MicroRNA Function in the Profibrogenic Interplay upon Chronic Liver Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
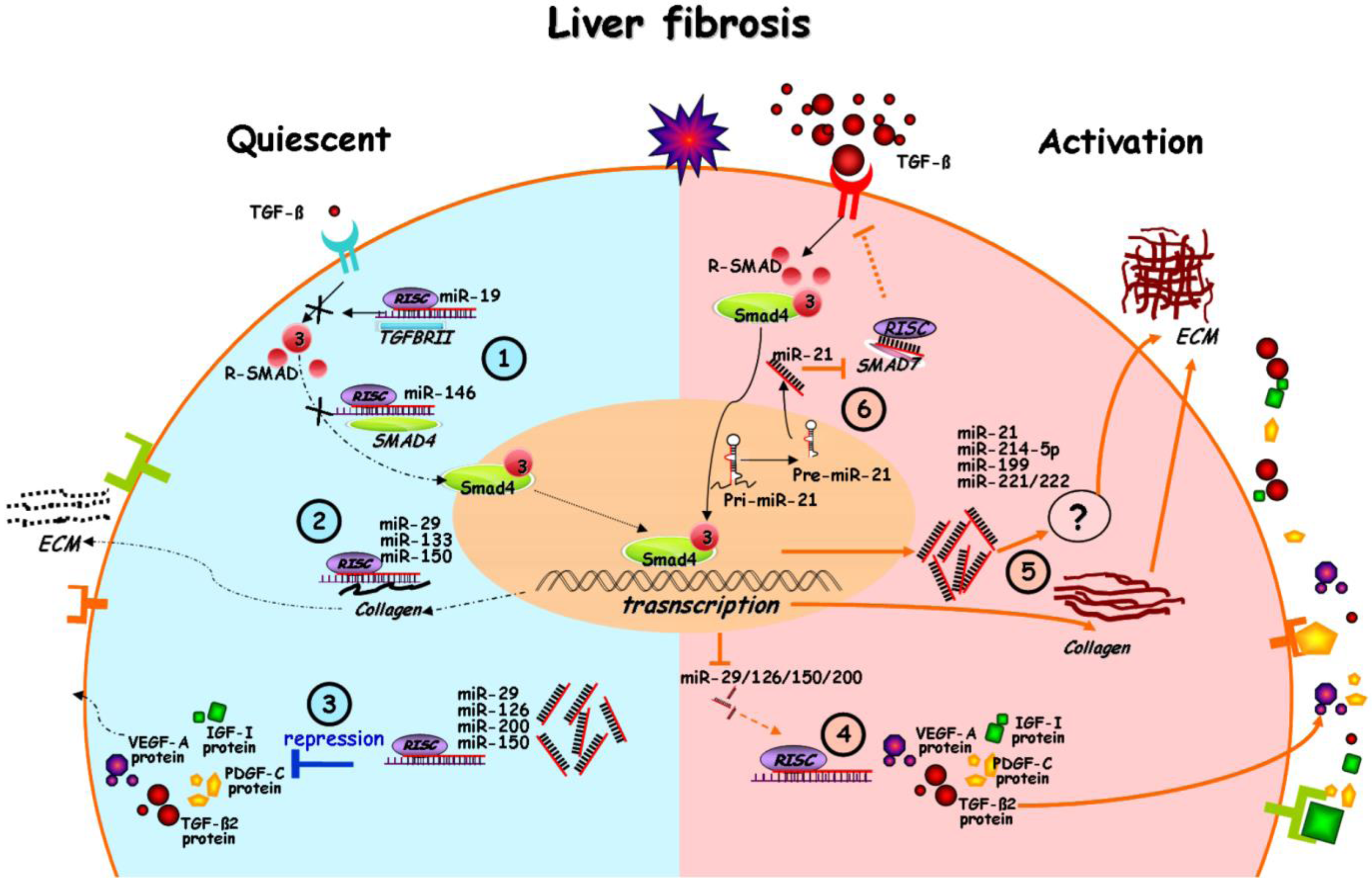
2. Altered miRNA Expression upon Liver Fibrosis
3. Linkage of miRNA Alteration to Myofibroblastic Transition

4. miRNA in the Interplay of Profibrogenic Pathways
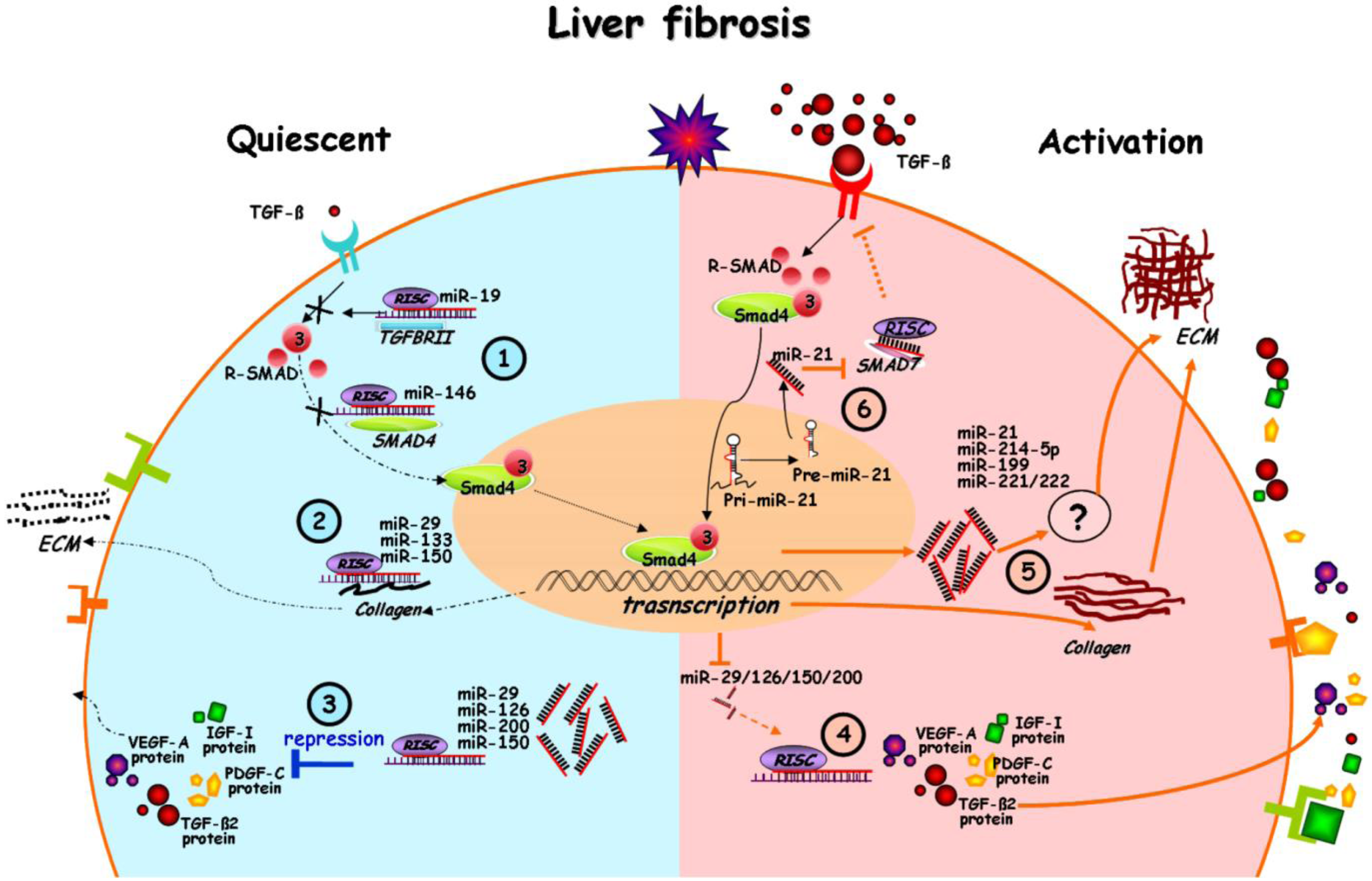
5. Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursors that bypass Drosha processing. Nature 2007, 448, 83–86. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar]
- Krek, A.; Grun, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef]
- Fabbri, M.; Ivan, M.; Cimmino, A.; Negrini, M.; Calin, G.A. Regulatory mechanisms of microRNAs involvement in cancer. Expert Opin. Biol. Ther. 2007, 7, 1009–1019. [Google Scholar] [CrossRef]
- Kong, Y.W.; Ferland-McCollough, D.; Jackson, T.J.; Bushell, M. microRNAs in cancer management. Lancet Oncol. 2012, 13, e249–e258. [Google Scholar] [CrossRef]
- Garbacki, N.; di Valentin, E.; Huynh-Thu, V.A.; Geurts, P.; Irrthum, A.; Crahay, C.; Arnould, T.; Deroanne, C.; Piette, J.; Cataldo, D.; et al. MicroRNAs profiling in murine models of acute and chronic asthma: A relationship with mRNAs targets. PLoS One 2011, 6, e16509. [Google Scholar] [CrossRef]
- Mallick, B.; Ghosh, Z.; Chakrabarti, J. MicroRNome analysis unravels the molecular basis of SARS infection in bronchoalveolar stem cells. PLoS One 2009, 4, e7837. [Google Scholar] [CrossRef]
- Recchiuti, A.; Krishnamoorthy, S.; Fredman, G.; Chiang, N.; Serhan, C.N. MicroRNAs in resolution of acute inflammation: Identification of novel resolvin D1-miRNA circuits. FASEB J. 2011, 25, 544–560. [Google Scholar] [CrossRef]
- Zhou, T.; Garcia, J.G.; Zhang, W. Integrating microRNAs into a system biology approach to acute lung injury. Transl. Res. 2011, 157, 180–190. [Google Scholar]
- Cheung, O.; Sanyal, A.J. Role of microRNAs in non-alcoholic steatohepatitis. Curr. Pharm. Des. 2010, 16, 1952–1957. [Google Scholar] [CrossRef]
- Haybaeck, J.; Zeller, N.; Heikenwalder, M. The parallel universe: microRNAs and their role in chronic hepatitis, liver tissue damage and hepatocarcinogenesis. Swiss Med. Wkly. 2011, 141, w13287. [Google Scholar]
- Noetel, A.; Kwiecinski, M.; Elfimova, N.; Huang, J.; Odenthal, M. microRNA are central players in anti- and profibrotic gene regulation during liver fibrosis. Front. Physiol. 2012, 3, 49. [Google Scholar]
