bedard, S.; Black, B.E.; Englander, S.W. Protein dynamics viewed by hydrogen exchange. Protein Sci. 2012, 21, 996–1005. [Google Scholar] [CrossRef] [PubMed]
- Schanda, P.; Forge, V.; Brutscher, B. Protein folding and unfolding studied at atomic resolution by fast two-dimensional NMR spectroscopy. Proc. Natl. Acad. Sci. USA 2007, 104, 11257–11262. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Milne, J.S.; Mayne, L.; Englander, S.W. Primary structure effects on peptide group hydrogen exchange. Proteins 1993, 17, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Lennon-Dumenil, A.M.; Bakker, A.H.; Wolf-Bryant, P.; Ploegh, H.L.; Lagaudriere-Gesbert, C. A closer look at proteolysis and MHC-class-II-restricted antigen presentation. Curr. Opin. Immunol. 2002, 14, 15–21. [Google Scholar] [CrossRef]
- Loughlin, W.A.; Tyndall, J.D.; Glenn, M.P.; Hill, T.A.; Fairlie, D.P. Update 1 of: Beta-strand mimetics. Chem. Rev. 2010, 110, PR32–PR69. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Grutsch, S.; Fuchs, J.E.; Freier, R.; Kofler, S.; Bibi, M.; Asam, C.; Wallner, M.; Ferreira, F.; Brandstetter, H.; Liedl, K.R.; et al. Ligand binding modulates the structural dynamics and compactness of the major birch pollen allergen. Biophys. J. 2014, 107, 2972–2981. [Google Scholar] [CrossRef] [PubMed]
- Stank, A.; Kokh, D.B.; Fuller, J.C.; Wade, R.C. Protein binding pocket dynamics. Acc. Chem. Res. 2016, 49, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Mittag, T.; Franzoni, L.; Cavazzini, D.; Schaffhausen, B.; Rossi, G.L.; Gunther, U.L. Retinol modulates site-specific mobility of apo-cellular retinol-binding protein to promote ligand binding. J. Am. Chem. Soc. 2006, 128, 9844–9848. [Google Scholar] [CrossRef] [PubMed]
- Perazzolo, C.; Verde, M.; Homans, S.W.; Bodenhausen, G. Evidence of chemical exchange in recombinant Major Urinary Protein and quenching thereof upon pheromone binding. J. Biomol. NMR 2007, 38, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Yang, D. Coexistence of multiple minor states of fatty acid binding protein and their functional relevance. Sci. Rep. 2016, 6, 34171. [Google Scholar] [CrossRef] [PubMed]
- Ragona, L.; Pagano, K.; Tomaselli, S.; Favretto, F.; Ceccon, A.; Zanzoni, S.; D’Onofrio, M.; Assfalg, M.; Molinari, H. The role of dynamics in modulating ligand exchange in intracellular lipid binding proteins. Biochim. Biophys. Acta 2014, 1844, 1268–1278. [Google Scholar] [CrossRef] [PubMed]
- Falconi, M.; Parrilli, L.; Battistoni, A.; Desideri, A. Flexibility in monomeric Cu,Zn superoxide dismutase detected by limited proteolysis and molecular dynamics simulation. Proteins 2002, 47, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Friedman, R.; Nachliel, E.; Gutman, M. Fatty acid binding proteins: same structure but different binding mechanisms? Molecular dynamics simulations of intestinal fatty acid binding protein. Biophys. J. 2006, 90, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Machado, Y.; Freier, R.; Scheiblhofer, S.; Thalhamer, T.; Mayr, M.; Briza, P.; Grutsch, S.; Ahammer, L.; Fuchs, J.E.; Wallnoefer, H.G.; et al. Fold stability during endolysosomal acidification is a key factor for allergenicity and immunogenicity of the major birch pollen allergen. J. Allergy Clin. Immunol. 2016, 137, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.; Fassina, G.; Vita, C.; Dalzoppo, D.; Zamai, M.; Zambonin, M. Correlation between sites of limited proteolysis and segmental mobility in thermolysin. Biochemistry 1986, 25, 1847–1851. [Google Scholar] [CrossRef] [PubMed]
- Carmicle, S.; Dai, G.; Steede, N.K.; Landry, S.J. Proteolytic sensitivity and helper T-cell epitope immunodominance associated with the mobile loop in Hsp10s. J. Biol. Chem. 2002, 277, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Carmicle, S.; Steede, N.K.; Landry, S.J. Antigen three-dimensional structure guides the processing and presentation of helper T-cell epitopes. Mol. Immunol. 2007, 44, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Wijesinha-Bettoni, R.; Alexeev, Y.; Johnson, P.; Marsh, J.; Sancho, A.I.; Abdullah, S.U.; Mackie, A.R.; Shewry, P.R.; Smith, L.J.; Mills, E.N. The structural characteristics of nonspecific lipid transfer proteins explain their resistance to gastroduodenal proteolysis. Biochemistry 2010, 49, 2130–2139. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, S.U.; Alexeev, Y.; Johnson, P.E.; Rigby, N.M.; Mackie, A.R.; Dhaliwal, B.; Mills, E.N. Ligand binding to an allergenic lipid transfer protein enhances conformational flexibility resulting in an increase in susceptibility to gastroduodenal Proteolysis. Sci. Rep. 2016, 6, 30279. [Google Scholar] [CrossRef] [PubMed]
