Genetic and Molecular Differences in Prostate Carcinogenesis between African American and Caucasian American Men

{kind=link}
Abstract
:1. Introduction
2. Androgen and Estrogen Levels
3. Androgen Receptor Gene Structure and Function
3.1. Androgen Receptor (AR) Gene Structure
3.2. Androgen Receptor Function
4. Genetic Variation
5. Epigenetic Changes
6. Cancer Genes
TMPRSS2-ERG Fusions
7. Conclusions
Acknowledgments
Conflict of Interest
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics. CA Cancer J. Clin 2013, 63, 11–30. [Google Scholar]
- Chornokur, G.; Dalton, K.; Borysova, M.E.; Kumar, N.B. Disparities at presentation, diagnosis, treatment, and survival in African American men affected by prostate cancer. Prostate 2011, 71, 985–997. [Google Scholar]
- Schwartz, K.; Powell, I.J.; Underwood, W., 3rd; George, J.; Yee, C.; Banerjee, M. Interplay of race, socioeconomic status, and treatment on survival of patients with prostate cancer. Urology 2009, 74, 1296–1302. [Google Scholar]
- Major, J.M.; Oliver, M.N.; Doubeni, C.A.; Hollenbeck, A.R.; Graubard, B.I.; Sinha, R. Socioeconomic status, healthcare density, and risk of prostate cancer among African American and Caucasian men in a large prospective study. Cancer Causes Control 2012, 23, 1185–1191. [Google Scholar]
- Sridhar, G.; Masho, S.W.; Adera, T.; Ramakrishnan, V.; Roberts, J.D. Do African American men have lower survival from prostate cancer compared with White men? A meta-analysis. Am. J. Mens. Health 2010, 4, 189–206. [Google Scholar]
- Cullen, J.; Brassell, S.; Chen, Y.; Porter, C.; L’Esperance, J.; Brand, T.; McLeod, D.G. Racial/ethnic patterns in prostate cancer outcomes in an active surveillance cohort. Prostate Cancer 2011, 2011. [Google Scholar] [CrossRef]
- Berger, A.D.; Satagopan, J.; Lee, P.; Taneja, S.S.; Osman, I. Differences in clinicopathologic features of prostate cancer between black and white patients treated in the 1990s and 2000s. Urology 2006, 67, 120–124. [Google Scholar]
- Kheirandish, P.; Chinegwundoh, F. Ethnic differences in prostate cancer. Br. J. Cancer 2011, 105, 481–485. [Google Scholar]
- Odedina, F.T.; Akinremi, T.O.; Chinegwundoh, F.; Roberts, R.; Yu, D.; Reams, R.R.; Freedman, M.L.; Rivers, B.; Green, B.L.; Kumar, N. Prostate cancer disparities in black men of African descent: A comparative literature review of prostate cancer burden among black men in the United States, Caribbean, United Kingdom, and West Africa. Infect. Agents Cancer 2009, 4. [Google Scholar] [CrossRef]
- Heath, E.I.; Kattan, M.W.; Powell, I.J.; Sakr, W.; Brand, T.C.; Rybicki, B.A.; Thompson, I.M.; Aronson, W.J.; Terris, M.K.; Kane, C.J.; et al. The effect of race/ethnicity on the accuracy of the 2001 Partin Tables for predicting pathologic stage of localized prostate cancer. Urology 2008, 71, 151–155. [Google Scholar]
- Moul, J.W.; Sesterhenn, I.A.; Connelly, R.R.; Douglas, T.; Srivastava, S.; Mostofi, F.K.; McLeod, D.G. Prostate-specific antigen values at the time of prostate cancer diagnosis in African-American men. JAMA 1995, 274, 1277–1281. [Google Scholar]
- Tewari, A.; Horninger, W.; Badani, K.K.; Hasan, M.; Coon, S.; Crawford, E.D.; Gamito, E.J.; Wei, J.; Taub, D.; Montie, J.; et al. Racial differences in serum prostate-specific (PSA) doubling time, histopathological variables and long-term PSA recurrence between African-American and white American men undergoing radical prostatectomy for clinically localized prostate cancer. BJU Int 2005, 96, 29–33. [Google Scholar]
- Wallace, T.A.; Prueitt, R.L.; Yi, M.; Howe, T.M.; Gillespie, J.W.; Yfantis, H.G.; Stephens, R.M.; Caporaso, N.E.; Loffredo, C.A.; Ambs, S. Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer Res 2008, 68, 927–936. [Google Scholar]
- Eichholz, A.; Ferraldeschi, R.; Attard, G.; de Bono, J.S. Putting the brakes on continued androgen receptor signaling in castration-resistant prostate cancer. Mol. Cell. Endocrinol 2012, 360, 68–75. [Google Scholar]
- Ryan, C.J.; Tindall, D.J. Androgen receptor rediscovered: The new biology and targeting the androgen receptor therapeutically. J. Clin. Oncol 2011, 29, 3651–3658. [Google Scholar]
- Lamont, K.R.; Tindall, D.J. Minireview: Alternative activation pathways for the androgen receptor in prostate cancer. Mol. Endocrinol 2011, 25, 897–907. [Google Scholar]