- Szabo, G.; Bala, S. MicroRNAs in liver disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 542–552. [Google Scholar] [CrossRef]
- Takahashi, K.; Yan, I.; Wen, H.J.; Patel, T. microRNAs in liver disease: From diagnostics to therapeutics. Clin. Biochem. 2013, 46, 946–952. [Google Scholar] [CrossRef]
- Wang, X.W.; Heegaard, N.H.; Orum, H. MicroRNAs in liver disease. Gastroenterology 2012, 142, 1431–1443. [Google Scholar] [CrossRef]
- Brenner, D.A. Molecular pathogenesis of liver fibrosis. Trans. Am. Clin. Climatol. Assoc. 2009, 120, 361–368. [Google Scholar]
- Lee, Y.; Friedman, S.L. Fibrosis in the liver: Acute protection and chronic disease. Prog. Mol. Biol. Transl. Sci. 2010, 97, 151–200. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Friedman, S.L. Fibrosis-dependent mechanisms of hepatocarcinogenesis. Hepatology 2012, 56, 769–775. [Google Scholar] [CrossRef]
- Lade, A.; Noon, L.A.; Friedman, S.L. Contributions of metabolic dysregulation and inflammation to nonalcoholic steatohepatitis, hepatic fibrosis, and cancer. Curr. Opin. Oncol. 2014, 26, 100–107. [Google Scholar] [CrossRef]
- Dooley, S.; ten Dijke, P. TGF-beta in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic steatohepatitis is associated with altered hepatic MicroRNA expression. Hepatology 2008, 48, 1810–1820. [Google Scholar] [CrossRef]
- Lakner, A.M.; Steuerwald, N.M.; Walling, T.L.; Ghosh, S.; Li, T.; McKillop, I.H.; Russo, M.W.; Bonkovsky, H.L.; Schrum, L.W. Inhibitory effects of microRNA 19b in hepatic stellate cell-mediated fibrogenesis. Hepatology 2012, 56, 300–310. [Google Scholar]
- Morita, K.; Taketomi, A.; Shirabe, K.; Umeda, K.; Kayashima, H.; Ninomiya, M.; Uchiyama, H.; Soejima, Y.; Maehara, Y. Clinical significance and potential of hepatic microRNA-122 expression in hepatitis C. Liver Int. 2011, 31, 474–484. [Google Scholar] [CrossRef]
- Roderburg, C.; Urban, G.W.; Bettermann, K.; Vucur, M.; Zimmermann, H.; Schmidt, S.; Janssen, J.; Koppe, C.; Knolle, P.; Castoldi, M.; et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology 2011, 53, 209–218. [Google Scholar] [CrossRef]
- Jopling, C. Liver-specific microRNA-122: Biogenesis and function. RNA Biol. 2012, 9, 137–142. [Google Scholar] [CrossRef]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar]
- Trebicka, J.; Anadol, E.; Elfimova, N.; Strack, I.; Roggendorf, M.; Viazov, S.; Wedemeyer, I.; Drebber, U.; Rockstroh, J.; Sauerbruch, T.; et al. Hepatic and serum levels of miR-122 after chronic HCV-induced fibrosis. J. Hepatol. 2013, 58, 234–239. [Google Scholar] [CrossRef]
