DNA Methylation and Cancer Diagnosis



{kind=link}
Abstract
:1. Introduction
1.1. DNA Methylation, a Physiological Process
1.2. DNA Methyltransferase Family and Establishment of DNA Methylation Profiles
1.3. DNA Methylation Alterations in Cancers and Preneoplastic Lesions
1.4. Altered Expression of DNMTs in Cancers
2. DNA Methylation Studies in Biological Samples
2.1. Most Common Approaches for DNA Methylation Studies
2.2. Variety of Biological Samples for DNA Methylation Studies
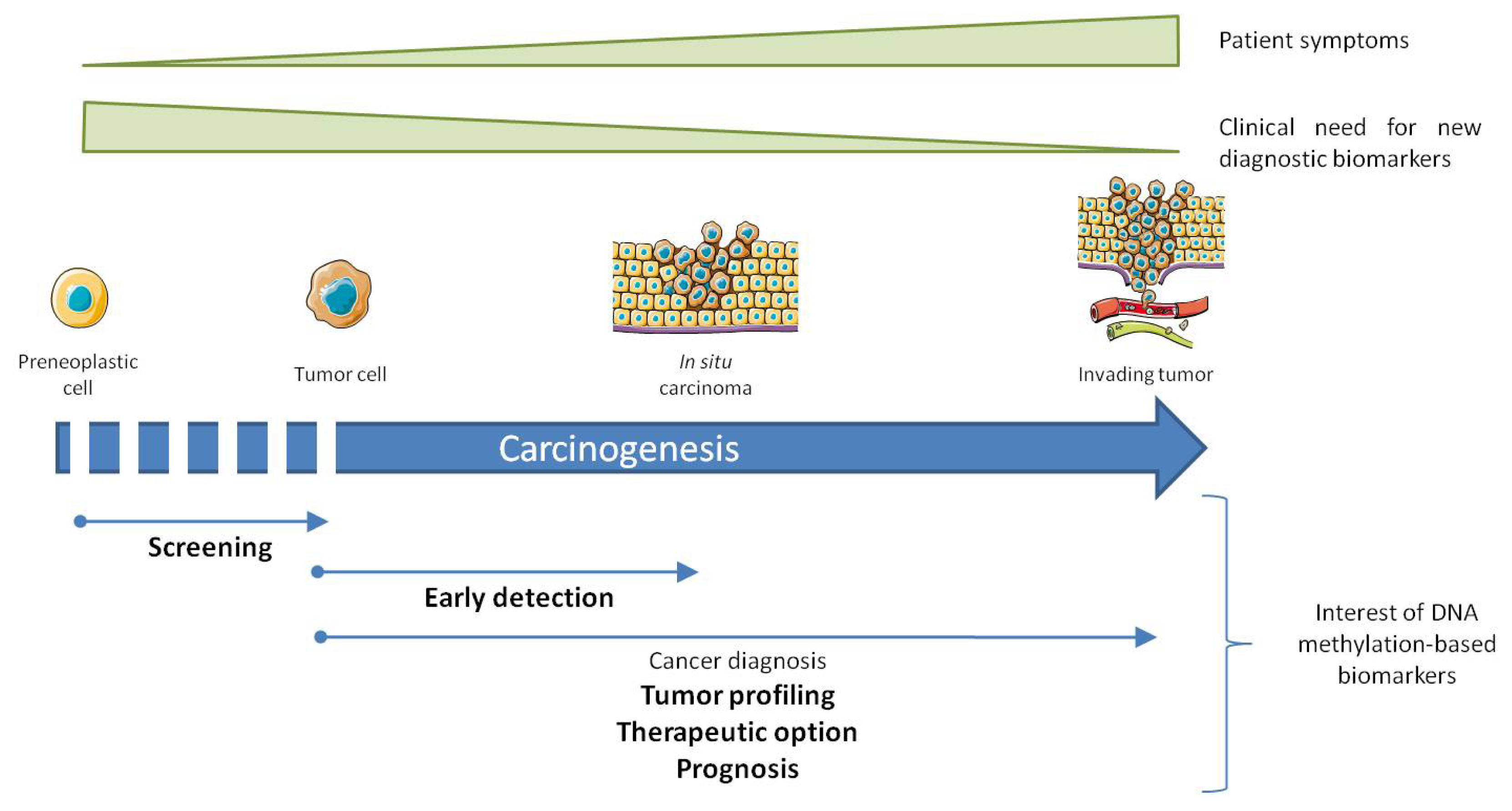
3. Altered DNA Methylation, Marks for Cancer Diagnosis
- - To improve existing tests using an existing biomarker;
- - To discover new biomarkers with high sensitivity and specificity;
- - To associate several biomarkers to compensate poor performance.