- Ghanadian, R.; Puah, C.M.; O’Donoghue, E.P. Serum testosterone and dihydrotestosterone in carcinoma of the prostate. Br. J. Cancer 1979, 39, 696–699. [Google Scholar]
- Noble, R.L. The development of prostatic adenocarcinoma in Nb rats following prolonger sex hormone administration. Cancer Res 1977, 37, 1929–1933. [Google Scholar]
- Bosland, M.C.; Ford, H.; Horton, L. Induction at high incidence of ductal prostate adenocarcinomas in NBL/Cr and Sprague-Dawley Hsd:SD rats treated with a combination of testosterone and estradiol-17 beta or diethylstilbestrol. Carcinogenesis 1995, 16, 1311–1317. [Google Scholar]
- Ho, S.M.; Lee, M.T.; Lam, H.M.; Leung, Y.K. Estrogens and prostate cancer: Etiology, mediators, prevention, and management. Endocrinol. Metab. Clin. North Am 2011, 40, 591–614. [Google Scholar]
- Gann, P.H.; Hennekens, C.H.; Ma, J.; Longcope, C.; Stampfer, M.J. Prospective study of sex hormone levels and risk of prostate cancer. J. Natl. Cancer Inst 1996, 88, 1118–1126. [Google Scholar]
- Roddam, A.W.; Allen, N.E.; Appleby, P.; Key, T.J. Endogenous sex hormones and prostate cancer: A collaborative analysis of 18 prospective studies. J. Natl. Cancer Inst 2008, 100, 170–183. [Google Scholar]
- Bosland, M.C.; Mahmoud, A.M. Hormones and prostate carcinogensis: Androgens and estrogens. J. Carcinog. 2011. [Google Scholar] [CrossRef]
- Massengill, J.C.; Sun, L.; Moul, J.W.; Wu, H.; McLeod, D.G.; Amling, C.; Lance, R.; Foley, J.; Sexton, W.; Kusuda, L.; et al. Pretreatment total testosterone level predicts pathological stage in patients with localized prostate cancer treated with radical prostatectomy. J. Urol 2003, 169, 1670–1675. [Google Scholar]
- Schatzl, G.; Madersbacher, S.; Haitel, A.; Gsur, A.; Preyer, M.; Haidinger, G.; Gassner, C.; Ochsner, M.; Marberger, M. Associations of serum testosterone with microvessel density, androgen receptor density and androgen receptor gene polymorphism in prostate cancer. J. Urol 2003, 169, 1312–1315. [Google Scholar]
- Yao, S.; Till, C.; Kristal, A.R.; Goodman, P.J.; Hsing, A.W.; Tangen, C.M.; Platz, E.A.; Stanczyk, F.Z.; Reichardt, J.K.; Tang, L.; et al. Serum estrogen levels and prostate cancer risk in the prostate cancer prevention trial: A nested case-control study. Cancer Causes Control 2011, 22, 1121–1131. [Google Scholar]
- Andriole, G.L.; Bostwick, D.G.; Brawley, O.W.; Gomella, L.G.; Marberger, M.; Montorsi, F.; Pettaway, C.A.; Tammela, T.L.; Teloken, C.; Tindall, D.J.; et al. Effect of dutasteride on the risk of prostate cancer. N. Engl. J. Med 2010, 363, 1192–1202. [Google Scholar]
- Thompson, I.M.; Goodman, P.J.; Tangen, C.M.; Lucia, M.S.; Miller, G.J.; Ford, L.G.; Lieber, M.M.; Cespedes, R.D.; Atkins, J.N.; Lippman, S.M.; et al. The influence of finasteride on the development of prostate cancer. N. Engl. J. Med 2003, 349, 215–224. [Google Scholar]
- Ellem, S.J.; Risbridger, G.P. Aromatase and regulating the estrogen:androgen ratio in the prostate gland. J. Steroid Biochem. Mol. Biol 2010, 118, 246–251. [Google Scholar]
- Price, D.; Stein, B.; Sieber, P.; Tutrone, R.; Bailen, J.; Goluboff, E.; Burzon, D.; Bostwick, D.; Steiner, M. Toremifene for the prevention of prostate cancer in men with high grade prostatic intraepithelial neoplasia: Results of a double-blind, placebo controlled, phase IIB clinical trial. J. Urol 2006, 176, 965–970. [Google Scholar]
- Taneja, S.S.; Morton, R.; Barnette, G.; Sieber, P.; Hancock, M.L.; Steiner, M. Prostate cancer diagnosis among men with isolated high-grade intraepithelial neoplasia enrolled onto a 3-year prospective phase III clinical trial of oral toramifene. J. Clin. Oncol 2013, 31, 523–529. [Google Scholar]
- Ross, R.; Bernstein, L.; Judd, H.; Hanisch, R.; Pike, M.; Henderson, B. Serum testosterone levels in healthy young black and white men. J. Natl. Cancer Inst 1986, 76, 45–48. [Google Scholar]
- Winters, S.J.; Brufsky, A.; Weissfeld, J.; Trump, D.L.; Dyky, M.A.; Hadeed, V. Testosterone, sex hormone-binding globulin, and body composition in young adult African American and Caucasian men. Metabolism 2001, 50, 1242–1247. [Google Scholar]
- Rohrmann, S.; Nelson, W.G.; Rifai, N.; Brown, T.R.; Dobs, A.; Kanarek, N.; Yager, J.D.; Platz, E.A. Serum estrogen, but not testosterone levels differ between black and white men in a nationally representative sample of Americans. J. Clin. Endocrinol. Metab 2007, 92, 2519–2525. [Google Scholar]