- Coulouarn, C.; Factor, V.M.; Andersen, J.B.; Durkin, M.E.; Thorgeirsson, S.S. Loss of miR-122 expression in liver cancer correlates with suppression of the hepatic phenotype and gain of metastatic properties. Oncogene 2009, 28, 3526–3536. [Google Scholar]
- Jopling, C.L. Targeting microRNA-122 to treat hepatitis C virus infection. Viruses 2010, 2, 1382–1393. [Google Scholar] [CrossRef]
- Roberts, A.P.; Lewis, A.P.; Jopling, C.L. The role of microRNAs in viral infection. Prog. Mol. Biol. Transl. Sci. 2011, 102, 101–139. [Google Scholar] [CrossRef]
- Lanford, R.E.; Hildebrandt-Eriksen, E.S.; Petri, A.; Persson, R.; Lindow, M.; Munk, M.E.; Kauppinen, S.; Orum, H. Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 2010, 327, 198–201. [Google Scholar] [CrossRef]
- Janssen, H.L.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV infection by targeting microRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar]
- Pogribny, I.P.; Starlard-Davenport, A.; Tryndyak, V.P.; Han, T.; Ross, S.A.; Rusyn, I.; Beland, F.A. Difference in expression of hepatic microRNAs miR-29c, miR-34a, miR-155 and miR-200b is associated with strain-specific susceptibility to dietary nonalcoholic steatohepatitis in mice. Lab. Investig. 2010, 90, 1437–1446. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Friedman, R.C.; Marquez, R.T.; Keck, K.; Kong, B.; Icardi, M.S.; Brown, K.E.; Burge, C.B.; Schmidt, W.N.; Wang, Y.; et al. Hepatitis C virus infection and hepatic stellate cell activation downregulate miR-29: miR-29 overexpression reduces hepatitis C viral abundance in culture. J. Infect. Dis. 2011, 203, 1753–1762. [Google Scholar]
- Mott, J.L.; Kobayashi, S.; Bronk, S.F.; Gores, G.J. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 2007, 26, 6133–6140. [Google Scholar] [CrossRef]
- Xiong, Y.; Fang, J.H.; Yun, J.P.; Yang, J.; Zhang, Y.; Jia, W.H.; Zhuang, S.M. Effects of microRNA-29 on apoptosis, tumorigenicity, and prognosis of hepatocellular carcinoma. Hepatology 2010, 51, 836–845. [Google Scholar]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar] [CrossRef]
- Cushing, L.; Kuang, P.P.; Qian, J.; Shao, F.; Wu, J.; Little, F.; Thannickal, V.J.; Cardoso, W.V.; Lu, J. miR-29 is a major regulator of genes associated with pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2011, 45, 287–294. [Google Scholar] [CrossRef]
