Targeting the Redox Balance in Inflammatory Skin Conditions

Abstract
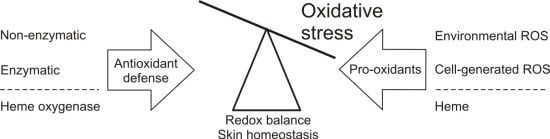
:
1. Introduction
2. Reactive Species Mediate Cellular Signaling
3. ROS and (Chronic) Inflammation of the Skin
3.1. Sunburn
3.2. Psoriasis
3.3. Burn Injury
4. Maintaining the Redox Balance in the Skin
4.1. Enzymatic Sources of Oxidants in the Skin
4.2. Non-Enzymatic Sources of Oxidants in the Skin
4.3. Antioxidant Systems in the Skin
5. Oxidative Stress as Therapeutic Target in Inflammatory Skin Conditions
5.1. Reduction of ROS Production
5.1.1. Nox Inhibition
5.1.2. Metal Scavenging Proteins
5.2. Increasing Enzymatic Antioxidant Defenses
5.2.1. Antioxidant Upregulation
5.2.2. Synthetic Antioxidants
5.2.3. Induction of the HO System
5.3. Increasing Non-Enzymatic Antioxidant Defenses
5.3.1. Oral and Topical Administration
5.3.2. Targeted Antioxidant Delivery
4. Concluding Remarks
Acknowledgments
Conflict of Interest
Abbreviations
| APE/Ref-1 | apurinic/apyrimidinic endonuclease/redox effector factor-1 |
| ARE | antioxidant responsive element |
| CO | carbon monoxide |
| CoQ10 | coenzyme Q10 |
| ERK | extracellular signal-regulated protein kinases |
| G6PD | glucose-6-phosphate dehydrogenase |
| GPx | glutathione peroxidase |
| GSH | reduced glutathione |
| H2O2 | hydrogen peroxide |
| HO• | hydroxyl radical |
| HIF-1 | hypoxia inducible factor-1 |
| HO | heme oxygenase |
| HRE | hypoxia response element |
| JNK | c-Jun N-terminal kinases |
| KO | knockout |
| LDL | low density lipoprotein |
| MAPK | mitogen-activated protein kinase |
| Mn | manganese |
| NAC | N-acetyl cysteine |
| NF-κB | nuclear factor-κB |
| Nox | NADPH oxidase |
| Nrf2 | NF-E2-related factor 2 |
| O2−• | superoxide anion |
| Prdx | peroxiredoxin |
| PTP | protein tyrosine phosphatase |
| ROS | reactive oxygen species |
| SOD | superoxide dismutase. |
References
- Contassot, E.; Beer, H.D.; French, L.E. Interleukin-1, inflammasomes, autoinflammation and the skin. Swiss Med. Wkly. 2012, 142. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol 2009, 27, 229–265. [Google Scholar]
- Feldmeyer, L.; Werner, S.; French, L.E.; Beer, H.D. Interleukin-1, inflammasomes and the skin. Eur. J. Cell Biol 2010, 89, 638–644. [Google Scholar]
- Nestle, F.O.; Di Meglio, P.; Qin, J.Z.; Nickoloff, B.J. Skin immune sentinels in health and disease. Nat. Rev. Immunol 2009, 9, 679–691. [Google Scholar]
- Fuchs, E.; Raghavan, S. Getting under the skin of epidermal morphogenesis. Nat. Rev. Genet 2002, 3, 199–209. [Google Scholar]
- Sorrell, J.M.; Caplan, A.I. Fibroblast heterogeneity: More than skin deep. J. Cell Sci 2004, 117, 667–675. [Google Scholar]
- Glaser, R.; Harder, J.; Lange, H.; Bartels, J.; Christophers, E.; Schroder, J.M. Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nat. Immunol 2005, 6, 57–64. [Google Scholar]
- Masters, S.L. Specific inflammasomes in complex diseases. Clin. Immunol. 2012. [Google Scholar] [CrossRef]
- Weber, A.; Wasiliew, P.; Kracht, M. Interleukin-1 (IL-1) pathway. Sci. Signal. 2010, 3. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol 2009, 7, 99–109. [Google Scholar]
- Yeretssian, G.; Labbe, K.; Saleh, M. Molecular regulation of inflammation and cell death. Cytokine 2008, 43, 380–390. [Google Scholar]
- Miao, E.A.; Leaf, I.A.; Treuting, P.M.; Mao, D.P.; Dors, M.; Sarkar, A.; Warren, S.E.; Wewers, M.D.; Aderem, A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol 2010, 11, 1136–1142. [Google Scholar]
- Feldmeyer, L.; Keller, M.; Niklaus, G.; Hohl, D.; Werner, S.; Beer, H.D. The inflammasome mediates UVB-induced activation and secretion of interleukin-1beta by keratinocytes. Curr. Biol 2007, 17, 1140–1145. [Google Scholar]
- Watanabe, H.; Gaide, O.; Petrilli, V.; Martinon, F.; Contassot, E.; Roques, S.; Kummer, J.A.; Tschopp, J.; French, L.E. Activation of the IL-1beta-processing inflammasome is involved in contact hypersensitivity. J. Investig. Dermatol 2007, 127, 1956–1963. [Google Scholar]
- Kupper, T.S.; Ballard, D.W.; Chua, A.O.; McGuire, J.S.; Flood, P.M.; Horowitz, M.C.; Langdon, R.; Lightfoot, L.; Gubler, U. Human keratinocytes contain mRNA indistinguishable from monocyte interleukin 1 alpha and beta mRNA. Keratinocyte epidermal cell-derived thymocyte-activating factor is identical to interleukin 1. J. Exp. Med 1986, 164, 2095–2100. [Google Scholar]
- Kondo, S.; Sauder, D.N.; Kono, T.; Galley, K.A.; McKenzie, R.C. Differential modulation of interleukin-1 alpha (IL-1 alpha) and interleukin-1 beta (IL-1 beta) in human epidermal keratinocytes by UVB. Exp. Dermatol 1994, 3, 29–39. [Google Scholar]
- Barker, J.N.; Mitra, R.S.; Griffiths, C.E.; Dixit, V.M.; Nickoloff, B.J. Keratinocytes as initiators of inflammation. Lancet 1991, 337, 211–214. [Google Scholar]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol 2010, 11, 136–140. [Google Scholar]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev.. Immunol 2010, 10, 210–215. [Google Scholar]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol 2011, 12, 222–230. [Google Scholar]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar]
- Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev 2002, 82, 47–95. [Google Scholar]
- Forman, H.J.; Fukuto, J.M.; Miller, T.; Zhang, H.; Rinna, A.; Levy, S. The chemistry of cell signaling by reactive oxygen and nitrogen species and 4-hydroxynonenal. Arch. Biochem. Biophys 2008, 477, 183–195. [Google Scholar]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar]
- Kamata, H.; Hirata, H. Redox regulation of cellular signalling. Cell. Signal 1999, 11, 1–14. [Google Scholar]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal 2008, 10, 1343–1374. [Google Scholar]
- Finkel, T. Signal transduction by reactive oxygen species. J. Cell. Biol 2011, 194, 7–15. [Google Scholar]
- Rhee, S.G.; Chang, T.S.; Bae, Y.S.; Lee, S.R.; Kang, S.W. Cellular regulation by hydrogen peroxide. J. Am. Soc. Nephrol 2003, 14, S211–S215. [Google Scholar]
- Haddad, J.J. Antioxidant and prooxidant mechanisms in the regulation of redox(y)-sensitive transcription factors. Cell. Signal 2002, 14, 879–897. [Google Scholar]
- Pastore, A.; Piemonte, F. S-Glutathionylation signaling in cell biology: Progress and prospects. Eur. J. Pharm. Sci 2012, 46, 279–292. [Google Scholar]
- Jones, D.P. Radical-free biology of oxidative stress. Am. J. Physiol. Cell Physiol 2008, 295, C849–C868. [Google Scholar]
- Fritz, G.; Grosch, S.; Tomicic, M.; Kaina, B. APE/Ref-1 and the mammalian response to genotoxic stress. Toxicology 2003, 193, 67–78. [Google Scholar]
- Rahman, I.; Marwick, J.; Kirkham, P. Redox modulation of chromatin remodeling: Impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expression. Biochem. Pharm 2004, 68, 1255–1267. [Google Scholar]
- England, K.; Cotter, T.G. Direct oxidative modifications of signalling proteins in mammalian cells and their effects on apoptosis. Redox Rep 2005, 10, 237–245. [Google Scholar]
- Bourdon, E.; Blache, D. The importance of proteins in defense against oxidation. Antioxid. Redox Signal 2001, 3, 293–311. [Google Scholar]
- Pantano, C.; Reynaert, N.L.; van der Vliet, A.; Janssen-Heininger, Y.M. Redox-sensitive kinases of the nuclear factor-kappaB signaling pathway. Antioxid. Redox Signal 2006, 8, 1791–1806. [Google Scholar]
- Shao, D.; Oka, S.; Brady, C.D.; Haendeler, J.; Eaton, P.; Sadoshima, J. Redox modification of cell signaling in the cardiovascular system. J. Mol. Cell. Cardiol 2012, 52, 550–558. [Google Scholar]
- Jacob, C.; Giles, G.I.; Giles, N.M.; Sies, H. Sulfur and selenium: The role of oxidation state in protein structure and function. Angew. Chem 2003, 42, 4742–4758. [Google Scholar]