- Orwoll, E.S.; Nielson, C.M.; Labrie, F.; Barrett-Connor, E.; Cauley, J.A.; Cummings, S.R.; Ensrud, K.; Karlsson, M.; Lau, E.; Leung, P.C.; et al. Evidence for geographical and racial variation in serum sex levels in older men. J. Clin. Endocrinol. Metab 2010, 95, E151–E160. [Google Scholar]
- Kubricht, W.S., 3rd; Williams, B.J.; Whately, T.; Pinckard, P.; Eastham, J.A. Serum testosterone levels in African-American and white men undergoing prostate biopsy. Urology 1999, 54, 1035–1038. [Google Scholar]
- Marks, L.S.; Hess, D.L.; Dorey, F.J.; Macairan, M.L. Prostate tissue testosterone and dihydrotestosterone in African-American and white men. Urology 2006, 68, 337–341. [Google Scholar]
- Kazemi-Esfarjani, P.; Trifiro, M.A.; Pinsky, L. Evidence for a repressive function of the long polyglutamine tract in the human androgen receptor: Possible pathogenetic relevance for the (CAG)n-expanded neuronopathies. Hum. Mol. Genet 1995, 4, 523–527. [Google Scholar]
- Edwards, A.; Hammond, H.A.; Jin, L.; Caskey, C.T.; Chakraborty, R. Genetic variation at five trimeric and tetrameric tandem repeat loci in human population groups. Genomics 1992, 12, 241–253. [Google Scholar]
- Chamberlain, N.L.; Driver, E.D.; Miesfeld, R.L. The length and location of CAG trinucleotide repeats in the androgen receptor N-terminal domain affect transactivation function. Nucleic Acids Res 1994, 22, 3181–3186. [Google Scholar]
- Beilin, J.; Ball, E.M.; Favaloro, J.M.; Zajac, J.D. Effect of the androgen receptor CAG repeat polymorphism on transcriptional activity: Specificity in prostate and non-prostate cell lines. J. Mol. Endocrinol 2000, 25, 85–96. [Google Scholar]
- Bennett, C.L.; Price, D.K.; Kim, S.; Lui, D.; Jovanovic, B.D.; Nathan, D.; Johnson, M.E.; Montgomery, J.S.; Cude, K.; Brockbank, J.C.; et al. Racial variation in CAG repeat lengths within the androgen receptor gene among prostate cancer patients of lower socioeconomic status. J. Clin. Oncol 2002, 20, 3599–3604. [Google Scholar]
- Powell, I.J.; Land, S.J.; Dey, J.; Heilbrun, L.K.; Hughes, M.R.; Sakr, W.; Everson, R.B. The impact of CAG repeats in exon 1 of the androgen receptor on disease progression after prostatectomy. Cancer 2005, 103, 528–537. [Google Scholar]
- Freedman, S.J.; Isaacs, W.B. Explaining racial differences in prostate cancer in the United States: Sociology or biology? Prostate 2005, 62, 243–252. [Google Scholar]
- Sartor, O.; Zheng, Q.; Eastham, J.A. Androgen receptor gene CAG repeat length varies in a race-specific fashion in men without prostate cancer. Urology 1999, 53, 378–380. [Google Scholar]
- Giovannucci, E.; Stampfer, M.J.; Krithivas, K.; Bown, M.; Dahl, D.; Brufsky, A.; Talcott, J.; Hennekens, C.H.; Kantoff, P.W. The CAG repeat within the androgen receptor gene and its relationship to prostate cancer. Proc. Natl. Acad. Sci. USA 1997, 94, 3320–3323. [Google Scholar]
- Ingles, S.A.; Ross, R.K.; Yu, M.C.; Irvine, R.A.; La Pera, G.; Haile, R.W.; Coetzee, G.A. Association of prostate cancer risk with genetic polymorphisms in vitamin D receptor and androgen receptor. J. Natl. Cancer Inst 1997, 89, 166–170. [Google Scholar]
- Giovannucci, E. Is the androgen receptor CAG repeat length significant for prostate cancer? Cancer Epidemiol. Biomarkers Prev 2002, 11, 985–986. [Google Scholar]
- Freedman, M.L.; Pearce, C.L.; Penney, K.L.; Hirschhorn, J.N.; Kolonel, L.N.; Henderson, B.E.; Altshuler, D. Systematic evaluation of genetic variation at the androgen receptor locus and risk of prostate cancer in a multiethnic cohort study. Am. J. Hum. Genet 2005, 76, 82–90. [Google Scholar]
- Price, D.K.; Chau, C.H.; Till, C.; Goodman, P.J.; Baum, C.E.; Ockers, S.B.; English, B.C.; Minasian, L.; Parnes, H.L.; Hsing, A.W.; et al. Androgen receptor CAG repeat length and association with prostate cancer risk: Results from the prostate cancer prevention trial. J. Urol. 2010, 184, 2297–2302. [Google Scholar]
- Gilligan, T.; Manola, J.; Sartor, O.; Weinrich, S.P.; Moul, J.W.; Kantoff, P.W. Absence of a correlation of androgen receptor gene CAG repeat length and prostate cancer risk in an African-American population. Clin. Prostate Cancer 2004, 3, 98–103. [Google Scholar]
- Lange, E.M.; Sarma, A.V.; Ray, A.; Wang, Y.; Ho, L.; Anderson, S.A.; Cunningham, J.M.; Cooney, K.A. The androgen receptor CAG and GGN repeat polymorphisms and prostate cancer susceptibility in African-American men: Results from the Flint Men’s Health Study. J. Hum. Genet 2008, 53, 220–226. [Google Scholar]