- Maurer, B.; Stanczyk, J.; Jungel, A.; Akhmetshina, A.; Trenkmann, M.; Brock, M.; Kowal-Bielecka, O.; Gay, R.E.; Michel, B.A.; Distler, J.H.; et al. MicroRNA-29, a key regulator of collagen expression in systemic sclerosis. Arthritis Rheum. 2010, 62, 1733–1743. [Google Scholar]
- Roderburg, C.; Luedde, M.; Vargas Cardenas, D.; Vucur, M.; Mollnow, T.; Zimmermann, H.W.; Koch, A.; Hellerbrand, C.; Weiskirchen, R.; Frey, N.; et al. miR-133a mediates TGF-beta-dependent derepression of collagen synthesis in hepatic stellate cells during liver fibrosis. J. Hepatol. 2013, 58, 736–742. [Google Scholar] [CrossRef]
- Meng, F.; Glaser, S.S.; Francis, H.; Yang, F.; Han, Y.; Stokes, A.; Staloch, D.; McCarra, J.; Liu, J.; Venter, J.; et al. Epigenetic regulation of miR-34a expression in alcoholic liver injury. Am. J. Pathol. 2012, 181, 804–817. [Google Scholar] [CrossRef]
- Murakami, Y.; Toyoda, H.; Tanaka, M.; Kuroda, M.; Harada, Y.; Matsuda, F.; Tajima, A.; Kosaka, N.; Ochiya, T.; Shimotohno, K. The progression of liver fibrosis is related with overexpression of the miR-199 and 200 families. PLoS One 2011, 6, e16081. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, T.; Enomoto, M.; Fujii, H.; Sekiya, Y.; Yoshizato, K.; Ikeda, K.; Kawada, N. MicroRNA-221/222 upregulation indicates the activation of stellate cells and the progression of liver fibrosis. Gut 2012, 61, 1600–1609. [Google Scholar]
- Sun, X.; He, Y.; Ma, T.T.; Huang, C.; Zhang, L.; Li, J. Participation of miR-200a in TGF-beta1-mediated hepatic stellate cell activation. Mol. Cell. Biochem. 2014, 388, 11–23. [Google Scholar] [CrossRef]
- Ramachandran, S.; Ilias Basha, H.; Sarma, N.J.; Lin, Y.; Crippin, J.S.; Chapman, W.C.; Mohanakumar, T. Hepatitis C virus induced miR200c down modulates FAP-1, a negative regulator of Src signaling and promotes hepatic fibrosis. PLoS One 2013, 8, e70744. [Google Scholar] [CrossRef]
- Vinall, R.L.; Ripoll, A.Z.; Wang, S.; Pan, C.X.; deVere White, R.W. MiR-34a chemosensitizes bladder cancer cells to cisplatin treatment regardless of p53-Rb pathway status. Int. J. Cancer 2012, 130, 2526–2538. [Google Scholar] [CrossRef]
- Yamakuchi, M.; Ferlito, M.; Lowenstein, C.J. miR-34a repression of SIRT1 regulates apoptosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13421–13426. [Google Scholar] [CrossRef]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef]