- Martyniuk, C.J.; Fang, B.; Koomen, J.M.; Gavin, T.; Zhang, L.; Barber, D.S.; Lopachin, R.M. Molecular mechanism of glyceraldehyde-3-phosphate dehydrogenase inactivation by alpha, beta-unsaturated carbonyl derivatives. Chem. Res. Toxicol 2011, 24, 2302–2311. [Google Scholar]
- Go, Y.M.; Duong, D.M.; Peng, J.; Jones, D.P. Protein cysteines map to functional networks according to steady-state level of oxidation. J. Proteomics Bioinform 2011, 4, 196–209. [Google Scholar]
- Forman, H.J.; Fukuto, J.M.; Torres, M. Redox signaling: Thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am. J. Physiol. Cell Physiol 2004, 287, C246–C256. [Google Scholar]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med 2008, 45, 549–561. [Google Scholar]
- Paulsen, C.E.; Carroll, K.S. Orchestrating redox signaling networks through regulatory cysteine switches. ACS Chem. Biol 2010, 5, 47–62. [Google Scholar]
- Chung, H.S.; Wang, S.B.; Venkatraman, V.; Murray, C.I.; Van Eyk, J.E. Cysteine oxidative posttranslational modifications: Emerging regulation in the cardiovascular system. Circ. Res 2013, 112, 382–392. [Google Scholar]
- Denu, J.M.; Dixon, J.E. Protein tyrosine phosphatases: Mechanisms of catalysis and regulation. Curr. Opin. Chem. Biol 1998, 2, 633–641. [Google Scholar]
- Ostman, A.; Frijhoff, J.; Sandin, A.; Bohmer, F.D. Regulation of protein tyrosine phosphatases by reversible oxidation. J. Biochem 2011, 150, 345–356. [Google Scholar]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences. J. Cell. Biol 2004, 164, 341–346. [Google Scholar]
- Frand, A.R.; Cuozzo, J.W.; Kaiser, C.A. Pathways for protein disulphide bond formation. Trends Cell. Biol 2000, 10, 203–210. [Google Scholar]
- Jones, D.P.; Go, Y.M.; Anderson, C.L.; Ziegler, T.R.; Kinkade, J.M., Jr; Kirlin, W.G. Cysteine/cystine couple is a newly recognized node in the circuitry for biologic redox signaling and control. FASEB J 2004, 18, 1246–1248. [Google Scholar]
- Chen, C.Y.; Willard, D.; Rudolph, J. Redox regulation of SH2-domain-containing protein tyrosine phosphatases by two backdoor cysteines. Biochemistry 2009, 48, 1399–1409. [Google Scholar]
- Caselli, A.; Marzocchini, R.; Camici, G.; Manao, G.; Moneti, G.; Pieraccini, G.; Ramponi, G. The inactivation mechanism of low molecular weight phosphotyrosine-protein phosphatase by H2O2. J. Biol. Chem 1998, 273, 32554–32560. [Google Scholar]
- Lee, S.R.; Yang, K.S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem 2002, 277, 20336–20342. [Google Scholar]
- Sohn, J.; Rudolph, J. Catalytic and chemical competence of regulation of cdc25 phosphatase by oxidation/reduction. Biochemistry 2003, 42, 10060–10070. [Google Scholar]
- Tonks, N.K. Redox redux: Revisiting PTPs and the control of cell signaling. Cell 2005, 121, 667–670. [Google Scholar]
- Klomsiri, C.; Karplus, P.A.; Poole, L.B. Cysteine-based redox switches in enzymes. Antioxid. Redox Signal 2011, 14, 1065–1077. [Google Scholar]
- Shackelford, R.E.; Heinloth, A.N.; Heard, S.C.; Paules, R.S. Cellular and molecular targets of protein S-glutathiolation. Antioxid. Redox Signal 2005, 7, 940–950. [Google Scholar]
- Shlomai, J. Redox control of protein-DNA interactions: From molecular mechanisms to significance in signal transduction, gene expression, and DNA replication. Antioxid. Redox Signal 2010, 13, 1429–1476. [Google Scholar]
- Dinkova-Kostova, A.T.; Holtzclaw, W.D.; Cole, R.N.; Itoh, K.; Wakabayashi, N.; Katoh, Y.; Yamamoto, M.; Talalay, P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA 2002, 99, 11908–11913. [Google Scholar]
- Shelton, M.D.; Mieyal, J.J. Regulation by reversible S-glutathionylation: Molecular targets implicated in inflammatory diseases. Mol. Cells 2008, 25, 332–346. [Google Scholar]
- Qanungo, S.; Starke, D.W.; Pai, H.V.; Mieyal, J.J.; Nieminen, A.L. Glutathione supplementation potentiates hypoxic apoptosis by S-glutathionylation of p65-NFkappaB. J. Biol. Chem 2007, 282, 18427–18436. [Google Scholar]
- Cross, J.V.; Templeton, D.J. Regulation of signal transduction through protein cysteine oxidation. Antioxid. Redox Signal 2006, 8, 1819–1827. [Google Scholar]
- Gilmore, T.D. Introduction to NF-kappaB: Players, pathways, perspectives. Oncogene 2006, 25, 6680–6684. [Google Scholar]
- Benhar, M.; Forrester, M.T.; Stamler, J.S. Protein denitrosylation: Enzymatic mechanisms and cellular functions. Nat. Rev. Mol. Cell Biol 2009, 10, 721–732. [Google Scholar]
- Benhar, M.; Forrester, M.T.; Hess, D.T.; Stamler, J.S. Regulated protein denitrosylation by cytosolic and mitochondrial thioredoxins. Science 2008, 320, 1050–1054. [Google Scholar]
- Lima, B.; Forrester, M.T.; Hess, D.T.; Stamler, J.S. S-nitrosylation in cardiovascular signaling. Circ. Res 2010, 106, 633–646. [Google Scholar]
- Evangelista, A.M.; Kohr, M.J.; Murphy, E. S-Nitrosylation: Specificity, occupancy, and interaction with other post-translational modifications. Antioxid. Redox Signal. 2013. [Google Scholar] [CrossRef]
- Becker, K.; Savvides, S.N.; Keese, M.; Schirmer, R.H.; Karplus, P.A. Enzyme inactivation through sulfhydryl oxidation by physiologic NO-carriers. Nat. Struct. Biol 1998, 5, 267–271. [Google Scholar]
- Kim, S.; Wing, S.S.; Ponka, P. S-nitrosylation of IRP2 regulates its stability via the ubiquitin-proteasome pathway. Mol. Cell. Biol 2004, 24, 330–337. [Google Scholar]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J 2009, 417, 1–13. [Google Scholar]
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cocheme, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci 2012, 125, 801–806. [Google Scholar]
- Murphy, M.P.; Holmgren, A.; Larsson, N.G.; Halliwell, B.; Chang, C.J.; Kalyanaraman, B.; Rhee, S.G.; Thornalley, P.J.; Partridge, L.; Gems, D.; et al. Unraveling the biological roles of reactive oxygen species. Cell Metab 2011, 13, 361–366. [Google Scholar]
- Hoehn, K.L.; Salmon, A.B.; Hohnen-Behrens, C.; Turner, N.; Hoy, A.J.; Maghzal, G.J.; Stocker, R.; van Remmen, H.; Kraegen, E.W.; Cooney, G.J.; et al. Insulin resistance is a cellular antioxidant defense mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 17787–17792. [Google Scholar]
- Zhou, L.; Aon, M.A.; Almas, T.; Cortassa, S.; Winslow, R.L.; O’Rourke, B. A reaction-diffusion model of ROS-induced ROS release in a mitochondrial network. PLoS Comput. Biol 2010, 6, e1000657. [Google Scholar]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol 2008, 4, 278–286. [Google Scholar]
- Hausladen, A.; Fridovich, I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J. Biol. Chem 1994, 269, 29405–29408. [Google Scholar]
- Fridovich, I. Superoxide anion radical (O2 −•), superoxide dismutases, and related matters. J. Biol. Chem 1997, 272, 18515–18517. [Google Scholar]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Kennedy, M.C. Mitochondrial aconitase is a source of hydroxyl radical. An electron spin resonance investigation. J. Biol. Chem 2000, 275, 14064–14069. [Google Scholar]
- James, A.M.; Collins, Y.; Logan, A.; Murphy, M.P. Mitochondrial oxidative stress and the metabolic syndrome. Trends Endocrinol. Metab 2012, 23, 429–434. [Google Scholar]
- Kietzmann, T.; Gorlach, A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin. Cell Dev. Biol 2005, 16, 474–486. [Google Scholar]
- Murphy, M.P. Modulating mitochondrial intracellular location as a redox signal. Sci. Signal. 2012, 5. [Google Scholar] [CrossRef]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar]