- Foradori, C.D.; Weiser, M.J.; Handa, R.J. Non-genomic actions of androgens. Front. Neuroendocrinol 2008, 29, 169–181. [Google Scholar]
- Gaston, K.E.; Desok, K.; Singh, S.; Ford, O.H.; Mohler, J.L. Racial differences in androgen receptor protein expression in men with clinically localized prostate cancer. J. Urol 2003, 170, 990–993. [Google Scholar]
- Sterbis, J.R.; Gao, C.; Furusato, B.; Chen, Y.; Shaheduzzaman, S.; Ravindranath, L.; Osborn, D.J.; Rosner, I.L.; Dobi, A.; McLeod, D.G.; et al. Higher expression of the androgen-regulared gene PSA/HK3 mRNA in prostate cancer tissues predicts biochemical recurrence-free survival. Clin. Cancer Res 2008, 14, 758–763. [Google Scholar]
- Dobi, A.; Furusato, B.; Shaheduzzaman, S.; Chen, Y.; Vahey, M.; Nydam, T.; Sesterhenn, I.A.; McLeod, D.G.; Petrovics, G.; Srivastava, S. ERG expression levels in prostate tumors reflect functional status of the androgen receptor (AR) as a consequence of fusion of ERG with AR regulated gene promoters. Open Cancer J 2010, 3, 101–108. [Google Scholar]
- Kim, H.S.; Moreira, D.M.; Jayachandran, J.; Gerber, L.; Banez, L.L.; Vollmer, R.T.; Lark, A.L.; Donovan, M.J.; Powell, D.; Khan, F.M.; et al. Prostate biopsies from black men express higher levels of aggressive disease biomarkers than prostate biopsies from white men. Prostate Cancer Prostatic Dis 2011, 14, 262–265. [Google Scholar]
- Choudury, A.D.; Eeles, R.; Freedland, S.J.; Issacs, W.B.; Pomerantz, M.M.; Schalken, J.A.; Tammela, T.L.; Visakorpi, T. The role of genetic markers in the management of prostate cancer. Eur. Urol 2012, 62, 577–587. [Google Scholar]
- Goh, C.L.; Saunders, E.J.; Leongamornlert, D.A.; Tymrakiewicz, M.; Thomas, K.; Selvadurai, E.D.; Woode-Amissah, R.; Dadaev, T.; Mahmud, N.; Castro, E.; et al. Clinical implications of family history of prostate cancer and genetic risk single nucleotide polymorphism (SNP) profiles in an active surveillance cohort. BJU Int. 2013. [Google Scholar] [CrossRef]
- Zeegers, M.P.; Jellema, A.; Ostrer, H. Empiric risk of prostate carcinoma for relatives of patients with prostate carcinoma: A meta-analysis. Cancer 2003, 97, 1894–1903. [Google Scholar]
- Pomerantz, M.M.; Freedman, M.L. Genetics of prostate cancer risk. Mt. Sinai J. Med 2010, 77, 643–654. [Google Scholar]
- Witte, J.S. Prostate cancer genomics: Toward a new understanding. Nat. Rev. Genet 2009, 10, 77–82. [Google Scholar]
- Sfano, K.S.; de Marzo, A.M. Prostate cancer and inflammation: The evidence. Histopathology 2012, 60, 199–215. [Google Scholar]
- Thomas, G.; Jacobs, K.B.; Yeager, M.; Kraft, P.; Wacholder, S.; Orr, N.; Yu, K.; Chatterjee, N.; Welch, R.; Hutchinson, A. Multiple loci identified in a genome-wide association study of prostate cancer. Nat. Genet 2008, 40, 310–315. [Google Scholar]
- Bensen, J.T.; Xu, Z.; Smith, G.J.; Mohler, J.L.; Fontham, E.T.; Taylor, J.A. Genetic polymorphism and prostate cancer aggressiveness: A case-only study of 1,536 GWAS and candidate SNPs in African-Americans and European-Americans. Prostate 2013, 73, 11–22. [Google Scholar]
- Amundadottir, L.T.; Sulem, P.; Gudmundsson, J.; Helgason, A.; Baker, A.; Agnarsson, B.A.; Sigurdsson, A.; Benediktsottir, K.R.; Cazier, J.B.; Sainz, J.; et al. A common variant associated with prostate cancer in European and African populations. Nat. Genet 2006, 38, 652–658. [Google Scholar]
- Freedman, M.L.; Haiman, C.A.; Patterson, N.; McDonald, G.J.; Tandon, A.; Waliszewska, A.; Penney, K.; Steen, R.G.; Ardlie, K.; John, E.M.; et al. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc. Natl. Acad. Sci. USA 2006, 103, 14068–14073. [Google Scholar]
- Haiman, C.A.; Patterson, N.; Freedman, M.L.; Myers, S.R.; Pike, M.C.; Waliszewska, A.; Neubauer, J.; Tandon, A.; Schirmer, C.; McDonald, G.J.; et al. Multiple regions within 8q24 independently affect risk for prostate cancer. Nat. Genet 2007, 39, 638–644. [Google Scholar]
- Zheng, S.L.; Sun, L.; Wiklund, F.; Smith, S.; Stattin, P.; Li, G.; Adami, H.O.; Hsu, F.C.; Zhu, Y.; Balter, K.; et al. Cumulative association of five genetic variants with prostate cancer. N. Engl. J. Med 2008, 358, 910–919. [Google Scholar]