- Lee, U.E.; Friedman, S.L. Mechanisms of hepatic fibrogenesis. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 195–206. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef]
- Li, D.; Friedman, S.L. Liver fibrogenesis and the role of hepatic stellate cells: New insights and prospects for therapy. J. Gastroenterol. Hepatol. 1999, 14, 618–633. [Google Scholar] [CrossRef]
- Dechene, A.; Sowa, J.P.; Gieseler, R.K.; Jochum, C.; Bechmann, L.P.; El Fouly, A.; Schlattjan, M.; Saner, F.; Baba, H.A.; Paul, A.; et al. Acute liver failure is associated with elevated liver stiffness and hepatic stellate cell activation. Hepatology 2010, 52, 1008–1016. [Google Scholar] [CrossRef]
- Guo, C.J.; Pan, Q.; Cheng, T.; Jiang, B.; Chen, G.Y.; Li, D.G. Changes in microRNAs associated with hepatic stellate cell activation status identify signaling pathways. FEBS J. 2009, 276, 5163–5176. [Google Scholar]
- Guo, C.J.; Pan, Q.; Jiang, B.; Chen, G.Y.; Li, D.G. Effects of upregulated expression of microRNA-16 on biological properties of culture-activated hepatic stellate cells. Apoptosis 2009, 14, 1331–1340. [Google Scholar]
- He, Y.; Huang, C.; Sun, X.; Long, X.R.; Lv, X.W.; Li, J. MicroRNA-146a modulates TGF-beta1-induced hepatic stellate cell proliferation by targeting SMAD4. Cell Signal. 2012, 24, 1923–1930. [Google Scholar] [CrossRef]
- Ji, J.; Zhang, J.; Huang, G.; Qian, J.; Wang, X.; Mei, S. Over-expressed microRNA-27a and -27b influence fat accumulation and cell proliferation during rat hepatic stellate cell activation. FEBS Lett. 2009, 583, 759–766. [Google Scholar] [CrossRef]
- Maubach, G.; Lim, M.C.; Chen, J.; Yang, H.; Zhuo, L. miRNA studies in in vitro and in vivo activated hepatic stellate cells. World J. Gastroenterol. 2011, 17, 2748–2773. [Google Scholar]
- Noetel, A.; Elfimova, N.; Altmuller, J.; Becker, C.; Becker, D.; Lahr, W.; Nurnberg, P.; Wasmuth, H.; Teufel, A.; Buttner, R.; et al. Next generation sequencing of the Ago2 interacting transcriptome identified chemokine family members as novel targets of neuronal microRNAs in hepatic stellate cells. J. Hepatol. 2013, 58, 335–341. [Google Scholar] [CrossRef]
- Zheng, J.; Lin, Z.; Dong, P.; Lu, Z.; Gao, S.; Chen, X.; Wu, C.; Yu, F. Activation of hepatic stellate cells is suppressed by microRNA-150. Int. J. Mol. Med. 2013, 32, 17–24. [Google Scholar]