- Patten, D.A.; Lafleur, V.N.; Robitaille, G.A.; Chan, D.A.; Giaccia, A.J.; Richard, D.E. Hypoxia-inducible factor-1 activation in nonhypoxic conditions: The essential role of mitochondrial-derived reactive oxygen species. Mol. Biol. Cell 2010, 21, 3247–3257. [Google Scholar]
- Ushio-Fukai, M.; Nakamura, Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett 2008, 266, 37–52. [Google Scholar]
- Tsai, Y.P.; Wu, K.J. Hypoxia-regulated target genes implicated in tumor metastasis. J. Biomed. Sci. 2012, 19. [Google Scholar] [CrossRef]
- Acker, T.; Fandrey, J.; Acker, H. The good, the bad and the ugly in oxygen-sensing: ROS, cytochromes and prolyl-hydroxylases. Cardiovasc. Res 2006, 71, 195–207. [Google Scholar]
- Al-Mehdi, A.B.; Pastukh, V.M.; Swiger, B.M.; Reed, D.J.; Patel, M.R.; Bardwell, G.C.; Pastukh, V.V.; Alexeyev, M.F.; Gillespie, M.N. Perinuclear mitochondrial clustering creates an oxidant-rich nuclear domain required for hypoxia-induced transcription. Sci. Signal. 2012, 5. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol 2007, 39, 44–84. [Google Scholar]
- Alfadda, A.A.; Sallam, R.M. Reactive oxygen species in health and disease. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef]
- Mailloux, R.J.; Harper, M.E. Mitochondrial proticity and ROS signaling: Lessons from the uncoupling proteins. Trends Endocrinol. Metab 2012, 23, 451–458. [Google Scholar]
- Lenaz, G. Mitochondria and reactive oxygen species. Which role in physiology and pathology? Adv. Exp. Med. Biol 2012, 942, 93–136. [Google Scholar]
- Garcia-Bailo, B.; El-Sohemy, A.; Haddad, P.S.; Arora, P.; Benzaied, F.; Karmali, M.; Badawi, A. Vitamins D, C, and E in the prevention of type 2 diabetes mellitus: Modulation of inflammation and oxidative stress. Biologics 2011, 5, 7–19. [Google Scholar]
- Nasti, T.H.; Timares, L. Inflammasome activation of IL-1 family mediators in response to cutaneous photodamage. Photochem. Photobiol 2012, 88, 1111–1125. [Google Scholar]
- Muthusamy, V.; Piva, T.J. The UV response of the skin: A review of the MAPK, NFkappaB and TNFalpha signal transduction pathways. Arch. Dermatol. Res 2010, 302, 5–17. [Google Scholar]
- Diaz, J.H.; Nesbitt, L.T., Jr. Sun exposure behavior and protection: Recommendations for travelers. J. Travel Med 2013, 20, 108–118. [Google Scholar]
- Aroun, A.; Zhong, J.L.; Tyrrell, R.M.; Pourzand, C. Iron, oxidative stress and the example of solar ultraviolet A radiation. Photochem. Photobiol. Sci 2012, 11, 118–134. [Google Scholar] [Green Version]
- Rastogi, R.P.; Richa; Kumar, A.; Tyagi, M.B.; Sinha, R.P. Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair. J. Nucleic Acids 2010, 2010. [Google Scholar] [CrossRef]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact 2006, 160, 1–40. [Google Scholar]
- Akasaka, E.; Takekoshi, S.; Horikoshi, Y.; Toriumi, K.; Ikoma, N.; Mabuchi, T.; Tamiya, S.; Matsuyama, T.; Ozawa, A. Protein oxidative damage and heme oxygenase in sunlight-exposed human skin: Roles of MAPK responses to oxidative stress. Tokai J. Exp. Clin. Med 2010, 35, 152–164. [Google Scholar]
- Hruza, L.L.; Pentland, A.P. Mechanisms of UV-induced inflammation. J. Investig. Dermatol 1993, 100, 35S–41S. [Google Scholar]
- Clydesdale, G.J.; Dandie, G.W.; Muller, H.K. Ultraviolet light induced injury: Immunological and inflammatory effects. Immunol. Cell Biol 2001, 79, 547–568. [Google Scholar]
- Bickers, D.R.; Athar, M. Oxidative stress in the pathogenesis of skin disease. J. Investig. Dermatol 2006, 126, 2565–2575. [Google Scholar]
- Vile, G.F.; Tanew-Ilitschew, A.; Tyrrell, R.M. Activation of NF-kappa B in human skin fibroblasts by the oxidative stress generated by UVA radiation. Photochem. Photobiol 1995, 62, 463–468. [Google Scholar]
- Vile, G.F.; Tyrrell, R.M. UVA radiation-induced oxidative damage to lipids and proteins in vitro and in human skin fibroblasts is dependent on iron and singlet oxygen. Free Radic. Biol. Med 1995, 18, 721–730. [Google Scholar]
- Scharffetter, K.; Wlaschek, M.; Hogg, A.; Bolsen, K.; Schothorst, A.; Goerz, G.; Krieg, T.; Plewig, G. UVA irradiation induces collagenase in human dermal fibroblasts in vitro and in vivo. Arch. Dermatol. Res 1991, 283, 506–511. [Google Scholar]
- Wlaschek, M.; Briviba, K.; Stricklin, G.P.; Sies, H.; Scharffetter-Kochanek, K. Singlet oxygen may mediate the ultraviolet A-induced synthesis of interstitial collagenase. J. Investig. Dermatol 1995, 104, 194–198. [Google Scholar]
- Ryter, S.W.; Tyrrell, R.M. Singlet molecular oxygen ((1)O2): A possible effector of eukaryotic gene expression. Free Radic. Biol. Med 1998, 24, 1520–1534. [Google Scholar]
- Podda, M.; Traber, M.G.; Weber, C.; Yan, L.J.; Packer, L. UV-irradiation depletes antioxidants and causes oxidative damage in a model of human skin. Free Radic. Biol. Med 1998, 24, 55–65. [Google Scholar]
- Armstrong, A.W.; Harskamp, C.T.; Armstrong, E.J. Psoriasis and the risk of diabetes mellitus: A systematic review and meta-analysis. JAMA Dermatol 2013, 149, 84–91. [Google Scholar]
- Azfar, R.S.; Seminara, N.M.; Shin, D.B.; Troxel, A.B.; Margolis, D.J.; Gelfand, J.M. Increased risk of diabetes mellitus and likelihood of receiving diabetes mellitus treatment in patients with psoriasis. Arch. Dermatol 2012, 148, 995–1000. [Google Scholar]
- Cheng, J.; Kuai, D.; Zhang, L.; Yang, X.; Qiu, B. Psoriasis increased the risk of diabetes: A meta-analysis. Arch. Dermatol. Res 2012, 304, 119–125. [Google Scholar]
- Maritim, A.C.; Sanders, R.A.; Watkins, J.B. 3rd Diabetes, oxidative stress, and antioxidants: A review. J. Biochem. Mol. Toxicol 2003, 17, 24–38. [Google Scholar]
- Enamandram, M.; Kimball, A.B. Psoriasis epidemiology: The interplay of genes and the environment. J. Investig. Dermatol 2013, 133, 287–289. [Google Scholar]
- Nickoloff, B.J.; Xin, H.; Nestle, F.O.; Qin, J.Z. The cytokine and chemokine network in psoriasis. Clin. Dermatol 2007, 25, 568–573. [Google Scholar]
- Lowes, M.A.; Bowcock, A.M.; Krueger, J.G. Pathogenesis and therapy of psoriasis. Nature 2007, 445, 866–873. [Google Scholar]
- Zhou, Q.; Mrowietz, U.; Rostami-Yazdi, M. Oxidative stress in the pathogenesis of psoriasis. Free Radic. Biol. Med 2009, 47, 891–905. [Google Scholar]
- Briganti, S.; Picardo, M. Antioxidant activity, lipid peroxidation and skin diseases. What’s new. J. Eur. Acad. Dermatol. Venereol 2003, 17, 663–669. [Google Scholar]
- Racz, E.; Prens, E.P. Molecular pathophysiology of psoriasis and molecular targets of antipsoriatic therapy. Expert Rev. Mol. Med 2009, 11, e38. [Google Scholar]
- Rocha-Pereira, P.; Santos-Silva, A.; Rebelo, I.; Figueiredo, A.; Quintanilha, A.; Teixeira, F. Dislipidemia and oxidative stress in mild and in severe psoriasis as a risk for cardiovascular disease. Clin. Chim. Acta 2001, 303, 33–39. [Google Scholar]
- Tekin, N.S.; Tekin, I.O.; Barut, F.; Sipahi, E.Y. Accumulation of oxidized low-density lipoprotein in psoriatic skin and changes of plasma lipid levels in psoriatic patients. Mediators Inflamm 2007, 2007, 78454. [Google Scholar]
- Simonetti, O.; Ferretti, G.; Salvi, A.; Offidani, A.M.; Bossi, G. Plasma lipid changes in psoriatic children. Dermatology 1992, 185, 96–100. [Google Scholar]
- Kokcam, I.; Naziroglu, M. Antioxidants and lipid peroxidation status in the blood of patients with psoriasis. Clin. Chim. Acta 1999, 289, 23–31. [Google Scholar]
- Drewa, G.; Krzyzynska-Malinowska, E.; Wozniak, A.; Protas-Drozd, F.; Mila-Kierzenkowska, C.; Rozwodowska, M.; Kowaliszyn, B.; Czajkowski, R. Activity of superoxide dismutase and catalase and the level of lipid peroxidation products reactive with TBA in patients with psoriasis. Med. Sci. Monit. 2002, 8, BR338–BR343. [Google Scholar]