- Zheng, S.L.; Sun, J.; Wiklund, F.; Gao, Z.; Stattin, P.; Purcell, L.D.; Adami, H.O.; Hsu, F.C.; Zhu, Y.; Adolfsson, J.; et al. Genetic Variants and family history predict prostate cancer similar to PSA. Clin Cancer Res 2009, 15, 1105–1111. [Google Scholar]
- Thompson, I.M.; Ankerst, D.P.; Chi, C.; Lucia, M.S.; Goodman, P.J.; Crawley, J.J.; Parnes, H.L.; Coltman, C.A., Jr. Operating characteristics of prostate-specific antigen in men with an initial PSA level of 3.0 ng/mL or lower. JAMA 2005, 294, 66–70. [Google Scholar]
- Hughes, L.; Ahu, F.; Ross, E.; Gross, L.; Uzzo, R.G.; Chen, D.Y.; Viterbo, R.; Rebbeck, T.R.; Giri, V.N. Assessing the clinical role of genetic markers of early-onset prostate cancer among high-risk men enrolled in prostate cancer early detection. Cancer Epidemiol. Biomarkers Prev 2012, 21, 53–60. [Google Scholar]
- Pomerantz, M.M.; Beckwith, C.A.; Regan, M.M.; Wyman, S.K.; Petrovics, G.; Chen, Y.; Hawksworth, D.J.; Schumacher, F.R.; Mucci, L.; Penney, K.L.; et al. Evaluation of the 8q24 prostate cancer risk locus and MYC expression. Cancer Res 2009, 69, 5568–5574. [Google Scholar]
- Troutman, S.M.; Sissung, T.M.; Cropp, C.D.; Venzon, D.J.; Spencer, S.D.; Adesunloye, B.A.; Huang, X.; Karzai, F.H.; Price, D.K.; Figg, W.D. Racial disparities in the association between variants on 8q24 and prostate cancer: A systematic review and meta-analysis. Oncologist 2012, 17, 312–320. [Google Scholar]
- Wasserman, N.F.; Aneas, I.; Nobrega, M.A. An 8q24 gene desert variant associated with prostate cancer risk confers differential in vivo activity to a MYC enhancer. Genome Res 2010, 20, 1191–1197. [Google Scholar]
- Fromont, G.; Godet, J.; Peyret, A.; Irani, J.; Celhay, O.; Rozet, F.; Cathelineau, X.; Cussenot, O. 8q24 amplification is associated with Myc expression and prostate cancer progression and is an independent predictor of recurrence after radical prostatectomy. Hum. Pathol. 2013. [Google Scholar] [CrossRef]
- Barnabas, N.; Xu, L.; Savera, A.; Hou, Z.; Barrack, E.R. Chromosome 8 markers of metastatic prostate cancer in African American men: Gain of the MIR151 gene and loss of the NKX3-1 gene. Prostate 2011, 71, 857–871. [Google Scholar]
- Ding, J.; Huang, S.; Wu, S.; Zhao, Y.; Liang, L.; Yan, M.; Ge, C.; Yao, J.; Chen, T.; Wan, D.; et al. Gain of miR-151 on chromosome 8q24.3 facilitates tumour cell migration and spreading through downregulating RhoGDIA. Nat. Cell Biol 2010, 12, 390–399. [Google Scholar]
- Huppi, K.; Pitt, J.J.; Wahlberg, B.M.; Caplen, N.J. The 8q24 gene desert: An oasis of non-coding transcriptional activity. Front. Genet. 2012. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Shrestha, Y.; Flavin, R.J.; Regan, M.M.; Penney, K.L.; Mucci, L.A.; Stampfer, M.J.; Hunter, D.J.; Chanock, S.J.; Schafer, E.J.; et al. Analysis of the 10q11 cancer risk locu implicates MSMB and NCOA4 in human prostate tumorigenesis. PLoS Genet. 2010, 11. [Google Scholar] [CrossRef]
- Whitaker, H.C.; Kote-Jarai, Z.; Ross-Adams, H.; Warren, A.Y.; Burge, J.; George, A.; Bancroft, E.; Jhavar, S.; Leongamornlert, D.; Tymrakiewicz, M.; et al. The rs10993994 risk allele for prostate cancer results in clinically relevant changes in microseminoprotein-beta expression in tissue and urine. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Haiman, C.A.; Chen, G.K.; Blot, W.J.; Strom, S.S.; Berndt, S.I.; Kittles, R.A.; Rybicki, B.A.; Isaacs, W.; Ingles, S.A.; Stanford, J.L.; et al. Genome-wide association study of prostate cancer in men of African ancestry identifies a susceptibility locus at 17q21. Nat. Genet 2011, 43, 570–573. [Google Scholar]
- Zeigler-Johnson, C.M.; Spangler, E.; Jalloh, M.; Gueye, M.; Rennert, H.; Rebbeck, T.R. Genetic susceptibility to prostate cancer in men of African descent: Implications for global disparities in incidence and outcomes. Can. J. Urol 2008, 15, 3872–3882. [Google Scholar]
- Sarma, A.V.; Dunn, R.L.; Lange, L.; Ray, A.; Wang, Y.; Lange, E.M.; Cooney, K.A. Genetic polymorphisms in CYP17, CYP3A4, CYP19A1, SRD5A2, IGF-1, and IGFBP-3 and prostate cancer risk in African-American Men: The Flint Men’s Health Study. Prostate 2008, 68, 296–305. [Google Scholar]
- Wright, J.L.; Kwon, E.M.; Lin, D.W.; Kolb, S.; Koopmeiners, J.S.; Feng, Z.; Ostrander, E.A.; Stanford, J.L. CYP17 polymorphisms and prostate cancer outcomes. Prostate 2010, 70, 1094–1101. [Google Scholar]