- Honda, N.; Jinnin, M.; Kira-Etoh, T.; Makino, K.; Kajihara, I.; Makino, T.; Fukushima, S.; Inoue, Y.; Okamoto, Y.; Hasegawa, M.; et al. miR-150 down-regulation contributes to the constitutive type I collagen overexpression in scleroderma dermal fibroblasts via the induction of integrin beta3. Am. J. Pathol. 2013, 182, 206–216. [Google Scholar] [CrossRef]
- Kwiecinski, M.; Noetel, A.; Elfimova, N.; Trebicka, J.; Schievenbusch, S.; Strack, I.; Molnar, L.; von Brandenstein, M.; Tox, U.; Nischt, R.; et al. Hepatocyte growth factor (HGF) inhibits collagen I and IV synthesis in hepatic stellate cells by miRNA-29 induction. PLoS One 2011, 6, e24568. [Google Scholar] [CrossRef]
- Sekiya, Y.; Ogawa, T.; Yoshizato, K.; Ikeda, K.; Kawada, N. Suppression of hepatic stellate cell activation by microRNA-29b. Biochem. Biophys. Res. Commun. 2011, 412, 74–79. [Google Scholar] [CrossRef]
- Iizuka, M.; Ogawa, T.; Enomoto, M.; Motoyama, H.; Yoshizato, K.; Ikeda, K.; Kawada, N. Induction of microRNA-214–5p in human and rodent liver fibrosis. Fibrogenesis Tissue Repair. 2012, 5, 12. [Google Scholar] [CrossRef]
- Denby, L.; Ramdas, V.; Lu, R.; Conway, B.R.; Grant, J.S.; Dickinson, B.; Aurora, A.B.; McClure, J.D.; Kipgen, D.; Delles, C.; et al. MicroRNA-214 antagonism protects against renal fibrosis. J. Am. Soc. Nephrol. 2014, 25, 65–80. [Google Scholar]
- Davis, B.N.; Hilyard, A.C.; Lagna, G.; Hata, A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature 2008, 454, 56–61. [Google Scholar] [CrossRef]
- Qin, W.; Chung, A.C.; Huang, X.R.; Meng, X.M.; Hui, D.S.; Yu, C.M.; Sung, J.J.; Lan, H.Y. TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 2011, 22, 1462–1474. [Google Scholar] [CrossRef]
- Ogawa, T.; Iizuka, M.; Sekiya, Y.; Yoshizato, K.; Ikeda, K.; Kawada, N. Suppression of type I collagen production by microRNA-29b in cultured human stellate cells. Biochem. Biophys. Res. Commun. 2010, 391, 316–321. [Google Scholar] [CrossRef]
- Kwiecinski, M.; Elfimova, N.; Noetel, A.; Tox, U.; Steffen, H.M.; Hacker, U.; Nischt, R.; Dienes, H.P.; Odenthal, M. Expression of platelet-derived growth factor-C and insulin-like growth factor I in hepatic stellate cells is inhibited by miR-29. Lab. Investig. 2012, 92, 978–987. [Google Scholar] [CrossRef]
- Corpechot, C.; Barbu, V.; Wendum, D.; Kinnman, N.; Rey, C.; Poupon, R.; Housset, C.; Rosmorduc, O. Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology 2002, 35, 1010–1021. [Google Scholar] [CrossRef]