- Yildirim, M.; Inaloz, H.S.; Baysal, V.; Delibas, N. The role of oxidants and antioxidants in psoriasis. J. Eur. Acad. Dermatol. Venereol 2003, 17, 34–36. [Google Scholar]
- Therond, P.; Gerbaud, P.; Dimon, S.; Anderson, W.B.; Evain-Broin, D.; Raynaud, F. Antioxidant enzymes in psoriatic fibroblasts and erythrocytes. J. Investig. Dermatol 1996, 106, 1325–1328. [Google Scholar]
- Frank, S.; Munz, B.; Werner, S. The human homologue of a bovine non-selenium glutathione peroxidase is a novel keratinocyte growth factor-regulated gene. Oncogene 1997, 14, 915–921. [Google Scholar]
- Ryu, J.; Park, S.G.; Park, B.C.; Choe, M.; Lee, K.S.; Cho, J.W. Proteomic analysis of psoriatic skin tissue for identification of differentially expressed proteins: Up-regulation of GSTP1, SFN and PRDX2 in psoriatic skin. Int. J. Mol. Med 2011, 28, 785–792. [Google Scholar]
- Armstrong, A.W.; Voyles, S.V.; Armstrong, E.J.; Fuller, E.N.; Rutledge, J.C. Angiogenesis and oxidative stress: Common mechanisms linking psoriasis with atherosclerosis. J. Dermatol. Sci 2011, 63, 1–9. [Google Scholar]
- Takahashi, H.; Ibe, M.; Nakamura, S.; Ishida-Yamamoto, A.; Hashimoto, Y.; Iizuka, H. Extracellular regulated kinase and c-Jun N-terminal kinase are activated in psoriatic involved epidermis. J. Dermatol. Sci 2002, 30, 94–99. [Google Scholar]
- Johansen, C.; Flindt, E.; Kragballe, K.; Henningsen, J.; Westergaard, M.; Kristiansen, K.; Iversen, L. Inverse regulation of the nuclear factor-kappaB binding to the p53 and interleukin-8 kappaB response elements in lesional psoriatic skin. J. Investig. Dermatol 2005, 124, 1284–1292. [Google Scholar]
- Yu, X.J.; Li, C.Y.; Dai, H.Y.; Cai, D.X.; Wang, K.Y.; Xu, Y.H.; Chen, L.M.; Zhou, C.L. Expression and localization of the activated mitogen-activated protein kinase in lesional psoriatic skin. Exp. Mol. Pathol 2007, 83, 413–418. [Google Scholar]
- Abdou, A.G.; Hanout, H.M. Evaluation of survivin and NF-kappaB in psoriasis, an immunohistochemical study. J. Cutan. Pathol 2008, 35, 445–451. [Google Scholar]
- Lizzul, P.F.; Aphale, A.; Malaviya, R.; Sun, Y.; Masud, S.; Dombrovskiy, V.; Gottlieb, A.B. Differential expression of phosphorylated NF-kappaB/RelA in normal and psoriatic epidermis and downregulation of NF-kappaB in response to treatment with etanercept. J. Investig. Dermatol 2005, 124, 1275–1283. [Google Scholar]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1. [Google Scholar] [CrossRef]
- Treumer, F.; Zhu, K.; Glaser, R.; Mrowietz, U. Dimethylfumarate is a potent inducer of apoptosis in human T cells. J. Investig. Dermatol 2003, 121, 1383–1388. [Google Scholar]
- Amigo, M.; Paya, M.; De Rosa, S.; Terencio, M.C. Antipsoriatic effects of avarol-3′-thiosalicylate are mediated by inhibition of TNF-alpha generation and NF-kappaB activation in mouse skin. Br. J. Pharmacol 2007, 152, 353–365. [Google Scholar]
- Amigo, M.; Schalkwijk, J.; Olthuis, D.; De Rosa, S.; Paya, M.; Terencio, M.C.; Lamme, E. Identification of avarol derivatives as potential antipsoriatic drugs using an in vitro model for keratinocyte growth and differentiation. Life Sci 2006, 79, 2395–2404. [Google Scholar]
- Teunissen, M.B.; Koomen, C.W.; de Waal Malefyt, R.; Wierenga, E.A.; Bos, J.D. Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J. Investig. Dermatol 1998, 111, 645–649. [Google Scholar]
- Bito, T.; Nishigori, C. Impact of reactive oxygen species on keratinocyte signaling pathways. J. Dermatol. Sci 2012, 68, 3–8. [Google Scholar]
- Kao, C.C.; Garner, W.L. Acute burns. Plast. Reconstr. Surg 2000, 101, 2482–2493. [Google Scholar]
- Latha, B.; Babu, M. The involvement of free radicals in burn injury: A review. Burns 2001, 27, 309–317. [Google Scholar]
- Kupper, T.S.; Deitch, E.A.; Baker, C.C.; Wong, W.C. The human burn wound as a primary source of interleukin-1 activity. Surgery 1986, 100, 409–415. [Google Scholar]
- Ward, P.A.; Till, G.O. Pathophysiologic events related to thermal injury of skin. J. Trauma 1990, 30, S75–S79. [Google Scholar]
- Till, G.O.; Beauchamp, C.; Menapace, D.; Tourtellotte, W., Jr; Kunkel, R.; Johnson, K.J.; Ward, P.A. Oxygen radical dependent lung damage following thermal injury of rat skin. J. Trauma 1983, 23, 269–277. [Google Scholar]
- Till, G.O.; Hatherill, J.R.; Tourtellotte, W.W.; Lutz, M.J.; Ward, P.A. Lipid peroxidation and acute lung injury after thermal trauma to skin. Evidence of a role for hydroxyl radical. Am. J. Pathol 1985, 119, 376–384. [Google Scholar]
- Gurbuz, V.; Corak, A.; Yegen, B.C.; Kurtel, H.; Alican, I. Oxidative organ damage in a rat model of thermal injury: The effect of cyclosporin A. Burns 1997, 23, 37–42. [Google Scholar]
- Jahovic, N.; Guzel, E.; Arbak, S.; Yegen, B.C. The healing-promoting effect of saliva on skin burn is mediated by epidermal growth factor (EGF): Role of the neutrophils. Burns 2004, 30, 531–538. [Google Scholar]
- Cetinkale, O.; Konukoglu, D.; Senel, O.; Kemerli, G.D.; Yazar, S. Modulating the functions of neutrophils and lipid peroxidation by FK506 in a rat model of thermal injury. Burns 1999, 25, 105–112. [Google Scholar]
- Haycock, J.W.; Ralston, D.R.; Morris, B.; Freedlander, E.; MacNeil, S. Oxidative damage to protein and alterations to antioxidant levels in human cutaneous thermal injury. Burns 1997, 23, 533–540. [Google Scholar]
- Tredget, E.E.; Yu, Y.M. The metabolic effects of thermal injury. World J. Surg 1992, 16, 68–79. [Google Scholar]
- Bertin-Maghit, M.; Goudable, J.; Dalmas, E.; Steghens, J.P.; Bouchard, C.; Gueugniaud, P.Y.; Petit, P.; Delafosse, B. Time course of oxidative stress after major burns. Intensive Care Med 2000, 26, 800–803. [Google Scholar]
- Horton, J.W. Free radicals and lipid peroxidation mediated injury in burn trauma: The role of antioxidant therapy. Toxicology 2003, 189, 75–88. [Google Scholar]
- Demling, R.; Ikegami, K.; Lalonde, C. Increased lipid peroxidation and decreased antioxidant activity correspond with death after smoke exposure in the rat. J. Burn Care Rehabil 1995, 16, 104–110. [Google Scholar]
- Hosnuter, M.; Gurel, A.; Babuccu, O.; Armutcu, F.; Kargi, E.; Isikdemir, A. The effect of CAPE on lipid peroxidation and nitric oxide levels in the plasma of rats following thermal injury. Burns 2004, 30, 121–125. [Google Scholar]
- Saez, J.C.; Ward, P.H.; Gunther, B.; Vivaldi, E. Superoxide radical involvement in the pathogenesis of burn shock. Circ. Shock 1984, 12, 229–239. [Google Scholar]
- Oldham, K.T.; Guice, K.S.; Till, G.O.; Ward, P.A. Activation of complement by hydroxyl radical in thermal injury. Surgery 1988, 104, 272–279. [Google Scholar]
- Sener, G.; Sehirli, A.O.; Satiroglu, H.; Keyer-Uysal, M.; Yegen, B.C. Melatonin improves oxidative organ damage in a rat model of thermal injury. Burns 2002, 28, 419–425. [Google Scholar]
- Saitoh, D.; Ookawara, T.; Fukuzuka, K.; Kawakami, M.; Sakamoto, T.; Ohno, H.; Okada, Y. Characteristics of plasma extracellular SOD in burned patients. Burns 2001, 27, 577–581. [Google Scholar]
- Saitoh, D.; Shirani, K.Z.; Cioffi, W.G.; Kizaki, T.; Ohno, H.; Okada, Y.; Mason, A.D., Jr; Pruitt, B.A., Jr. Changes in the tissue and plasma superoxide dismutase (SOD) levels in a burned rat model. Tohoku J. Exp. Med 2001, 193, 27–36. [Google Scholar]
- Cetinkale, O.; Belce, A.; Konukoglu, D.; Senyuva, C.; Gumustas, M.K.; Tas, T. Evaluation of lipid peroxidation and total antioxidant status in plasma of rats following thermal injury. Burns 1997, 23, 114–116. [Google Scholar]