- Taioli, E.; Sears, V.; Watson, A.; Flores-Obando, R.E.; Jackson, M.D.; Ukoli, F.A.; de Syllos Colus, I.M.; Fernandez, P.; McFarlance-Anderson, N.; Ostrander, E.A.; et al. Polymorphisms in CYP17 and CYP3A4 and prostate cancer in men of African descent. Prostate 2013, 73, 668–676. [Google Scholar]
- Whitman, E.J.; Pomerantz, M.; Chen, Y.; Chamberlin, M.M.; Furusato, B.; Gao, C.; Ali, A.; Ravindranath, L.; Dobi, A.; Sesterhenn, I.A.; et al. Prostate cancer risk allele specific for African descent associates with pathologic stage at prostatectomy. Cancer Epidemiol. Biomarkers Prev 2010, 19, 1–8. [Google Scholar]
- Bao, B.Y.; Pao, J.B.; Huang, C.N.; Pu, Y.S.; Chang, T.Y.; Lan, Y.H.; Lu, T.L.; Lee, H.Z.; Chen, L.M.; Ting, W.C.; et al. Significant associations of prostate cancer susceptibility variants with survival in patients treated with androgen-deprivation therapy. Int. J. Cancer 2012, 130, 876–884. [Google Scholar]
- Cooper, C.S.; Foster, C.S. Concepts of epigenetics in prostate cancer development. Br. J. Cancer 2009, 100, 240–245. [Google Scholar]
- Jeronimo, C.; Bastian, P.J.; Bjartell, A.; Carbone, G.M.; Catto, J.W.; Clark, S.J.; Henrique, R.; Nelson, W.G.; Shariat, S.F. Epigenetics in prostate cancer: Biologic and relevance. Eur. Urol 2011, 60, 753–766. [Google Scholar]
- Nelson, W.G.; Yegnasubramanian, S.; Agoston, A.T.; Bastian, P.J.; Lee, B.H.; Nakayama, M.; de Marzo, A.M. Abnormal DNA methylation, epigenetics, and prostate cancer. Front. Biosci 2007, 12, 4254–4266. [Google Scholar]
- Kwabi-Addo, B.; Chung, W.; Shen, L.; Ittman, M.; Wheeler, T.; Jelinek, J.; Issa, J.P. Age-related DNA methylation changes in normal human prostate tissues. Clin. Cancer Res 2007, 13, 3796–3802. [Google Scholar]
- Nakayama, M.; Bennett, C.J.; Hicks, J.L.; Epstein, J.I.; Platz, E.A.; Nelson, W.G.; de Marzo, A.M. Hypermethylation of the human glutathione S-transferase-pi gene (GSTP1) CpG island is present in a subset of proliferative inflammatory atrophy lesions but not in normal or hyperplastic epithelium of the prostate: A detailed study using laser-capture microdissection. Am. J. Pathol 2003, 163, 923–933. [Google Scholar]
- Kwabi-Addo, B.; Wang, S.; Chung, W.; Jelinek, J.; Patierno, S.R.; Wang, B.; Andrawis, R.; Lee, N.H.; Apprey, V.; Issa, J.; et al. Identification of differentially methylated genes in normal prostate tissues from African American and Caucasion Men. Clinc. Cancer Res 2010, 16, 3539–3547. [Google Scholar]
- Bastian, P.J.; Yegnasubramanian, S.; Palapattu, G.S.; Rogers, C.G.; Lin, X.; de Marzo, A.M.; Nelson, W.G. Molecular biomarker in prostate cancer: The role of CpG island hypermethylation. Eur. Urol 2004, 46, 698–708. [Google Scholar]
- Hopkins, T.G.; Burns, P.A.; Routledge, M.N. DNA methylation of GSTP1 as biomarker in diagnosis of prostate cancer. Urology 2007, 69, 11–16. [Google Scholar]
- Beke, L.; Nuytten, M.; van Eynde, A.; Beullens, M.; Bollen, M. The gene encoding the prostatic tumor suppressor PSP94 is a target for repression by the polycomb group protein EZH2. Oncogene 2007, 26, 4590–4595. [Google Scholar]
- Iljin, K.; Wolf, M.; Edgren, H.; Gupta, S.; Kilpinen, S.; Skotheim, R.I.; Peltola, M.; Smit, F.; Verhaegh, G.; Schalken, J.; et al. TMPRSS2 fusions with oncogenic ETS factors in prostate cancer involve unbalanced genomic rearrangements and are associated with HDAC1 and epigenetic reprogramming. Cancer Res 2006, 66, 10242–10246. [Google Scholar]
- Rose, A.E.; Satagopan, J.M.; Oddoux, C.; Zhou, Q.; Xu, R.; Olshen, A.B.; Yu, J.Z.; Dash, A.; Jean-Gilles, J.; Reuter, V.; et al. Copy number and gene expression differences between African American and Caucasian American prostate cancer. J. Transl. Med. 2010, 8. [Google Scholar] [CrossRef]
- Ross, S.A. Evidence for the relationship between diet and cancer. Exp. Oncol 2010, 32, 137–142. [Google Scholar]
- Joshi, A.D.; John, E.M.; Koo, J.; Ingles, S.A.; Stern, M.C. Fish intake, cooking practices, and risk of prostate cancer: Results from a multi-ethnic case-control study. Cancer Causes Control 2012, 23, 405–420. [Google Scholar]
- Stott-Miller, M.; Neuhouser, M.L.; Stanford, J.L. Consumption of deep-fried foods and risk of prostate cancer. Prostate 2013, 73, 960–969. [Google Scholar]
- Berguin, I.M.; Min, Y.; Wu, R.; Wu, J.; Perry, D.; Cline, J.M.; Thomas, M.J.; Thornburg, T.; Kulik, G.; Smith, A. Modulation of prostate cancer genetic risk by omega-3 and omega-6 fatty acids. J. Clin. Invest 2007, 117, 1866–1875. [Google Scholar]