3.1. Cyclin Dependent Kinase Inhibitor, P16 ink4a
3.2. O6-Methylguanine-DNA-Methyltransferase, MGMT
3.3. Glutathione S-Transferase Pi 1, GSTP1
3.4. MutL Homolog 1, MLH1
3.5. Breast Cancer Type 1 Susceptibility Protein, BRCA1
3.6. Septin 9, SEPT9
3.7. MicroRNA Encoding Genes
3.8. Hypomethylated Genes in Cancer
3.9. Imprinted Genes
4. DNA Methylation as Biomarkers
4.1. In Cancer Diagnosis
4.2. In Early Detection of Cancer and Screening of High Risk Population
4.3. As an Advanced Diagnostic Tool
5. Principal Requirements to Develop Diagnostic Biomarkers
5.1. Preclinical Requirements
5.2. Validation
5.3. Clinical Requirements
6. Current Drugs/DNA Methylation Inhibitors and Clinical Trials, DNA Methylation Inhibitors
6.1. Nucleosidic DNA Methylation Inhibitors
6.1.1. Azacytidine
6.1.2. Decitabine
6.1.3. Zebularine
6.2. Non-Nucleoside DNA Methylation Inhibitors
6.2.1. Hydralazine
6.2.2. Procainamide Derivatives
6.2.3. Flavonoids
6.2.4. Other Inhibitors
7. Conclusions
Acknowledgements
Conflicts of Interest
References
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev 2002, 16, 6–21. [Google Scholar]
- Gopalakrishnan, S.; van Emburgh, B.O.; Robertson, K.D. DNA methylation in development and human disease. Mutat. Res 2008, 647, 30–38. [Google Scholar]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet 2012, 13, 484–492. [Google Scholar]
- Chen, T.; Li, E. Structure and function of eukaryotic DNA methyltransferases. Curr. Top. Dev. Biol 2004, 60, 55–89. [Google Scholar]
- Kinney, S.R.M.; Pradhan, S. Regulation of expression and activity of DNA (cytosine-5) methyltransferases in mammalian cells. Prog. Mol. Biol. Transl. Sci 2011, 101, 311–333. [Google Scholar]
- Bachman, K.E.; Rountree, M.R.; Baylin, S.B. Dnmt3a and Dnmt3b are transcriptional repressors that exhibit unique localization properties to heterochromatin. J. Biol. Chem 2001, 276, 32282–32287. [Google Scholar]
- Jeong, S.; Liang, G.; Sharma, S.; Lin, J.C.; Choi, S.H.; Han, H.; Yoo, C.B.; Egger, G.; Yang, A.S.; Jones, P.A. Selective anchoring of DNA methyltransferases 3A and 3B to nucleosomes containing methylated DNA. Mol. Cell. Biol 2009, 29, 5366–5376. [Google Scholar]
- Sharma, S.; de Carvalho, D.D.; Jeong, S.; Jones, P.A.; Liang, G. Nucleosomes containing methylated DNA stabilize DNA methyltransferases 3A/3B and ensure faithful epigenetic inheritance. PLoS Genet 2011, 7, e1001286. [Google Scholar]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar]
- Chedin, F.; Lieber, M.R.; Hsieh, C.L. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc. Natl. Acad. Sci. USA 2002, 99, 16916–16921. [Google Scholar]
- Okano, M.; Xie, S.; Li, E. Dnmt2 is not required for de novo and maintenance methylation of viral DNA in embryonic stem cells. Nucleic Acids Res 1998, 26, 2536–2540. [Google Scholar]
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 2006, 311, 395–398. [Google Scholar]
- Tuorto, F.; Liebers, R.; Musch, T.; Schaefer, M.; Hofmann, S.; Kellner, S.; Frye, M.; Helm, M.; Stoecklin, G.; Lyko, F. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat. Struct. Mol. Biol 2012, 19, 900–905. [Google Scholar]
- Jurkowski, T.P.; Shanmugam, R.; Helm, M.; Jeltsch, A. Mapping the tRNA binding site on the surface of human DNMT2 methyltransferase. Biochemistry 2012, 51, 4438–4444. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar]
- Berdasco, M.; Esteller, M. Aberrant epigenetic landscape in cancer: How cellular identity goes awry. Dev. Cell 2010, 19, 698–711. [Google Scholar]
- Wild, L.; Flanagan, J.M. Genome-wide hypomethylation in cancer may be a passive consequence of transformation. Biochim. Biophys. Acta 2010, 1806, 50–57. [Google Scholar]
- Pastor, W.A.; Aravind, L.; Rao, A. TETonic shift: Biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol 2013, 14, 341–356. [Google Scholar]
- Hashimoto, H.; Liu, Y.; Upadhyay, A.K.; Chang, Y.; Howerton, S.B.; Vertino, P.M.; Zhang, X.; Cheng, X. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res 2012, 40, 4841–4849. [Google Scholar]
- Valinluck, V.; Sowers, L.C. Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res 2007, 67, 946–950. [Google Scholar]
- He, Y.-F.; Li, B.-Z.; Li, Z.; Liu, P.; Wang, Y.; Tang, Q.; Ding, J.; Jia, Y.; Chen, Z.; Li, L.; et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011, 333, 1303–1307. [Google Scholar]
- Song, C.-X.; Clark, T.A.; Lu, X.Y.; Kislyuk, A.; Dai, Q.; Turner, S.W.; He, C.; Korlach, J. Sensitive and specific single-molecule sequencing of 5-hydroxymethylcytosine. Nat. Methods 2012, 9, 75–77. [Google Scholar]
- Sun, M.; Song, C.X.; Huang, H.; Frankenberger, C.A.; Sankarasharma, D.; Gomes, S.; Chen, P.; Chen, J.; Chada, K.K.; He, C.; et al. HMGA2/TET1/HOXA9 signaling pathway regulates breast cancer growth and metastasis. Proc. Natl. Acad. Sci. USA 2013, 110, 9920–9925. [Google Scholar]
- Hsu, C.-H.; Peng, K.L.; Kang, M.L.; Chen, Y.R.; Yang, Y.C.; Tsai, C.H.; Chu, C.S.; Jeng, Y.M.; Chen, Y.T.; Lin, F.M.; et al. TET1 suppresses cancer invasion by activating the tissue inhibitors of metalloproteinases. Cell Rep 2012, 2, 568–579. [Google Scholar]