- Steiling, H.; Munz, B.; Werner, S.; Brauchle, M. Different types of ROS-scavenging enzymes are expressed during cutaneous wound repair. Exp. Cell Res 1999, 247, 484–494. [Google Scholar]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar]
- Agarwal, A.; Banerjee, A.; Banerjee, U.C. Xanthine oxidoreductase: A journey from purine metabolism to cardiovascular excitation-contraction coupling. Crit. Rev. Biotechnol 2011, 31, 264–280. [Google Scholar]
- Zhang, R.; Brennan, M.L.; Shen, Z.; MacPherson, J.C.; Schmitt, D.; Molenda, C.E.; Hazen, S.L. Myeloperoxidase functions as a major enzymatic catalyst for initiation of lipid peroxidation at sites of inflammation. J. Biol. Chem 2002, 277, 46116–46122. [Google Scholar]
- Fleming, I.; Michaelis, U.R.; Bredenkotter, D.; Fisslthaler, B.; Dehghani, F.; Brandes, R.P.; Busse, R. Endothelium-derived hyperpolarizing factor synthase (Cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ. Res 2001, 88, 44–51. [Google Scholar]
- Tabima, D.M.; Frizzell, S.; Gladwin, M.T. Reactive oxygen and nitrogen species in pulmonary hypertension. Free Radic. Biol. Med 2012, 52, 1970–1986. [Google Scholar]
- Andrew, P.J.; Mayer, B. Enzymatic function of nitric oxide synthases. Cardiovasc. Res 1999, 43, 521–531. [Google Scholar]
- Xia, Y.; Tsai, A.L.; Berka, V.; Zweier, J.L. Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J. Biol. Chem 1998, 273, 25804–25808. [Google Scholar]
- Roe, N.D.; Ren, J. Nitric oxide synthase uncoupling: A therapeutic target in cardiovascular diseases. Vasc. Pharmacol 2012, 57, 168–172. [Google Scholar]
- Altenhofer, S.; Kleikers, P.W.; Radermacher, K.A.; Scheurer, P.; Rob Hermans, J.J.; Schiffers, P.; Ho, H.; Wingler, K.; Schmidt, H.H. The NOX toolbox: Validating the role of NADPH oxidases in physiology and disease. Cell. Mol. Life Sci 2012, 69, 2327–2343. [Google Scholar]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Radic. Biol. Med 2009, 47, 1239–1253. [Google Scholar]
- Takac, I.; Schroder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem 2011, 286, 13304–13313. [Google Scholar]
- Jiang, F.; Zhang, Y.; Dusting, G.J. NADPH oxidase-mediated redox signaling: Roles in cellular stress response, stress tolerance, and tissue repair. Pharmacol. Rev 2011, 63, 218–242. [Google Scholar]
- Lambeth, J.D.; Kawahara, T.; Diebold, B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med 2007, 43, 319–331. [Google Scholar]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev 2007, 87, 245–313. [Google Scholar]
- Chamulitrat, W.; Stremmel, W.; Kawahara, T.; Rokutan, K.; Fujii, H.; Wingler, K.; Schmidt, H.H.; Schmidt, R. A constitutive NADPH oxidase-like system containing gp91phox homologs in human keratinocytes. J. Investig. Dermatol 2004, 122, 1000–1009. [Google Scholar]
- Sen, C.K.; Khanna, S.; Babior, B.M.; Hunt, T.K.; Ellison, E.C.; Roy, S. Oxidant-induced vascular endothelial growth factor expression in human keratinocytes and cutaneous wound healing. J. Biol. Chem 2002, 277, 33284–33290. [Google Scholar]
- Chiu, C.; Maddock, D.A.; Zhang, Q.; Souza, K.P.; Townsend, A.R.; Wan, Y. TGF-beta-induced p38 activation is mediated by Rac1-regulated generation of reactive oxygen species in cultured human keratinocytes. Int. J. Mol. Med 2001, 8, 251–255. [Google Scholar]
- Chamulitrat, W.; Schmidt, R.; Tomakidi, P.; Stremmel, W.; Chunglok, W.; Kawahara, T.; Rokutan, K. Association of gp91phox homolog Nox1 with anchorage-independent growth and MAP kinase-activation of transformed human keratinocytes. Oncogene 2003, 22, 6045–6053. [Google Scholar]
- Steinbeck, M.J.; Khan, A.U.; Karnovsky, M.J. Extracellular production of singlet oxygen by stimulated macrophages quantified using 9,10-diphenylanthracene and perylene in a polystyrene film. J. Biol. Chem 1993, 268, 15649–15654. [Google Scholar]
- Tsiftsoglou, A.S.; Tsamadou, A.I.; Papadopoulou, L.C. Heme as key regulator of major mammalian cellular functions: Molecular, cellular, and pharmacological aspects. Pharmacol. Ther 2006, 111, 327–345. [Google Scholar]
- Wagener, F.A.; Volk, H.D.; Willis, D.; Abraham, N.G.; Soares, M.P.; Adema, G.J.; Figdor, C.G. Different faces of the heme-heme oxygenase system in inflammation. Pharmacol. Rev 2003, 55, 551–571. [Google Scholar]
- Ryter, S.W.; Tyrrell, R.M. The heme synthesis and degradation pathways: Role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med 2000, 28, 289–309. [Google Scholar]
- Halliwell, B.; Gutteridge, J.M. Oxygen toxicity, oxygen radicals, transition metals and disease. Biochem. J 1984, 219, 1–14. [Google Scholar]
- Kumar, S.; Bandyopadhyay, U. Free heme toxicity and its detoxification systems in human. Toxicol. Lett 2005, 157, 175–188. [Google Scholar]
- Thomas, D.D.; Espey, M.G.; Vitek, M.P.; Miranda, K.M.; Wink, D.A. Protein nitration is mediated by heme and free metals through Fenton-type chemistry: An alternative to the NO/O2– reaction. Proc. Natl. Acad. Sci. USA 2002, 99, 12691–12696. [Google Scholar]
- Jeney, V.; Balla, J.; Yachie, A.; Varga, Z.; Vercellotti, G.M.; Eaton, J.W.; Balla, G. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002, 100, 879–887. [Google Scholar]
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercellotti, G.M. Hemin: A possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arterioscler. Thromb 1991, 11, 1700–1711. [Google Scholar]
- Balla, J.; Jacob, H.S.; Balla, G.; Nath, K.; Eaton, J.W.; Vercellotti, G.M. Endothelial-cell heme uptake from heme proteins: Induction of sensitization and desensitization to oxidant damage. Proc. Natl. Acad. Sci. USA 1993, 90, 9285–9289. [Google Scholar]
- Wagener, F.A.; Abraham, N.G.; van, K.Y.; de, W.T.; Figdor, C.G. Heme-induced cell adhesion in the pathogenesis of sickle-cell disease and inflammation. Trends Pharmacol. Sci 2001, 22, 52–54. [Google Scholar]
- Wagener, F.A.; da Silva, J.L.; Farley, T.; de, W.T.; Kappas, A.; Abraham, N.G. Differential effects of heme oxygenase isoforms on heme mediation of endothelial intracellular adhesion molecule 1 expression. J. Pharmacol. Exp. Ther 1999, 291, 416–423. [Google Scholar]
- Wagener, F.A.; Feldman, E.; de, W.T.; Abraham, N.G. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc. Soc. Exp. Biol. Med 1997, 216, 456–463. [Google Scholar]
- Tolosano, E.; Fagoonee, S.; Hirsch, E.; Berger, F.G.; Baumann, H.; Silengo, L.; Altruda, F. Enhanced splenomegaly and severe liver inflammation in haptoglobin/hemopexin double-null mice after acute hemolysis. Blood 2002, 100, 4201–4208. [Google Scholar]
- Ma, J.L.; Yang, P.Y.; Rui, Y.C.; Lu, L.; Kang, H.; Zhang, J. Hemin modulates cytokine expressions in macrophage-derived foam cells via heme oxygenase-1 induction. J. Pharmacol. Sci 2007, 103, 261–266. [Google Scholar]
- Cambos, M.; Bazinet, S.; Abed, E.; Sanchez-Dardon, J.; Bernard, C.; Moreau, R.; Olivier, M.; Scorza, T. The IL-12p70/IL-10 interplay is differentially regulated by free heme and hemozoin in murine bone-marrow-derived macrophages. Int J. Parasitol 2010, 40, 1003–1012. [Google Scholar]
- Cambos, M.; Scorza, T. Robust erythrophagocytosis leads to macrophage apoptosis via a hemin-mediated redox imbalance: Role in hemolytic disorders. J. Leukoc. Biol 2011, 89, 159–171. [Google Scholar]
- Wagener, F.A.; van Beurden, H.E.; von den Hoff, J.W.; Adema, G.J.; Figdor, C.G. The heme-heme oxygenase system: A molecular switch in wound healing. Blood 2003, 102, 521–528. [Google Scholar]
- Harris, E.D. Regulation of antioxidant enzymes. FASEB J 1992, 6, 2675–2683. [Google Scholar]
- Yoshikawa, S.; Muramoto, K.; Shinzawa-Itoh, K. The O(2) reduction and proton pumping gate mechanism of bovine heart cytochrome c oxidase. Biochim. Biophys. Acta 2011, 1807, 1279–1286. [Google Scholar]