- Price, R.S.; Cavazos, D.A.; de Angel, R.E.; Hursting, D.A.; deGraffenried, L.A. Obesity-related systemtic factors promote an invasive phenotype in prostate cancer cells. Prostate Cancer Prostatic Dis 2012, 15, 135–143. [Google Scholar]
- Liu, J.; Hickson, D.A.; Musani, S.K.; Talegawkar, S.A.; Carithers, T.C.; Tucker, K.L.; Fox, C.S.; Taylor, H.A. Dietary patterns, abdominal visceral adipose tissue, and cardiometabolic risk factors in African Americans: The Jackson heart study. Obesity 2013, 21, 644–651. [Google Scholar]
- Prensner, J.R.; Rubin, M.A.; Wei, J.T.; Chinnaiyan, A.M.; Beyond, P.S.A. The next generation of prostate cancer biomarkers. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Rubin, M.A.; Maher, C.A.; Chinnaiyan, A.M. Common gene rearrangements in prostate cancer. J. Clin. Oncol 2011, 29, 3659–3668. [Google Scholar]
- Sreenath, T.L.; Dobi, A.; Petrovics, G.; Srivastava, S. Oncogenic activation of ERG: A predominant mechanism in prostate cancer. J. Carcinog 2011, 11, 10–21. [Google Scholar]
- Petrovics, G.; Liu, A.; Shaheduzzaman, S.; Furasato, B.; Sun, C.; Chen, Y.; Nau, M.; Ravindranath, L.; Chen, Y.; Dobi, A.; et al. Frequent overexpression of ETS-related gene-1 (ERG1) in prostate cancer transcriptome. Oncogene 2005, 24, 3847–3852. [Google Scholar]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar]
- Lin, C.; Yang, L.; Tanasa, B.; Hutt, K.; Ju, B.G.; Ohgi, K.; Zhang, J.; Rose, D.W.; Fu, X.D.; Glass, C.K.; et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 2009, 139, 1069–1083. [Google Scholar]
- Haffner, M.C.; Aryee, M.J.; Toubaji, A.; Esopi, D.M.; Albadine, R.; Gurel, B.; Isaacs, W.B.; Bova, G.S.; Liu, W.; Xu, J.; et al. Androgen-induced TOP2B-mediated double-strand breaks and prostate cancer gene rearrangements. Nat. Genet 2010, 42, 668–675. [Google Scholar]
- Tomlins, S.A.; Bjartell, A.; Chinnaiyan, A.M.; Jenster, G.; Nam, R.K.; Rubin, M.A.; Schalken, J.A. ETS gene fusions in prostate cancer: From discovery to daily clinical practice. Eur. Urol 2009, 56, 275–286. [Google Scholar]
- St. John, J.; Powell, K.; Conley-LaComb, M.K.; Chinni, S.R. TMPRSS2-ERG fusion gene expression in prostate tumor cells and its clinical and biological significance in prostate cancer progression. J. Cancer Sci. Ther 2012, 4, 94–101. [Google Scholar]
- Dobi, A.; Sreenath, T.; Srivastava, S. Androgen Dependent Oncogenic Activation of ETS Transcription Factors by Recurrent Gene Fusions in Prostate Cancer. Biological and Clinical Implications. In Androgen-Responsive Genes in Prostate Cancer; Wang, Z., Ed.; Springer: New York, NY, USA, 2013. [Google Scholar]
- Nam, R.K.; Sugar, L.; Yang, W.; Srivastava, S.; Klotz, L.H.; Yang, L.-Y.; Stanimirovic, A.; Encioiu, E.; Neill, M.; Loblaw, D.A.; et al. Expression of the TMPRSS2:ERG fusion gene predicts cancer recurrence after surgery for localized prostate cancer. Br. J. Cancer 2007, 97, 1690–1695. [Google Scholar]
- Attard, G.; Clark, J.; Ambroisine, L.; Fisher, G.; Kovacs, G.; Flohr, P.; Berney, D.; Foster, C.S.; Fletcher, A.; Gerald, W.L.; et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene 2008, 27, 253–263. [Google Scholar]
- Darnel, A.D.; Lafarque, C.J.; Vollmer, R.T.; Corcos, J.; Bismar, T.A. TMPRSS2-ERG fusion is frequently observed in Gleason pattern 3 prostate cancer in a Canadian cohort. Cancer Biol. Ther 2009, 8, 125–130. [Google Scholar]
- Rajput, A.B.; Miller, M.A.; de Luca, A.; Boyd, N.; Leung, S.; Hurtado-Coll, A.; Fazli, L.; Jones, E.C.; Palmer, J.B.; Gleave, M.E.; et al. Frequency of the TMPRSS2:ERG fusion is increased in moderate to poorly differentiated prostate cancers. J. Clin. Pathol 2007, 60, 1238–1243. [Google Scholar]
- Pettersson, A.; Graff, R.E.; Bauer, S.R.; Pitt, M.J.; Lis, R.T.; Stack, E.C.; Martin, N.E.; Kunz, L.; Penney, K.L.; Ligon, A.; et al. The TMPRSS2:ERG rearrangement, ERG expression, and prostate cancer outcomes: A cohort study and meta-analysis. Cancer Epidemiol. Biomarkers Prev 2012, 21, 1497–1509. [Google Scholar]
- Park, K.; Tomlins, S.A.; Mudaliar, K.M.; Chiu, Y.L.; Esgueva, R.; Mehra, R.; Suleman, K.; Varambally, S.; Brenner, J.C.; MacDonald, T.; et al. Antibody-based detection of ERG rearrangement-positive prostate cancer. Neoplasia 2010, 12, 590–598. [Google Scholar]