- Liu, C.; Liu, L.; Chen, X.; Shen, J.; Shan, J.; Xu, Y.; Yang, Z.; Wu, L.; Xia, F.; Bie, P.; et al. Decrease of 5-Hydroxymethylcytosine is associated with progression of hepatocellular carcinoma through downregulation of TET1. PLoS One 2013, 8, e62828. [Google Scholar]
- Maul, R.W.; Gearhart, P.J. AID and somatic hypermutation. Adv. Immunol 2010, 105, 159–191. [Google Scholar]
- Bhutani, N.; Brady, J.J.; Damian, M.; Sacco, A.; Corbel, S.Y.; Blau, H.M. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature 2010, 463, 1042–1047. [Google Scholar]
- Métivier, R.; Gallais, R.; Tiffoche, C.; Le Péron, C.; Jurkowska, R.Z.; Carmouche, R.P.; Ibberson, D.; Barath, P.; Demay, F.; Reid, G.; et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature 2008, 452, 45–50. [Google Scholar]
- Kalari, S.; Pfeifer, G.P. Identification of driver and passenger DNA methylation in cancer by epigenomic analysis. Adv. Genet 2010, 70, 277–308. [Google Scholar]
- Peng, D.F.; Kanai, Y.; Sawada, M.; Ushijima, S.; Hiraoka, N.; Kosuge, T.; Hirohashi, S. Increased DNA methyltransferase 1 (DNMT1) protein expression in precancerous conditions and ductal carcinomas of the pancreas. Cancer Sci 2005, 96, 403–408. [Google Scholar]
- Belinsky, S.A.; Nikula, K.J.; Baylin, S.B.; Issa, J.P. Increased cytosine DNA-methyltransferase activity is target-cell-specific and an early event in lung cancer. Proc. Natl. Acad. Sci. USA 1996, 93, 4045–4050. [Google Scholar]
- Lopatina, N.G.; Vanyushin, B.F.; Cronin, G.M.; Poirier, L.A. Elevated expression and altered pattern of activity of DNA methyltransferase in liver tumors of rats fed methyl-deficient diets. Carcinogenesis 1998, 19, 1777–1781. [Google Scholar]
- Sato, N.; Fukushima, N.; Hruban, R.H.; Goggins, M. CpG island methylation profile of pancreatic intraepithelial neoplasia. Mod. Pathol 2007, 21, 238–244. [Google Scholar]
- Hanoun, N.; Delpu, Y.; Suriawinata, A.A.; Bournet, B.; Bureau, C.; Selves, J.; Tsongalis, G.J.; Dufresne, M.; Buscail, L.; Cordelier, P.; et al. The silencing of microRNA 148a production by DNA hypermethylation is an early event in pancreatic carcinogenesis. Clin. Chem 2010, 56, 1107–1118. [Google Scholar]
- House, M.G.; Guo, M.; Iacobuzio-Donahue, C.; Herman, J.G. Molecular progression of promoter methylation in intraductal papillary mucinous neoplasms (IPMN) of the pancreas. Carcinogenesis 2003, 24, 193–198. [Google Scholar]
- Lee, J.-H.; Park, S.-.J.; Abraham, S.C.; Seo, J.-.S.; Nam, J.-.H.; Choi, C.; Juhng, S.-.W.; Rashid, A.; Hamilton, S.R.; Wu, T.-.T. Frequent CpG island methylation in precursor lesions and early gastric adenocarcinomas. Oncogene 2004, 23, 4646–4654. [Google Scholar]
- Brooks, J.D.; Weinstein, M.; Lin, X.; Sun, Y.; Pin, S.S.; Bova, G.S.; Epstein, J.I.; Isaacs, W.B.; Nelson, W.G. CG island methylation changes near the GSTP1 gene in prostatic intraepithelial neoplasia. Cancer Epidemiol. Biomark. Prev 1998, 7, 531–536. [Google Scholar]
- Robertson, K.D.; Uzvolgyi, E.; Liang, G.; Talmadge, C.; Sumegi, J.; Gonzales, F.A.; Jones, P.A. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: Coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res 1999, 27, 2291–2298. [Google Scholar]
- El-Deiry, W.S.; Nelkin, B.D.; Celano, P.; Yen, R.-W.C.; Falco, J.P.; Hamilton, S.R.; Baylin, S.B. High expression of the DNA methyltransferase gene characterizes human neoplastic cells and progression stages of colon cancer. Proc. Natl. Acad. Sci. USA 1991, 88, 3470–3474. [Google Scholar]
- Girault, I.; Tozlu, S.; Lidereau, R.; Bièche, I. Expression Analysis of DNA Methyltransferases 1, 3A, and 3B in Sporadic Breast Carcinomas. Clin. Cancer Res 2003, 9, 4415–4422. [Google Scholar]
- Mizuno, S.; Chijiwa, T.; Okamura, T.; Akashi, K.; Fukumaki, Y.; Niho, Y.; Sasaki, H. Expression of DNA methyltransferases DNMT1,3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood 2001, 97, 1172–1179. [Google Scholar]
- Robertson, K.D. DNA methylation, methyltransferases, and cancer. Oncogene 2001, 20, 3139–3155. [Google Scholar]
- Gaidzik, V.I.; Paschka, P.; Späth, D.; Habdank, M.; Köhne, C.-H.; Germing, U.; von Lilienfeld-Toal, M.; Held, G.; Horst, H.-A.; Haase, D.; et al. TET2 Mutations in Acute Myeloid Leukemia (AML): Results from a comprehensive genetic and clinical analysis of the AML study group. J. Clin. Oncol 2012, 30, 1350–1357. [Google Scholar]
- Simó-Riudalbas, L.; Melo, S.A.; Esteller, M. DNMT3B gene amplification predicts resistance to DNA demethylating drugs. Genes Chromosomes Cancer 2011, 50, 527–534. [Google Scholar]
- Lopez de Silanes, I.; Gorospe, M.; Taniguchi, H.; Abdelmohsen, K.; Srikantan, S.; Alaminos, M.; Berdasco, M.; Urdinguio, R.G.; Fraga, M.F.; Jacinto, F.V.; et al. The RNA-binding protein HuR regulates DNA methylation through stabilization of DNMT3b mRNA. Nucleic Acids Res 2009, 37, 2658–2671. [Google Scholar]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med 2010, 363, 2424–2433. [Google Scholar]
- Singer, J.; Stellwagen, R.H.; Roberts-Ems, J.; Riggs, A.D. 5-Methylcytosine content of rat hepatoma DNA substituted with bromodeoxyuridine. J. Biol. Chem 1977, 252, 5509–5513. [Google Scholar]