- Martinez-Finley, E.J.; Chakraborty, S.; Fretham, S.J.; Aschner, M. Cellular transport and homeostasis of essential and nonessential metals. Metallomics 2012, 4, 593–605. [Google Scholar]
- Jomova, K.; Valko, M. Advances in metal-induced oxidative stress and human disease. Toxicology 2011, 283, 65–87. [Google Scholar]
- Ratnam, D.V.; Ankola, D.D.; Bhardwaj, V.; Sahana, D.K.; Kumar, M.N. Role of antioxidants in prophylaxis and therapy: A pharmaceutical perspective. J. Control. Release 2006, 113, 189–207. [Google Scholar]
- Packer, L.; Valacchi, G. Antioxidants and the response of skin to oxidative stress: Vitamin E as a key indicator. Skin Pharmacol. Appl. Skin Physiol 2002, 15, 282–290. [Google Scholar]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med 2002, 33, 337–349. [Google Scholar]
- Zamocky, M.; Jakopitsch, C.; Furtmuller, P.G.; Dunand, C.; Obinger, C. The peroxidase-cyclooxygenase superfamily: Reconstructed evolution of critical enzymes of the innate immune system. Proteins 2008, 72, 589–605. [Google Scholar]
- Brigelius-Flohe, R. Glutathione peroxidases and redox-regulated transcription factors. Biol. Chem 2006, 387, 1329–1335. [Google Scholar]
- Arthur, J.R. The glutathione peroxidases. Cell. Mol. Life Sci 2000, 57, 1825–1835. [Google Scholar]
- De Haan, J.B.; Bladier, C.; Griffiths, P.; Kelner, M.; O’Shea, R.D.; Cheung, N.S.; Bronson, R.T.; Silvestro, M.J.; Wild, S.; Zheng, S.S.; et al. Mice with a homozygous null mutation for the most abundant glutathione peroxidase, Gpx1, show increased susceptibility to the oxidative stress-inducing agents paraquat and hydrogen peroxide. J. Biol. Chem 1998, 273, 22528–22536. [Google Scholar]
- Auf dem Keller, U.; Kumin, A.; Braun, S.; Werner, S. Reactive oxygen species and their detoxification in healing skin wounds. J. Investig. Dermatol. Symp. Proc 2006, 11, 106–111. [Google Scholar]
- De Haan, J.B.; Crack, P.J.; Flentjar, N.; Iannello, R.C.; Hertzog, P.J.; Kola, I. An imbalance in antioxidant defense affects cellular function: The pathophysiological consequences of a reduction in antioxidant defense in the glutathione peroxidase-1 (Gpx1) knockout mouse. Redox Rep 2003, 8, 69–79. [Google Scholar]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins-molecular mechanisms and health significance: From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 2013. [Google Scholar] [CrossRef]
- Manevich, Y.; Sweitzer, T.; Pak, J.H.; Feinstein, S.I.; Muzykantov, V.; Fisher, A.B. 1-Cys peroxiredoxin overexpression protects cells against phospholipid peroxidation-mediated membrane damage. Proc. Natl. Acad. Sci. USA 2002, 99, 11599–11604. [Google Scholar]
- Wang, Y.; Manevich, Y.; Feinstein, S.I.; Fisher, A.B. Adenovirus-mediated transfer of the 1-cys peroxiredoxin gene to mouse lung protects against hyperoxic injury. Am. J. Physiol. Lung Cell. Mol. Physiol 2004, 286, L1188–L1193. [Google Scholar]
- Kumin, A.; Huber, C.; Rulicke, T.; Wolf, E.; Werner, S. Peroxiredoxin 6 is a potent cytoprotective enzyme in the epidermis. Am. J. Pathol 2006, 169, 1194–1205. [Google Scholar]
- Pak, J.H.; Manevich, Y.; Kim, H.S.; Feinstein, S.I.; Fisher, A.B. An antisense oligonucleotide to 1-cys peroxiredoxin causes lipid peroxidation and apoptosis in lung epithelial cells. J. Biol. Chem 2002, 277, 49927–49934. [Google Scholar]
- Mo, Y.; Feinstein, S.I.; Manevich, Y.; Zhang, Q.; Lu, L.; Ho, Y.S.; Fisher, A.B. 1-Cys peroxiredoxin knock-out mice express mRNA but not protein for a highly related intronless gene. FEBS Lett 2003, 555, 192–198. [Google Scholar]
- Wang, X.; Phelan, S.A.; Forsman-Semb, K.; Taylor, E.F.; Petros, C.; Brown, A.; Lerner, C.P.; Paigen, B. Mice with targeted mutation of peroxiredoxin 6 develop normally but are susceptible to oxidative stress. J. Biol. Chem 2003, 278, 25179–25190. [Google Scholar]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal 2008, 10, 179–206. [Google Scholar]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr 2004, 134, 489–492. [Google Scholar]
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012, 64, 362–369. [Google Scholar]
- Kletzien, R.F.; Harris, P.K.; Foellmi, L.A. Glucose-6-phosphate dehydrogenase: A “housekeeping” enzyme subject to tissue-specific regulation by hormones, nutrients, and oxidant stress. FASEB J 1994, 8, 174–181. [Google Scholar]
- Pandolfi, P.P.; Sonati, F.; Rivi, R.; Mason, P.; Grosveld, F.; Luzzatto, L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J 1995, 14, 5209–5215. [Google Scholar]
- Rosenstraus, M.; Chasin, L.A. Isolation of mammalian cell mutants deficient in glucose-6-phosphate dehydrogenase activity: Linkage to hypoxanthine phosphoribosyl transferase. Proc. Natl. Acad. Sci. USA 1975, 72, 493–497. [Google Scholar]
- Serpillon, S.; Floyd, B.C.; Gupte, R.S.; George, S.; Kozicky, M.; Neito, V.; Recchia, F.; Stanley, W.; Wolin, M.S.; Gupte, S.A. Superoxide production by NAD(P)H oxidase and mitochondria is increased in genetically obese and hyperglycemic rat heart and aorta before the development of cardiac dysfunction. The role of glucose-6-phosphate dehydrogenase-derived NADPH. Am. J. Physiol. Heart .Circ. Physiol 2009, 297, H153–H162. [Google Scholar]
- Haines, D.D.; Lekli, I.; Teissier, P.; Bak, I.; Tosaki, A. Role of haeme oxygenase-1 in resolution of oxidative stress-related pathologies: Focus on cardiovascular, lung, neurological and kidney disorders. Acta Physiol 2012, 204, 487–501. [Google Scholar]
- Abraham, N.G.; Kappas, A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev 2008, 60, 79–127. [Google Scholar]
- Maines, M.D. Heme oxygenase: Function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J 1988, 2, 2557–2568. [Google Scholar]
- Tenhunen, R.; Marver, H.S.; Schmid, R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA 1968, 61, 748–755. [Google Scholar]
- Wagener, F.A.; Scharstuhl, A.; Tyrrell, R.M.; Von den Hoff, J.W.; Jozkowicz, A.; Dulak, J.; Russel, F.G.; Kuijpers-Jagtman, A.M. The heme-heme oxygenase system in wound healing; Implications for scar formation. Curr. Drug Targets 2010, 11, 1571–1585. [Google Scholar]
- Lundvig, D.M.; Immenschuh, S.; Wagener, F.A. Heme oxygenase, inflammation, and fibrosis: The good, the bad, and the ugly? Front. Pharmacol 2012, 3, 81. [Google Scholar]
- Grochot-Przeczek, A.; Dulak, J.; Jozkowicz, A. Haem oxygenase-1: Non-canonical roles in physiology and pathology. Clin. Sci 2012, 122, 93–103. [Google Scholar]
- Alam, J.; Shibahara, S.; Smith, A. Transcriptional activation of the heme oxygenase gene by heme and cadmium in mouse hepatoma cells. J. Biol. Chem 1989, 264, 6371–6375. [Google Scholar]
- Applegate, L.A.; Noel, A.; Vile, G.; Frenk, E.; Tyrrell, R.M. Two genes contribute to different extents to the heme oxygenase enzyme activity measured in cultured human skin fibroblasts and keratinocytes: Implications for protection against oxidant stress. Photochem. Photobiol 1995, 61, 285–291. [Google Scholar]
- Cisowski, J.; Loboda, A.; Jozkowicz, A.; Chen, S.; Agarwal, A.; Dulak, J. Role of heme oxygenase-1 in hydrogen peroxide-induced VEGF synthesis: Effect of HO-1 knockout. Biochem. Biophys. Res. Commun 2005, 326, 670–676. [Google Scholar]
- Tosaki, A.; Das, D.K. The role of heme oxygenase signaling in various disorders. Mol. Cell. Biochem 2002, 232, 149–157. [Google Scholar]
- Tyrrell, R.M. Solar ultraviolet A radiation: An oxidizing skin carcinogen that activates heme oxygenase-1. Antioxid. Redox Signal 2004, 6, 835–840. [Google Scholar]