- Furusato, B.; Tan, S.H.; Young, D.; Dobi, A.; Sun, C.; Mohamed, A.A.; Thangapazham, R.; Chen, Y.; McMaster, G.; Sreenath, T.; et al. ERG oncoprotein expression in prostate cancer: Clonal progression of ERG-positive tumor cells and potential for ERG-based stratification. Prostate Cancer Prostatic Dis 2010, 13, 228–237. [Google Scholar]
- Braun, M.; Goltz, D.; Shaikhibrahim, Z.; Vogel, W.; Bohm, D.; Scheble, V.; Sotlar, K.; Fend, F.; Tan, S.H.; Dobi, A.; et al. ERG protein expression and genomic rearrangement status in primary and metastatic prostate cancer—A comparative study of two monoclonal antibodies. Prostate Cancer Prostatic Dis 2012, 15, 165–169. [Google Scholar]
- Magi-Galluzzi, C.; Tsusuki, T.; Elson, P.; Simmerman, K.; LaFarque, C.; Esqueva, R.; Klein, E.; Rubin, M.A.; Zhou, M. TMPRSS2-ERG gene fusion prevalence and class are significantly different in prostate cancer of Caucasian, African-American and Japanese patients. Prostate 2011, 71, 489–497. [Google Scholar]
- Rosen, P.; Pfister, D.; Young, D.; Petrovics, G.; Chen, Y.; Cullen, J.; Bohm, D.; Perner, S.; Dobi, A.; McLeod, D.G.; et al. Differences in frequency of ERG oncoprotein expression between index tumors of Caucasian and African American patients with prostate cancer. Urology 2012, 80, 749–753. [Google Scholar]
- Hu, Y.; Dobi, A.; Sreenath, T.; Cook, C.; Tadase, A.Y.; Ravindranath, L.; Cullen, J.; Furusato, B.; Chen, Y.; Thanqapazham, R.L.; et al. Delineation of TMPRSS2-ERG splice variants in prostate cancer. Clin. Cancer Res 2008, 14, 4719–4725. [Google Scholar]
- Rice, K.R.; Chen, Y.; Ali, A.; Whitman, E.J.; Blase, A.; Ibrahim, M.; Elsamanoudi, S.; Brassell, S.; Furusato, B.; Stingle, N.; et al. Evaluation of the ETS-related gene mRNA in urine for the detection of prostate cancer. Clin. Cancer Res 2010, 16, 1572–1576. [Google Scholar]
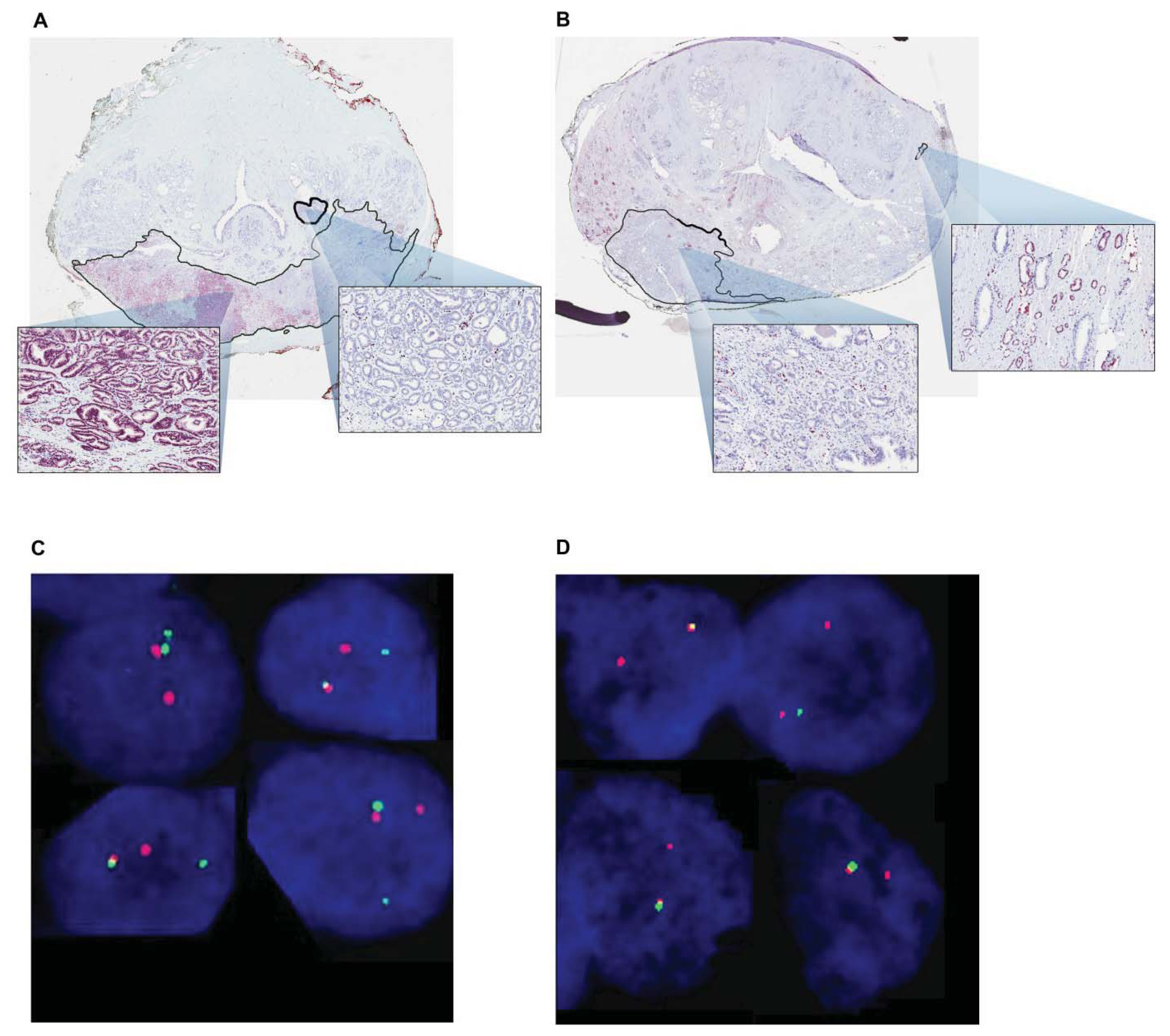
- Farrell, J.; Young, D.; Chen, Y.; Cullen, J.; Petrovics, G.; McLeod, D.G.; Sesterhenn, I.A.; Srivastava, S. The Prevalence of ERG Oncoprotein Expression in High Grade Prostate Cancer in African American and Caucasian American Patients. Proceedings of the 2012 Society of Urologic Oncology Annual Meeting, Bethesda, MD, USA, 28–30 November 2012.
- Koh, C.M.; Bieberich, C.J.; Dang, C.V.; Nelson, W.G.; Yegnasubramanian, S.; de Marzo, A.M. MYC and prostate cancer. Genes Cancer 2010, 1, 617–628. [Google Scholar]
- Grant, W.B.; Peiris, A.N. Differences in vitamin D status may account for unexplained disparities in cancer survival rates between African and white Americans. Dermatoendocrinology 2012, 4, 85–94. [Google Scholar]
- Washington, M.N.; Weigel, N.L. 1α,25-Dihydroxyvitamin D3 inhibits growth of VCaP prostate cancer cells despite inducing the growth-promoting TMPRSS2:ERG gene fusion. Endocrinology 2010, 151, 1409–1417. [Google Scholar]
- Hendrickson, W.K.; Flavin, R.; Kasperzyk, J.L.; Fiorentino, M.; Fang, F.; Lis, R.; Fiore, C.; Penney, K.L.; Ma, J.; Kantoff, P.W.; et al. Vitamin D receptor protein expression in tumor tissue and prostate cancer progression. J. Clin. Oncol 2011, 29, 2378–2385. [Google Scholar]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar]
- Garraway, L.A.; Lander, E.S. Lessons from the cancer genome. Cell 2013, 153, 17–37. [Google Scholar]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [Green Version]
- Barbieri, C.E.; Demichelis, F.; Rubin, M.A. Molecular genetics of prostate cancer: Emerging appreciation of genetic complexity. Histopathology 2012, 60, 187–198. [Google Scholar]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Farrell, J.; Petrovics, G.; McLeod, D.G.; Srivastava, S. Genetic and Molecular Differences in Prostate Carcinogenesis between African American and Caucasian American Men. Int. J. Mol. Sci. 2013, 14, 15510-15531. https://doi.org/10.3390/ijms140815510
Farrell J, Petrovics G, McLeod DG, Srivastava S. Genetic and Molecular Differences in Prostate Carcinogenesis between African American and Caucasian American Men. International Journal of Molecular Sciences. 2013; 14(8):15510-15531. https://doi.org/10.3390/ijms140815510
Chicago/Turabian StyleFarrell, James, Gyorgy Petrovics, David G. McLeod, and Shiv Srivastava. 2013. "Genetic and Molecular Differences in Prostate Carcinogenesis between African American and Caucasian American Men" International Journal of Molecular Sciences 14, no. 8: 15510-15531. https://doi.org/10.3390/ijms140815510