- Fraga, M.F.; Rodríguez, R.; Cañal, M.J. Rapid quantification of DNA methylation by high performance capillary electrophoresis. Electrophoresis 2000, 21, 2990–2994. [Google Scholar]
- Ramsahoye, B.H. Nearest-neighbor analysis. Methods Mol. Biol 2002, 200, 9–15. [Google Scholar]
- Guerrero-Preston, R.; Santella, R.M.; Blanco, A.; Desai, M.; Berdasco, M.; Fraga, M. Global DNA hypomethylation in liver cancer cases and controls: A phase I preclinical biomarker development study. Epigenetics 2007, 2, 223–226. [Google Scholar]
- Costello, J.F.; Smiraglia, D.J.; Plass, C. Restriction landmark genome scanning. Methods 2002, 27, 144–149. [Google Scholar]
- Frigola, J.; Ribas, M.; Risques, R.-A.; Peinado, M.A. Methylome profiling of cancer cells by amplification of inter-methylated sites (AIMS). Nucleic Acids Res 2002, 30, e28. [Google Scholar]
- Liang, G.; Chan, M.F.; Tomigahara, Y.; Tsai, Y.C.; Gonzales, F.A.; Li, E.; Laird, P.W.; Jones, P.A. Cooperativity between DNA methyltransferases in the maintenance methylation of repetitive elements. Mol. Cell. Biol 2002, 22, 480–491. [Google Scholar]
- Frommer, M.; McDonald, L.E.; Millar, D.S.; Collis, C.M.; Watt, F.; Grigg, G.W.; Molloy, P.L.; Paul, C.L. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl. Acad. Sci. USA 1992, 89, 1827–1831. [Google Scholar]
- Herman, J.G.; Graff, J.R.; Myöhänen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: A novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar]
- Yamamoto, N.; Nakayama, T.; Kajita, M.; Miyake, T.; Iwamoto, T.; Kim, S.J.; Sakai, A.; Ishihara, H.; Tamaki, Y.; Noguchi, S. Detection of aberrant promoter methylation of GSTP1, RASSF1A, and RARβ2 in serum DNA of patients with breast cancer by a newly established one-step methylation-specific PCR assay. Breast Cancer Res. Treat 2012, 132, 165–173. [Google Scholar]
- Xiong, Z.; Laird, P.W. COBRA: A sensitive and quantitative DNA methylation assay. Nucleic Acids Res 1997, 25, 2532–2534. [Google Scholar]
- Eads, C.A.; Danenberg, K.D.; Kawakami, K.; Saltz, L.B.; Blake, C.; Shibata, D.; Danenberg, P.V.; Laird, P.W. MethyLight: A high-throughput assay to measure DNA methylation. Nucleic Acids Res 2000, 28, E32. [Google Scholar]
- Tost, J.; Gut, I.G. DNA methylation analysis by pyrosequencing. Nat. Protoc 2007, 2, 2265–2275. [Google Scholar]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.-.L.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-wide high-resolution mapping and functional analysis of DNA methylation in arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar]
- Taylor, K.H.; Kramer, R.S.; Davis, J.W.; Guo, J.; Duff, D.J.; Xu, D.; Caldwell, C.W.; Shi, H. Ultradeep bisulfite sequencing analysis of DNA methylation patterns in multiple gene promoters by 454 sequencing. Cancer Res 2007, 67, 8511–8518. [Google Scholar]
- Kulis, M.; Heath, S.; Bibikova, M.; Queirós, A.C.; Navarro, A.; Clot, G.; Martínez-Trillos, A.; Castellano, G.; Brun-Heath, I.; Pinyol, M.; et al. Epigenomic analysis detects widespread gene-body DNA hypomethylation in chronic lymphocytic leukemia. Nat. Genet 2012, 44, 1236–1242. [Google Scholar]
- Bibikova, M.; Lin, Z.; Zhou, L.; Chudin, E.; Garcia, E.W.; Wu, B.; Doucet, D.; Thomas, N.J.; Wang, Y.; Vollmer, E.; et al. High-throughput DNA methylation profiling using universal bead arrays. Genome Res 2006, 16, 383–393. [Google Scholar]
- Dedeurwaerder, S.; Desmedt, C.; Calonne, E.; Singhal, S.K.; Haibe-Kains, B.; Defrance, M.; Michiels, S.; Volkmar, M.; Deplus, R.; Luciani, J.; et al. DNA methylation profiling reveals a predominant immune component in breast cancers. EMBO Mol. Med 2011, 3, 726–741. [Google Scholar]
- Liloglou, T.; Field, J.K. Detection of DNA methylation changes in body fluids. Adv. Genet 2010, 71, 177–207. [Google Scholar]
- Carvalho, A.L.; Henrique, R.; Jeronimo, C.; Nayak, C.S.; Reddy, A.N.; Hoque, M.O.; Chang, S.; Brait, M.; Jiang, W.-.W.; Kim, M.M.; et al. Detection of promoter hypermethylation in salivary rinses as a biomarker for head and neck squamous cell carcinoma surveillance. Clin. Cancer Res. 2011, 17, 4782–4789. [Google Scholar]
- Anglim, P.P.; Alonzo, T.A.; Laird-Offringa, I.A. DNA methylation-based biomarkers for early detection of non-small cell lung cancer: An update. Mol. Cancer 2008, 7, 81. [Google Scholar]
- Yan, L.; McFaul, C.; Howes, N.; Leslie, J.; Lancaster, G.; Wong, T.; Threadgold, J.; Evans, J.; Gilmore, I.; Smart, H.; et al. Molecular analysis to detect pancreatic ductal adenocarcinoma in high-risk groups. Gastroenterology 2005, 128, 2124–2130. [Google Scholar]
- Matsubayashi, H.; Canto, M.; Sato, N.; Klein, A.; Abe, T.; Yamashita, K.; Yeo, C.J.; Kalloo, A.; Hruban, R.; Goggins, M. DNA methylation alterations in the pancreatic juice of patients with suspected pancreatic disease. Cancer Res 2006, 66, 1208–1217. [Google Scholar]
- Wagner, P.D.; Verma, M.; Srivastava, S. Challenges for biomarkers in cancer detection. Ann. N. Y. Acad. Sci 2004, 1022, 9–16. [Google Scholar]
- Rocco, J.W.; Sidransky, D. p16(MTS-1/CDKN2/INK4a) in cancer progression. Exp. Cell Res 2001, 264, 42–55. [Google Scholar]