- Yoshiji, H.; Kuriyama, S.; Yoshii, J.; Ikenaka, Y.; Noguchi, R.; Hicklin, D.J.; Wu, Y.; Yanase, K.; Namisaki, T.; Yamazaki, M.; et al. Vascular endothelial growth factor and receptor interaction is a prerequisite for murine hepatic fibrogenesis. Gut 2003, 52, 1347–1354. [Google Scholar]
- Denby, L.; Ramdas, V.; McBride, M.W.; Wang, J.; Robinson, H.; McClure, J.; Crawford, W.; Lu, R.; Hillyard, D.Z.; Khanin, R.; et al. miR-21 and miR-214 are consistently modulated during renal injury in rodent models. Am. J. Pathol. 2011, 179, 661–672. [Google Scholar] [CrossRef]
- Marquez, R.T.; Bandyopadhyay, S.; Wendlandt, E.B.; Keck, K.; Hoffer, B.A.; Icardi, M.S.; Christensen, R.N.; Schmidt, W.N.; McCaffrey, A.P. Correlation between microRNA expression levels and clinical parameters associated with chronic hepatitis C viral infection in humans. Lab. Investig. 2010, 90, 1727–1736. [Google Scholar] [CrossRef]
- Wei, J.; Feng, L.; Li, Z.; Xu, G.; Fan, X. MicroRNA-21 activates hepatic stellate cells via PTEN/Akt signaling. Biomed. Pharmacother. 2013, 67, 387–392. [Google Scholar]
- Ji, J.; Yu, L.; Yu, Z.; Forgues, M.; Uenishi, T.; Kubo, S.; Wakasa, K.; Zhou, J.; Fan, J.; Tang, Z.Y.; et al. Development of a miR-26 companion diagnostic test for adjuvant interferon-alpha therapy in hepatocellular carcinoma. Int. J. Biol. Sci. 2013, 9, 303–312. [Google Scholar] [CrossRef]
- Ferracin, M.; Veronese, A.; Negrini, M. Micromarkers: miRNAs in cancer diagnosis and prognosis. Expert Rev. Mol. Diagn. 2010, 10, 297–308. [Google Scholar] [CrossRef]
- Kosaka, N.; Iguchi, H.; Ochiya, T. Circulating microRNA in body fluid: A new potential biomarker for cancer diagnosis and prognosis. Cancer Sci. 2010, 101, 2087–2092. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Huang, J.; Yu, X.; Fries, J.W.U.; Zhang, L.; Odenthal, M. MicroRNA Function in the Profibrogenic Interplay upon Chronic Liver Disease. Int. J. Mol. Sci. 2014, 15, 9360-9371. https://doi.org/10.3390/ijms15069360
Huang J, Yu X, Fries JWU, Zhang L, Odenthal M. MicroRNA Function in the Profibrogenic Interplay upon Chronic Liver Disease. International Journal of Molecular Sciences. 2014; 15(6):9360-9371. https://doi.org/10.3390/ijms15069360
Chicago/Turabian StyleHuang, Jia, Xiaojie Yu, Jochen W. U. Fries, Li'ang Zhang, and Margarete Odenthal. 2014. "MicroRNA Function in the Profibrogenic Interplay upon Chronic Liver Disease" International Journal of Molecular Sciences 15, no. 6: 9360-9371. https://doi.org/10.3390/ijms15069360