- Wagener, F.A.; Eggert, A.; Boerman, O.C.; Oyen, W.J.; Verhofstad, A.; Abraham, N.G.; Adema, G.; van Kooyk, Y.; de Witte, T.; Figdor, C.G. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood 2001, 98, 1802–1811. [Google Scholar]
- Wagener, F.A.; Dankers, A.C.; van Summeren, F.; Scharstuhl, A.; van den Heuvel, J.J.; Koenderink, J.B.; Pennings, S.W.; Russel, F.G.; Masereeuw, R. Heme oxygenase-1 and breast cancer resistance protein protect against heme-induced toxicity. Curr. Pharm. Des 2013, 19, 2698–2707. [Google Scholar]
- Hayashi, S.; Takamiya, R.; Yamaguchi, T.; Matsumoto, K.; Tojo, S.J.; Tamatani, T.; Kitajima, M.; Makino, N.; Ishimura, Y.; Suematsu, M. Induction of heme oxygenase-1 suppresses venular leukocyte adhesion elicited by oxidative stress: Role of bilirubin generated by the enzyme. Circ. Res 1999, 85, 663–671. [Google Scholar]
- Vachharajani, T.J.; Work, J.; Issekutz, A.C.; Granger, D.N. Heme oxygenase modulates selectin expression in different regional vascular beds. Am. J. Physiol 2000, 278, H1613–H1617. [Google Scholar]
- Takahashi, T.; Shimizu, H.; Morimatsu, H.; Inoue, K.; Akagi, R.; Morita, K.; Sassa, S. Heme oxygenase-1: A fundamental guardian against oxidative tissue injuries in acute inflammation. Mini Rev. Med. Chem 2007, 7, 745–753. [Google Scholar]
- Takahashi, T.; Shimizu, H.; Morimatsu, H.; Maeshima, K.; Inoue, K.; Akagi, R.; Matsumi, M.; Katayama, H.; Morita, K. Heme oxygenase-1 is an essential cytoprotective component in oxidative tissue injury induced by hemorrhagic shock. J. Clin. Biochem. Nutr 2009, 44, 28–40. [Google Scholar]
- Baranano, D.E.; Rao, M.; Ferris, C.D.; Snyder, S.H. Biliverdin reductase: A major physiologic cytoprotectant. Proc. Natl. Acad. Sci. USA 2002, 99, 16093–16098. [Google Scholar]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar]
- Kim, S.Y.; Park, S.C. Physiological antioxidative network of the bilirubin system in aging and age-related diseases. Front. Pharmacol 2012, 3, 45. [Google Scholar]
- Dore, S.; Snyder, S.H. Neuroprotective action of bilirubin against oxidative stress in primary hippocampal cultures. Ann. N. Y. Acad. Sci 1999, 890, 167–172. [Google Scholar]
- Dore, S.; Takahashi, M.; Ferris, C.D.; Zakhary, R.; Hester, L.D.; Guastella, D.; Snyder, S.H. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. USA 1999, 96, 2445–2450. [Google Scholar]
- Lanone, S.; Bloc, S.; Foresti, R.; Almolki, A.; Taille, C.; Callebert, J.; Conti, M.; Goven, D.; Aubier, M.; Dureuil, B.; et al. Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: Implications for protection against endotoxic shock in rats. FASEB J 2005, 19, 1890–1892. [Google Scholar]
- Jiang, F.; Roberts, S.J.; Datla, S.; Dusting, G.J. NO modulates NADPH oxidase function via heme oxygenase-1 in human endothelial cells. Hypertension 2006, 48, 950–957. [Google Scholar]
- Matsumoto, H.; Ishikawa, K.; Itabe, H.; Maruyama, Y. Carbon monoxide and bilirubin from heme oxygenase-1 suppresses reactive oxygen species generation and plasminogen activator inhibitor-1 induction. Mol. Cell. Biochem 2006, 291, 21–28. [Google Scholar]
- McDonagh, A.F. The biliverdin-bilirubin antioxidant cycle of cellular protection: Missing a wheel? Free Radic. Biol. Med 2010, 49, 814–820. [Google Scholar]
- Jomova, K.; Valko, M. Importance of iron chelation in free radical-induced oxidative stress and human disease. Curr. Pharm. Des 2011, 17, 3460–3473. [Google Scholar]
- Harrison, P.M.; Arosio, P. The ferritins: Molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta 1996, 1275, 161–203. [Google Scholar]
- Torti, F.M.; Torti, S.V. Regulation of ferritin genes and protein. Blood 2002, 99, 3505–3516. [Google Scholar]
- Orino, K.; Watanabe, K. Molecular, physiological and clinical aspects of the iron storage protein ferritin. Vet. J 2008, 178, 191–201. [Google Scholar]
- Villacorta, L.; Azzi, A.; Zingg, J.M. Regulatory role of vitamins E and C on extracellular matrix components of the vascular system. Mol. Asp. Med 2007, 28, 507–537. [Google Scholar]
- Fan, J.; Frey, R.S.; Rahman, A.; Malik, A.B. Role of neutrophil NADPH oxidase in the mechanism of tumor necrosis factor-alpha -induced NF-kappa B activation and intercellular adhesion molecule-1 expression in endothelial cells. J. Biol. Chem 2002, 277, 3404–3411. [Google Scholar]
- Kono, H.; Rusyn, I.; Yin, M.; Gabele, E.; Yamashina, S.; Dikalova, A.; Kadiiska, M.B.; Connor, H.D.; Mason, R.P.; Segal, B.H.; et al. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J. Clin. Investig 2000, 106, 867–872. [Google Scholar]
- Sadikot, R.T.; Zeng, H.; Yull, F.E.; Li, B.; Cheng, D.S.; Kernodle, D.S.; Jansen, E.D.; Contag, C.H.; Segal, B.H.; Holland, S.M.; et al. p47phox deficiency impairs NF-kappa B activation and host defense in Pseudomonas pneumonia. J. Immunol 2004, 172, 1801–1808. [Google Scholar]
- Sato, K.; Kadiiska, M.B.; Ghio, A.J.; Corbett, J.; Fann, Y.C.; Holland, S.M.; Thurman, R.G.; Mason, R.P. In vivo lipid-derived free radical formation by NADPH oxidase in acute lung injury induced by lipopolysaccharide: A model for ARDS. FASEB J 2002, 16, 1713–1720. [Google Scholar]
- Rahman, A.; Kefer, J.; Bando, M.; Niles, W.D.; Malik, A.B. E-selectin expression in human endothelial cells by TNF-alpha-induced oxidant generation and NF-kappaB activation. Am. J. Physiol 1998, 275, L533–L544. [Google Scholar]
- Li, J.M.; Fan, L.M.; Christie, M.R.; Shah, A.M. Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascular endothelial cells: Role of p47phox phosphorylation and binding to TRAF4. Mol. Cell. Biol 2005, 25, 2320–2330. [Google Scholar]
- Gu, Y.; Xu, Y.C.; Wu, R.F.; Souza, R.F.; Nwariaku, F.E.; Terada, L.S. TNFalpha activates c-Jun amino terminal kinase through p47(phox). Exp. Cell Res 2002, 272, 62–74. [Google Scholar]
- Lambeth, J.D.; Krause, K.H.; Clark, R.A. NOX enzymes as novel targets for drug development. Semin. Immunopathol 2008, 30, 339–363. [Google Scholar]
- Jaquet, V.; Scapozza, L.; Clark, R.A.; Krause, K.H.; Lambeth, J.D. Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid. Redox Signal 2009, 11, 2535–2552. [Google Scholar]
- Zhang, W.J.; Wei, H.; Frei, B. Genetic deficiency of NADPH oxidase does not diminish, but rather enhances, LPS-induced acute inflammatory responses in vivo. Free Radic. Biol. Med 2009, 46, 791–798. [Google Scholar]
- Marriott, H.M.; Jackson, L.E.; Wilkinson, T.S.; Simpson, A.J.; Mitchell, T.J.; Buttle, D.J.; Cross, S.S.; Ince, P.G.; Hellewell, P.G.; Whyte, M.K.; et al. Reactive oxygen species regulate neutrophil recruitment and survival in pneumococcal pneumonia. Am. J. Respir. Crit. Care Med 2008, 177, 887–895. [Google Scholar]
- Liochev, S.I.; Fridovich, I. The role of O2.– in the production of HO.: In vitro and in vivo. Free Radic. Biol. Med 1994, 16, 29–33. [Google Scholar]
- Kakhlon, O.; Cabantchik, Z.I. The labile iron pool: Characterization, measurement, and participation in cellular processes(1). Free Radic. Biol. Med 2002, 33, 1037–1046. [Google Scholar]
- Fleming, R.E.; Ponka, P. Iron overload in human disease. N. Engl. J. Med 2012, 366, 348–359. [Google Scholar]
- Larsen, R.; Gouveia, Z.; Soares, M.P.; Gozzelino, R. Heme cytotoxicity and the pathogenesis of immune-mediated inflammatory diseases. Front. Pharmacol 2012, 3, 77. [Google Scholar]
- Kell, D.B. Towards a unifying, systems biology understanding of large-scale cellular death and destruction caused by poorly liganded iron: Parkinson’s, Huntington’s, Alzheimer’s, prions, bactericides, chemical toxicology and others as examples. Arch. Toxicol 2010, 84, 825–889. [Google Scholar]