- Zou, H.-Z.; Yu, B.-M.; Wang, Z.-W.; Sun, J.-Y.; Cang, H.; Gao, F.; Li, D.H.; Zhao, R.; Feng, G.-G.; Yi, J. Detection of Aberrant p16 Methylation in the Serum of Colorectal Cancer Patients. Clin. Cancer Res 2002, 8, 188–191. [Google Scholar]
- Wong, I.H.N.; Lo, Y.M.D.; Zhang, J.; Liew, C.-T.; Ng, M.H.L.; Wong, N.; Lai, P.B.S.; Lau, W.Y.; Hjelm, N.M.; Johnson, P.J. Detection of Aberrant p16 Methylation in the Plasma and Serum of Liver Cancer Patients. Cancer Res 1999, 59, 71–73. [Google Scholar]
- Belinsky, S.A.; Nikula, K.J.; Palmisano, W.A.; Michels, R.; Saccomanno, G.; Gabrielson, E.; Baylin, S.B.; Herman, J.G. Aberrant methylation of p16INK4a is an early event in lung cancer and a potential biomarker for early diagnosis. Proc. Natl. Acad. Sci. USA 1998, 95, 11891–11896. [Google Scholar]
- Kaina, B.; Margison, G.P.; Christmann, M.; Targeting, O. 6-methylguanine-DNA methyltransferase with specific inhibitors as a strategy in cancer therapy. Cell. Mol. Life Sci 2010, 67, 3663–3681. [Google Scholar]
- Esteller, M.; Garcia-Foncillas, J.; Andion, E.; Goodman, S.N.; Hidalgo, O.F.; Vanaclocha, V.; Baylin, S.B.; Herman, J.G. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N. Engl. J. Med 2000, 343, 1350–1354. [Google Scholar]
- Shen, L.; Kondo, Y.; Rosner, G.L.; Xiao, L.; Hernandez, N.S.; Vilaythong, J.; Houlihan, P.S.; Krouse, R.S.; Prasad, A.R.; Einspahr, J.G.; et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J. Natl. Cancer Inst. 2005, 97, 1330–1338. [Google Scholar]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar]
- Nakamichi, I.; Tomita, Y.; Zhang, B.; Sugiyama, H.; Kanakura, Y.; Fukuhara, S.; Hino, M.; Kanamaru, A.; Ogawa, H.; Aozasa, K. Correlation between promoter hypermethylation of GSTP1 and response to chemotherapy in diffuse large B cell lymphoma. Ann. Hematol 2007, 86, 557–564. [Google Scholar]
- Miyake, T.; Nakayama, T.; Naoi, Y.; Yamamoto, N.; Otani, Y.; Kim, S.J.; Shimazu, K.; Shimomura, A.; Maruyama, N.; Tamaki, Y.; et al. GSTP1 expression predicts poor pathological complete response to neoadjuvant chemotherapy in ER-negative breast cancer. Cancer Sci. 2012, 103, 913–920. [Google Scholar]
- Zhang, Y.; Qu, X.; Jing, W.; Hu, X.; Yang, X.; Hou, K.; Teng, Y.; Zhang, J.; Liu, Y. GSTP1 determines cis-platinum cytotoxicity in gastric adenocarcinoma MGC803 cells: Regulation by promoter methylation and extracellular regulated kinase signaling. Anticancer Drugs 2009, 20, 208–214. [Google Scholar]
- Harden, S.V.; Guo, Z.; Epstein, J.I.; Sidransky, D. Quantitative Gstp1 methylation clearly distinguishes benign prostatic tissue and limited prostate adenocarcinoma. J. Urol 2003, 169, 1138–1142. [Google Scholar]
- Saxena, A.; Dhillon, V.S.; Shahid, M.; Khalil, H.S.; Rani, M.; Prasad, D.A.S.T.; Hedau, S.; Hussain, A.; Naqvi, R.A.; Deo, S.V.S.; et al. GSTP1 methylation and polymorphism increase the risk of breast cancer and the effects of diet and lifestyle in breast cancer patients. Exp. Ther. Med 2012, 4, 1097–1103. [Google Scholar]
- Hashad, D.I.; Hashad, M.M.E.I.; Talaat, I.M.; Ibrahim, M.A. Role of glutathione-S-transferase P1 hypermethylation in molecular detection of prostate cancer. Genet Test Mol. Biomark 2011, 15, 667–670. [Google Scholar]
- Fukui, K. DNA Mismatch Repair in Eukaryotes and Bacteria. J. Nucleic Acids 2010, 2010, 1–16. [Google Scholar]
- Kantelinen, J.; Kansikas, M.; Korhonen, M.K.; Ollila, S.; Heinimann, K.; Kariola, R.; Nyström, M. MutSbeta exceeds MutSalpha in dinucleotide loop repair. Br. J. Cancer 2010, 102, 1068–1073. [Google Scholar]
- Menigatti, M.; Di Gregorio, C.; Borghi, F.; Sala, E.; Scarselli, A.; Pedroni, M.; Foroni, M.; Benatti, P.; Roncucci, L.; Ponz de Leon, M.; et al. Methylation pattern of different regions of the MLH1 promoter and silencing of gene expression in hereditary and sporadic colorectal cancer. Genes Chromosomes Cancer 2001, 31, 357–361. [Google Scholar]
- Bischoff, J.; Ignatov, A.; Semczuk, A.; Schwarzenau, C.; Ignatov, T.; Krebs, T.; Küster, D.; Przadka-Rabaniuk, D.; Roessner, A.; Costa, S.D.; et al. hMLH1 promoter hypermethylation and MSI status in human endometrial carcinomas with and without metastases. Clin. Exp. Metastasis 2012, 29, 889–900. [Google Scholar]
- Ozdemir, F.; Altinisik, J.; Karateke, A.; Coksuer, H.; Buyru, N. Methylation of tumor suppressor genes in ovarian cancer. Exp. Ther. Med 2012, 4, 1092–1096. [Google Scholar]
- Esteller, M.; Corn, P.G.; Baylin, S.B.; Herman, J.G. A gene hypermethylation profile of human cancer. Cancer Res 2001, 61, 3225–3229. [Google Scholar]
- Venkitaraman, A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182. [Google Scholar]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter Hypermethylation and BRCA1 Inactivation in Sporadic Breast and Ovarian Tumors. J. Natl. Cancer Inst. 2000, 92, 564–569. [Google Scholar]
- Dobrovic, A.; Simpfendorfer, D. Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res 1997, 57, 3347–3350. [Google Scholar]
- Lofton-Day, C.; Model, F.; DeVos, T.; Tetzner, R.; Distler, J.; Schuster, M.; Song, X.; Lesche, R.; Liebenberg, V.; Ebert, M.; et al. DNA methylation biomarkers for blood-based colorectal cancer screening. Clin. Chem 2008, 54, 414–423. [Google Scholar]