- Apostolovic, D.; Stanic-Vucinic, D.; de Jongh, H.H.; de Jong, G.A.; Mihailovic, J.; Radosavljevic, J.; Radibratovic, M.; Nordlee, J.A.; Baumert, J.L.; Milcic, M.; et al. Conformational stability of digestion-resistant peptides of peanut conglutins reveals the molecular basis of their allergenicity. Sci. Rep. 2016, 6, 29249. [Google Scholar] [CrossRef] [PubMed]
- Wallner, M.; Himly, M.; Neubauer, A.; Erler, A.; Hauser, M.; Asam, C.; Mutschlechner, S.; Ebner, C.; Briza, P.; Ferreira, F. The influence of recombinant production on the immunologic behavior of birch pollen isoallergens. PLoS ONE 2009, 4, e8457. [Google Scholar] [CrossRef] [PubMed]
- Ahammer, L.; Grutsch, S.; Wallner, M.; Ferreira, F.; Tollinger, M. NMR resonance assignments and dynamics of a hypoallergenic isoform of the birch pollen allergen Bet v 1. Biomol. NMR Assign. 2017. accepted for publication. [Google Scholar]
- Tollinger, M.; Skrynnikov, N.R.; Mulder, F.A.A.; Forman-Kay, J.D.; Kay, L.E. Slow dynamics in folded and unfolded states of an SH3 domain. J. Am. Chem. Soc. 2001, 123, 11341–11352. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.F.; Vallurupalli, P.; Kay, L.E. An improved 15N relaxation dispersion experiment for the measurement of millisecond time-scale dynamics in proteins. J. Phys. Chem. B 2008, 112, 5898–5904. [Google Scholar] [CrossRef] [PubMed]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Carver, J.P.; Richards, R.E. General 2-Site Solution for Chemical Exchange Produced Dependence of T2 Upon Carr-Purcell Pulse Separation. J. Magn. Reson. 1972, 6, 89–96. [Google Scholar] [CrossRef]
- Case, D.; Berryman, J.; Betz, R.; Cerutti, D.; Cheatham, T.I.; Darden, T.; Duke, R.; Giese, T.; Gohlke, H.; Goetz, A.; et al. AMBER 2015; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins 2009, 75, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE). v2014.0901; Chemical Computing Group Inc.: Montreal, QC, Canada, 2014. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef] [PubMed]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical-integration of cartesian equations of motion of a system with constraints-molecular-dynamics of N-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Wallnoefer, H.G.; Handschuh, S.; Liedl, K.R.; Fox, T. Stabilizing of a globular protein by a highly complex water network: A molecular dynamics simulation study on factor Xa. J. Phys. Chem. B 2010, 114, 7405–7412. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Adelman, S.A.; Doll, J.D. Generalized langevin equation approach for atom-solid-surface scattering-general formulation for classical scattering off harmonic solids. J. Chem. Phys. 1976, 64, 2375–2388. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, J.E.; Huber, R.G.; Waldner, B.J.; Kahler, U.; von Grafenstein, S.; Kramer, C.; Liedl, K.R. Dynamics govern specificity of a protein-protein interface: substrate recognition by thrombin. PLoS ONE 2015, 10, e0140713. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bet v 1.0101 | Bet v 1.0102 | Bet v 1.0101 | Bet v 1.0102 | |
|---|---|---|---|---|
| T (K) | 300.0 | 300.0 | 360.0 | 360.0 |
| SASA (Å2) a | 8210 | 8338 | 8376 | 8607 |
| nHB b | 82.7 | 78.5 | 76.5 | 75.4 |
| Avg. B (Å2) c | 20.8 | 34.6 | 48.3 | 92.5 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grutsch, S.; Fuchs, J.E.; Ahammer, L.; Kamenik, A.S.; Liedl, K.R.; Tollinger, M. Conformational Flexibility Differentiates Naturally Occurring Bet v 1 Isoforms. Int. J. Mol. Sci. 2017, 18, 1192. https://doi.org/10.3390/ijms18061192
Grutsch S, Fuchs JE, Ahammer L, Kamenik AS, Liedl KR, Tollinger M. Conformational Flexibility Differentiates Naturally Occurring Bet v 1 Isoforms. International Journal of Molecular Sciences. 2017; 18(6):1192. https://doi.org/10.3390/ijms18061192
Chicago/Turabian StyleGrutsch, Sarina, Julian E. Fuchs, Linda Ahammer, Anna S. Kamenik, Klaus R. Liedl, and Martin Tollinger. 2017. "Conformational Flexibility Differentiates Naturally Occurring Bet v 1 Isoforms" International Journal of Molecular Sciences 18, no. 6: 1192. https://doi.org/10.3390/ijms18061192