- Bennett, K.L.; Karpenko, M.; Lin, M.; Claus, R.; Arab, K.; Dyckhoff, G.; Plinkert, P.; Herpel, E.; Smiraglia, D.; Plass, C. Frequently methylated tumor suppressor genes in head and neck squamous cell carcinoma. Cancer Res 2008, 68, 4494–4499. [Google Scholar]
- Connolly, D.; Yang, Z.; Castaldi, M.; Simmons, N.; Oktay, M.H.; Coniglio, S.; Fazzarim, M.J.; Verdier-Pinard, P.; Montagna, C. Septin 9 isoform expression, localization and epigenetic changes during human and mouse breast cancer progression. Breast Cancer Res. 2011, 13, R76. [Google Scholar]
- Grützmann, R.; Molnar, B.; Pilarsky, C.; Habermann, J.K.; Schlag, P.M.; Saeger, H.D.; Miehlke, S.; Stolz, T.; Model, F.; Roblick, U.J.; et al. Sensitive detection of colorectal cancer in peripheral blood by septin 9 DNA methylation assay. PLoS One 2008, 3, e3759. [Google Scholar]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA expression profiles classify human cancers. Nature 2005, 435, 834–838. [Google Scholar]
- Iorio, M.V.; Visone, R.; Leva, G.D.; Donati, V.; Petrocca, F.; Casalini, P.; Taccioli, C.; Volinia, S.; Liu, C.-G.; Alder, H.; et al. MicroRNA signatures in human ovarian cancer. Cancer Res 2007, 67, 8699–8707. [Google Scholar]
- Weber, B.; Stresemann, C.; Brueckner, B.; Lyko, F. Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle 2007, 6, 1001–1005. [Google Scholar]
- Saito, Y.; Liang, G.; Egger, G.; Friedman, J.M.; Chuang, J.C.; Coetzee, G.A.; Jones, P.A. Specific activation of microRNA-127 with downregulation of the proto-oncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell 2006, 9, 435–443. [Google Scholar]
- Lehmann, U.; Hasemeier, B.; Christgen, M.; Müller, M.; Römermann, D.; Länger, F.; Kreipe, H. Epigenetic inactivation of microRNA gene hsa-mir-9-1 in human breast cancer. J. Pathol 2008, 214, 17–24. [Google Scholar]
- Wong, K.Y.; Huang, X.; Chim, C.S. DNA methylation of microRNA genes in multiple myeloma. Carcinogenesis 2012, 33, 1629–1638. [Google Scholar]
- Cordelier, P.; Torrisani, J. MicroRNAs in Pancreatic Cancer: Potential Interests as Biomarkers and Therapeutic Tools. In MicroRNAs in Cancer Translational Research; Cho, W.C.S., Ed.; Springer: Dordrecht, The Netherlands, 2011; pp. 287–307. [Google Scholar]
- Sato, N.; Fukushima, N.; Matsubayashi, H.; Goggins, M. Identification of maspin and S100P as novel hypomethylation targets in pancreatic cancer using global gene expression profiling. Oncogene 2004, 23, 1531–1538. [Google Scholar]
- Wang, Q.; Williamson, M.; Bott, S.; Brookman-Amissah, N.; Freeman, A.; Nariculam, J.; Hubank, M.J.F.; Ahmed, A.; Masters, J.R. Hypomethylation of WNT5A, CRIP1 and S100P in prostate cancer. Oncogene 2007, 26, 6560–6565. [Google Scholar]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Chan, A.T.; Schernhammer, E.S.; Giovannucci, E.L.; Fuchs, C.S. A cohort study of tumoral LINE-1 hypomethylation and prognosis in colon cancer. J. Natl. Cancer Inst 2008, 100, 1734–1738. [Google Scholar]
- Koerner, M.V.; Barlow, D.P. Genomic imprinting—an epigenetic gene-regulatory model. Curr. Opin. Genet. Develop 2010, 20, 164–170. [Google Scholar]
- Jelinic, P.; Shaw, P. Loss of imprinting and cancer. J. Pathol 2007, 211, 261–268. [Google Scholar]
- Mulero-Navarro, S.; Esteller, M. Epigenetic biomarkers for human cancer: The time is now. Crit. Rev. Oncol. Hematol 2008, 68, 1–11. [Google Scholar]
- McCluskey, L.L.; Chen, C.; Delgadillo, E.; Felix, J.C.; Muderspach, L.I.; Dubeau, L. Differences inp16Gene methylation and expression in benign and malignant ovarian tumors. Gynecol. Oncol 1999, 72, 87–92. [Google Scholar]
- Fujiwara, K.; Fujimoto, N.; Tabata, M.; Nishii, K.; Matsuo, K.; Hotta, K.; Kozuki, T.; Aoe, M.; Kiura, K.; Ueoka, H.; et al. Identification of epigenetic aberrant promoter methylation in serum DNA is useful for early detection of lung cancer. Clin. Cancer Res 2005, 11, 1219–1225. [Google Scholar]
- Müller, H.M.; Oberwalder, M.; Fiegl, H.; Morandell, M.; Goebel, G.; Zitt, M.; Mühlthaler, M.; Öfner, D.; Margreiter, R.; Widschwendter, M. Methylation changes in faecal DNA: A marker for colorectal cancer screening? Lancet 2004, 363, 1283–1285. [Google Scholar]
- Stumpel, D.J.P.M.; Schneider, P.; van Roon, E.H.J.; Boer, J.M.; de Lorenzo, P.; Valsecchi, M.G.; de Menezes, R.X.; Pieters, R.; Stam, R.W. Specific promoter methylation identifies different subgroups of MLL-rearranged infant acute lymphoblastic leukemia, influences clinical outcome, and provides therapeutic options. Blood 2009, 114, 5490–5498. [Google Scholar]
- Figueroa, M.E.; Lugthart, S.; Li, Y.; Erpelinck-Verschueren, C.; Deng, X.; Christos, P.J.; Schifano, E.; Booth, J.; van Putten, W.; Skrabanek, L.; et al. DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 2010, 17, 13–27. [Google Scholar]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar]
- Uhlmann, K.; Rohde, K.; Zeller, C.; Szymas, J.; Vogel, S.; Marczinek, K.; Thiel, G.; Nürnberg, P.; Laird, P.W. Distinct methylation profiles of glioma subtypes. Int. J. Cancer 2003, 106, 52–59. [Google Scholar]
- Hinoue, T.; Weisenberger, D.J.; Lange, C.P.E.; Shen, H.; Byun, H.-M.; Van Den Berg, D.; Malik, S.; Pan, F.; Noushmehr, H.; van Dijk, C.M.; et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res 2012, 22, 271–282. [Google Scholar]
- Ransohoff, D.F. Rules of evidence for cancer molecular-marker discovery and validation. Nat. Rev. Cancer 2004, 4, 309–314. [Google Scholar]
- Mikeska, T.; Bock, C.; Do, H.; Dobrovic, A. DNA methylation biomarkers in cancer: Progress towards clinical implementation. Expert Rev. Mol. Diagn 2012, 12, 473–487. [Google Scholar]
- Gros, C.; Fahy, J.; Halby, L.; Dufau, I.; Erdmann, A.; Gregoire, J.-M.; Ausseil, F.; Vispé, S.; Arimondo, P.B. DNA methylation inhibitors in cancer: Recent and future approaches. Biochimie 2012, 94, 2280–2296. [Google Scholar]
- Santi, D.V.; Norment, A.; Garrett, C.E. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc. Natl. Acad. Sci. USA 1984, 81, 6993–6997. [Google Scholar]
- Cheng, J.C.; Yoo, C.B.; Weisenberger, D.J.; Chuang, J.; Wozniak, C.; Liang, G.; Marquez, V.E.; Greer, S.; Orntoft, T.F.; Thykjaer, T.; et al. Preferential response of cancer cells to zebularine. Cancer Cell 2004, 6, 151–158. [Google Scholar]
- Lübbert, M. DNA methylation inhibitors in the treatment of leukemias, myelodysplastic syndromes and hemoglobinopathies: Clinical results and possible mechanisms of action. Curr. Top. Microbiol. Immunol 2000, 249, 135–164. [Google Scholar]
- Robak, T. New nucleoside analogs for patients with hematological malignancies. Expert Opin. Investig. Drugs 2011, 20, 343–359. [Google Scholar]
- Tsai, H.-C.; Li, H.; Van Neste, L.; Cai, Y.; Robert, C.; Rassool, F.V.; Shin, J.J.; Harbom, K.M.; Beaty, R.; Pappou, E.; et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell 2012, 21, 430–446. [Google Scholar]
- Juergens, R.A.; Wrangle, J.; Vendetti, F.P.; Murphy, S.C.; Zhao, M.; Coleman, B.; Sebree, R.; Rodgers, K.; Hooker, C.M.; Franco, N.; et al. Combination epigenetic therapy has efficacy in patients with refractory advanced non-small cell lung cancer. Cancer Discov 2011, 1, 598–607. [Google Scholar]
- Filì, C.; Malagola, M.; Follo, M.Y.; Finelli, C.; Iacobucci, I.; Martinelli, G.; Cattina, F.; Clissa, C.; Candoni, A.; Fanin, R.; et al. Prospective phase II study on 5-days azacitidine for treatment of symptomatic and/or erythropoietin unresponsive patients with low/INT-1–risk myelodysplastic syndromes. Clin. Cancer Res 2013, 19, 3297–3308. [Google Scholar]
- Passweg, J.R.; Pabst, T.; Blum, S.; Bargetzi, M.; Li, Q.; Heim, D.; Stussi, G.; Gregor, M.; Leoncini, L.; Meyer-Monard, S.; et al. Azacytidine for acute myeloid leukemia in elderly or frail patients: A phase II trial (SAKK 30/07). Leuk. Lymphoma 2013. [Google Scholar] [CrossRef]
- Gore, S.D.; Fenaux, P.; Santini, V.; Bennett, J.M.; Silverman, L.R.; Seymour, J.F.; Hellstrom-Lindberg, E.; Swern, A.S.; Beach, C.L.; List, A.F.; et al. A multivariate analysis of the relationship between response and survival among patients with higher-risk myelodysplastic syndromes treated within azacitidine or conventional care regimens in the randomized AZA-001 trial. Haematologica 2013. [Google Scholar] [CrossRef]
- Bryan, J.; Kantarjian, H.; Garcia-Manero, G.; Jabbour, E. Pharmacokinetic evaluation of decitabine for the treatment of leukemia. Expert Opin. Drug Metab. Toxicol 2011, 7, 661–672. [Google Scholar]
- Matoušová, M.; Votruba, I.; Otmar, M.; Tloušt’ová, E.; Günterová, J.; Mertlíková-Kaiserová, H. 2′-deoxy-5,6-dihydro-5-azacytidine—a less toxic alternative of 2′-deoxy-5-azacytidine: A comparative study of hypomethylating potential. Epigenetics 2011, 6, 769–776. [Google Scholar]
- Chuang, J.C.; Warner, S.L.; Vollmer, D.; Vankayalapati, H.; Redkar, S.; Bearss, D.J.; Qiu, X.; Yoo, C.B.; Jones, P.A. S110, a 5-Aza-2′-deoxycytidine-containing dinucleotide, is an effective DNA methylation inhibitor in vivo and can reduce tumor growth. Mol. Cancer Ther 2010, 9, 1443–1450. [Google Scholar]
- Schroeder, G.K.; Zhou, L.; Snider, M.J.; Chen, X.; Wolfenden, R. Flight of a cytidine deaminase complex with an imperfect transition state analogue inhibitor: Mass spectrometric evidence for the presence of a trapped water molecule. Biochemistry 2012, 51, 6476–6486. [Google Scholar]
- Zhou, L.; Cheng, X.; Connolly, B.A.; Dickman, M.J.; Hurd, P.J.; Hornby, D.P. Zebularine: A novel DNA methylation inhibitor that forms a covalent complex with DNA methyltransferases. J. Mol. Biol 2002, 321, 591–599. [Google Scholar]
- Scott, S.A.; Lakshimikuttysamma, A.; Sheridan, D.P.; Sanche, S.E.; Geyer, C.R.; DeCoteau, J.F. Zebularine inhibits human acute myeloid leukemia cell growth in vitro in association with p15INK4B demethylation and reexpression. Exp. Hematol 2007, 35, 263–273. [Google Scholar]
- Yoo, C.B.; Chuang, J.C.; Byun, H.-M.; Egger, G.; Yang, A.S.; Dubeau, L.; Long, T.; Laird, P.W.; Marquez, V.E.; Jones, P.A. Long-term epigenetic therapy with oral zebularine has minimal side effects and prevents intestinal tumors in mice. Cancer Prev. Res 2008, 1, 233–240. [Google Scholar]
- Flotho, C.; Claus, R.; Batz, C.; Schneider, M.; Sandrock, I.; Ihde, S.; Plass, C.; Niemeyer, C.M.; Lübbert, M. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia 2009, 23, 1019–1028. [Google Scholar]
- Chik, F.; Szyf, M. Effects of specific DNMT gene depletion on cancer cell transformation and breast cancer cell invasion; toward selective DNMT inhibitors. Carcinogenesis 2011, 32, 224–232. [Google Scholar]
- Fahy, J.; Jeltsch, A.; Arimondo, P.B. DNA methyltransferase inhibitors in cancer: A chemical and therapeutic patent overview and selected clinical studies. Expert Opin. Ther. Pat 2012, 22, 1427–1442. [Google Scholar]
- Cornacchia, E.; Golbus, J.; Maybaum, J.; Strahler, J.; Hanash, S.; Richardson, B. Hydralazine and procainamide inhibit T cell DNA methylation and induce autoreactivity. J. Immunol 1988, 140, 2197–2200. [Google Scholar]
- Segura-Pacheco, B.; Trejo-Becerril, C.; Perez-Cardenas, E.; Taja-Chayeb, L.; Mariscal, I.; Chavez, A.; Acuña, C.; Salazar, A.M.; Lizano, M.; Dueñas-Gonzalez, A. Reactivation of tumor suppressor genes by the cardiovascular drugs hydralazine and procainamide and their potential use in cancer therapy. Clin. Cancer Res 2003, 9, 1596–1603. [Google Scholar]
- Zambrano, P.; Segura-Pacheco, B.; Perez-Cardenas, E.; Cetina, L.; Revilla-Vazquez, A.; Taja-Chayeb, L.; Chavez-Blanco, A.; Angeles, E.; Cabrera, G.; Sandoval, K.; et al. A phase I study of hydralazine to demethylate and reactivate the expression of tumor suppressor genes. BMC Cancer 2005, 5, 44. [Google Scholar]
- Chuang, J.C.; Yoo, C.B.; Kwan, J.M.; Li, T.W.H.; Liang, G.; Yang, A.S.; Jones, P.A. Comparison of biological effects of non-nucleoside DNA methylation inhibitors versus 5-aza-2′-deoxycytidine. Mol. Cancer Ther 2005, 4, 1515–1520. [Google Scholar]
- Fenster, P.E.; Comess, K.A.; Marsh, R.; Katzenberg, C.; Hager, W.D. Conversion of atrial fibrillation to sinus rhythm by acute intravenous procainamide infusion. Am. Heart J 1983, 106, 501–504. [Google Scholar]
- Villar-Garea, A.; Fraga, M.F.; Espada, J.; Esteller, M. Procaine is a DNA-demethylating agent with growth-inhibitory effects in human cancer cells. Cancer Res 2003, 63, 4984–4989. [Google Scholar]
- Lee, B.H.; Yegnasubramanian, S.; Lin, X.; Nelson, W.G. Procainamide is a specific inhibitor of DNA methyltransferase 1. J. Biol. Chem 2005, 280, 40749–40756. [Google Scholar]
- Stresemann, C.; Brueckner, B.; Musch, T.; Stopper, H.; Lyko, F. Functional diversity of DNA methyltransferase inhibitors in human cancer cell lines. Cancer Res 2006, 66, 2794–2800. [Google Scholar]
- Halby, L.; Champion, C.; Sénamaud-Beaufort, C.; Ajjan, S.; Drujon, T.; Rajavelu, A.; Ceccaldi, A.; Jurkowska, R.; Lequin, O.; Nelson, W.G.; et al. Rapid synthesis of new DNMT inhibitors derivatives of procainamide. Chembiochem 2012, 13, 157–165. [Google Scholar]
- Lin, Y.-S.; Shaw, A.Y.; Wang, S.-G.; Hsu, C.-C.; Teng, I.-W.; Tseng, M.-J.; Huang, T.H.; Chen, C.-S.; Leu, Y.-W.; Hsiao, S.-H. Identification of novel DNA methylation inhibitors via a two-component reporter gene system. J. Biomed. Sci 2011, 18, 3. [Google Scholar]
- Fang, M.Z.; Chen, D.; Sun, Y.; Jin, Z.; Christman, J.K.; Yang, C.S. Reversal of hypermethylation and reactivation of p16INK4a, RARbeta, and MGMT genes by genistein and other isoflavones from soy. Clin. Cancer Res 2005, 11, 7033–7041. [Google Scholar]
- Lee, W.J.; Shim, J.-Y.; Zhu, B.T. Mechanisms for the inhibition of DNA methyltransferases by tea catechins and bioflavonoids. Mol. Pharmacol 2005, 68, 1018–1030. [Google Scholar]
- Wang, Y.; Li, Y.; Liu, X.; Cho, W.C. Genetic and epigenetic studies for determining molecular targets of natural product anticancer agents. Curr. Cancer Drug Targets 2013, 13, 506–518. [Google Scholar]
- Plummer, R.; Vidal, L.; Griffin, M.; Lesley, M.; de Bono, J.; Coulthard, S.; Sludden, J.; Siu, L.L.; Chen, E.X.; Oza, A.M.; et al. Phase I study of MG98, an oligonucleotide antisense inhibitor of human DNA methyltransferase 1, given as a 7-day infusion in patients with advanced solid tumors. Clin. Cancer Res 2009, 15, 3177–3183. [Google Scholar]
- Klisovic, R.B.; Stock, W.; Cataland, S.; Klisovic, M.I.; Liu, S.; Blum, W.; Green, M.; Odenike, O.; Godley, L.; Burgt, J.V.; et al. A phase I biological study of MG98, an oligodeoxynucleotide antisense to DNA methyltransferase 1, in patients with high-risk myelodysplasia and acute myeloid leukemia. Clin. Cancer Res 2008, 14, 2444–2449. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Delpu, Y.; Cordelier, P.; Cho, W.C.; Torrisani, J. DNA Methylation and Cancer Diagnosis. Int. J. Mol. Sci. 2013, 14, 15029-15058. https://doi.org/10.3390/ijms140715029
Delpu Y, Cordelier P, Cho WC, Torrisani J. DNA Methylation and Cancer Diagnosis. International Journal of Molecular Sciences. 2013; 14(7):15029-15058. https://doi.org/10.3390/ijms140715029
Chicago/Turabian StyleDelpu, Yannick, Pierre Cordelier, William C. Cho, and Jérôme Torrisani. 2013. "DNA Methylation and Cancer Diagnosis" International Journal of Molecular Sciences 14, no. 7: 15029-15058. https://doi.org/10.3390/ijms140715029
APA StyleDelpu, Y., Cordelier, P., Cho, W. C., & Torrisani, J. (2013). DNA Methylation and Cancer Diagnosis. International Journal of Molecular Sciences, 14(7), 15029-15058. https://doi.org/10.3390/ijms140715029