- Perron, N.R.; Brumaghim, J.L. A review of the antioxidant mechanisms of polyphenol compounds related to iron binding. Cell Biochem. Biophys 2009, 53, 75–100. [Google Scholar]
- Sestili, P.; Guidarelli, A.; Dacha, M.; Cantoni, O. Quercetin prevents DNA single strand breakage and cytotoxicity caused by tert-butylhydroperoxide: Free radical scavenging versus iron chelating mechanism. Free Radic. Biol. Med 1998, 25, 196–200. [Google Scholar]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal 2011, 15, 1583–1606. [Google Scholar]
- Marklund, S.L. Extracellular superoxide dismutase and other superoxide dismutase isoenzymes in tissues from nine mammalian species. Biochem. J 1984, 222, 649–655. [Google Scholar]
- Moysan, A.; Marquis, I.; Gaboriau, F.; Santus, R.; Dubertret, L.; Morliere, P. Ultraviolet A-induced lipid peroxidation and antioxidant defense systems in cultured human skin fibroblasts. J. Investig. Dermatol 1993, 100, 692–698. [Google Scholar]
- Poswig, A.; Wenk, J.; Brenneisen, P.; Wlaschek, M.; Hommel, C.; Quel, G.; Faisst, K.; Dissemond, J.; Briviba, K.; Krieg, T.; et al. Adaptive antioxidant response of manganese-superoxide dismutase following repetitive UVA irradiation. J. Investig. Dermatol 1999, 112, 13–18. [Google Scholar]
- Choung, B.Y.; Byun, S.J.; Suh, J.G.; Kim, T.Y. Extracellular superoxide dismutase tissue distribution and the patterns of superoxide dismutase mRNA expression following ultraviolet irradiation on mouse skin. Exp. Dermatol 2004, 13, 691–699. [Google Scholar]
- Delanian, S.; Baillet, F.; Huart, J.; Lefaix, J.L.; Maulard, C.; Housset, M. Successful treatment of radiation-induced fibrosis using liposomal Cu/Zn superoxide dismutase: Clinical trial. Radiother. Oncol 1994, 32, 12–20. [Google Scholar]
- Lefaix, J.L.; Delanian, S.; Leplat, J.J.; Tricaud, Y.; Martin, M.; Nimrod, A.; Baillet, F.; Daburon, F. Successful treatment of radiation-induced fibrosis using Cu/Zn-SOD and Mn-SOD: An experimental study. Int. J. Radiat. Oncol. Biol. Phys 1996, 35, 305–312. [Google Scholar]
- Luo, J.D.; Wang, Y.Y.; Fu, W.L.; Wu, J.; Chen, A.F. Gene therapy of endothelial nitric oxide synthase and manganese superoxide dismutase restores delayed wound healing in type 1 diabetic mice. Circulation 2004, 110, 2484–2493. [Google Scholar]
- Ceradini, D.J.; Yao, D.; Grogan, R.H.; Callaghan, M.J.; Edelstein, D.; Brownlee, M.; Gurtner, G.C. Decreasing intracellular superoxide corrects defective ischemia-induced new vessel formation in diabetic mice. J. Biol. Chem 2008, 283, 10930–10938. [Google Scholar]
- Ha, H.Y.; Kim, Y.; Ryoo, Z.Y.; Kim, T.Y. Inhibition of the TPA-induced cutaneous inflammation and hyperplasia by EC-SOD. Biochem. Biophys. Res. Commun 2006, 348, 450–458. [Google Scholar]
- Kim, Y.; Kim, B.H.; Lee, H.; Jeon, B.; Lee, Y.S.; Kwon, M.J.; Kim, T.Y. Regulation of skin inflammation and angiogenesis by EC-SOD via HIF-1alpha and NF-kappaB pathways. Free Radic. Biol. Med 2011, 51, 1985–1995. [Google Scholar]
- Lee, Y.S.; Cheon, I.S.; Kim, B.H.; Kwon, M.J.; Lee, H.W.; Kim, T.Y. Loss of extracellular superoxide dismutase induces severe IL-23-mediated skin inflammation in mice. J. Investig. Dermatol 2013, 133, 732–741. [Google Scholar]
- Kwon, M.J.; Jeon, Y.J.; Lee, K.Y.; Kim, T.Y. Superoxide dismutase 3 controls adaptive immune responses and contributes to the inhibition of ovalbumin-induced allergic airway inflammation in mice. Antioxid. Redox Signal 2012, 17, 1376–1392. [Google Scholar]
- Nozik-Grayck, E.; Suliman, H.B.; Piantadosi, C.A. Extracellular superoxide dismutase. Int. J. Biochem. Cell Biol 2005, 37, 2466–2471. [Google Scholar]
- Qin, Z.; Reszka, K.J.; Fukai, T.; Weintraub, N.L. Extracellular superoxide dismutase (ecSOD) in vascular biology: An update on exogenous gene transfer and endogenous regulators of ecSOD. Transl. Res 2008, 151, 68–78. [Google Scholar]
- Carlsson, L.M.; Marklund, S.L.; Edlund, T. The rat extracellular superoxide dismutase dimer is converted to a tetramer by the exchange of a single amino acid. Proc. Natl. Acad. Sci. USA 1996, 93, 5219–5222. [Google Scholar]
- Yamaguchi, M.; Zhou, C.; Heistad, D.D.; Watanabe, Y.; Zhang, J.H. Gene transfer of extracellular superoxide dismutase failed to prevent cerebral vasospasm after experimental subarachnoid hemorrhage. Stroke 2004, 35, 2512–2517. [Google Scholar]
- Batinic-Haberle, I.; Reboucas, J.S.; Spasojevic, I. Superoxide dismutase mimics: Chemistry, pharmacology, and therapeutic potential. Antioxid. Redox Signal 2010, 13, 877–918. [Google Scholar]
- Day, B.J. Catalase and glutathione peroxidase mimics. Biochem. Pharmacol 2009, 77, 285–296. [Google Scholar]
- Doctrow, S.R.; Huffman, K.; Marcus, C.B.; Tocco, G.; Malfroy, E.; Adinolfi, C.A.; Kruk, H.; Baker, K.; Lazarowych, N.; Mascarenhas, J.; et al. Salen-manganese complexes as catalytic scavengers of hydrogen peroxide and cytoprotective agents: Structure-activity relationship studies. J. Med. Chem 2002, 45, 4549–4558. [Google Scholar]
- Day, B.J. Catalytic antioxidants: A radical approach to new therapeutics. Drug Discov. Today 2004, 9, 557–566. [Google Scholar]
- Rong, Y.; Doctrow, S.R.; Tocco, G.; Baudry, M. EUK-134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate-induced neuropathology. Proc. Natl. Acad. Sci. USA 1999, 96, 9897–9902. [Google Scholar]
- Liu, R.; Liu, I.Y.; Bi, X.; Thompson, R.F.; Doctrow, S.R.; Malfroy, B.; Baudry, M. Reversal of age-related learning deficits and brain oxidative stress in mice with superoxide dismutase/catalase mimetics. Proc. Natl. Acad. Sci. USA 2003, 100, 8526–8531. [Google Scholar]
- Zhang, H.J.; Doctrow, S.R.; Xu, L.; Oberley, L.W.; Beecher, B.; Morrison, J.; Oberley, T.D.; Kregel, K.C. Redox modulation of the liver with chronic antioxidant enzyme mimetic treatment prevents age-related oxidative damage associated with environmental stress. FASEB J 2004, 18, 1547–1549. [Google Scholar]
- Clausen, A.; Doctrow, S.; Baudry, M. Prevention of cognitive deficits and brain oxidative stress with superoxide dismutase/catalase mimetics in aged mice. Neurobiol. Aging 2010, 31, 425–433. [Google Scholar]
- Liesa, M.; Luptak, I.; Qin, F.; Hyde, B.B.; Sahin, E.; Siwik, D.A.; Zhu, Z.; Pimentel, D.R.; Xu, X.J.; Ruderman, N.B.; et al. Mitochondrial transporter ATP binding cassette mitochondrial erythroid is a novel gene required for cardiac recovery after ischemia/reperfusion. Circulation 2011, 124, 806–813. [Google Scholar]
- Mahmood, J.; Jelveh, S.; Calveley, V.; Zaidi, A.; Doctrow, S.R.; Hill, R.P. Mitigation of radiation-induced lung injury by genistein and EUK-207. Int. J. Radiat. Biol 2011, 87, 889–901. [Google Scholar]
- Doctrow, S.R.; Lopez, A.; Schock, A.M.; Duncan, N.E.; Jourdan, M.M.; Olasz, E.B.; Moulder, J.E.; Fish, B.L.; Mader, M.; Lazar, J.; et al. A synthetic superoxide dismutase/catalase mimetic EUK-207 mitigates radiation dermatitis and promotes wound healing in irradiated rat skin. J. Investig. Dermatol 2013, 133, 1088–1096. [Google Scholar]
- Soares, M.P.; Bach, F.H. Heme oxygenase-1: From biology to therapeutic potential. Trends Mol. Med 2009, 15, 50–58. [Google Scholar]
- Saikawa, Y.; Kaneda, H.; Yue, L.; Shimura, S.; Toma, T.; Kasahara, Y.; Yachie, A.; Koizumi, S. Structural evidence of genomic exon-deletion mediated by Alu-Alu recombination in a human case with heme oxygenase-1 deficiency. Hum. Mutat 2000, 16, 178–179. [Google Scholar]
- Radhakrishnan, N.; Yadav, S.P.; Sachdeva, A.; Pruthi, P.K.; Sawhney, S.; Piplani, T.; Wada, T.; Yachie, A. Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol./Oncol 2011, 33, 74–78. [Google Scholar]
- Exner, M.; Minar, E.; Wagner, O.; Schillinger, M. The role of heme oxygenase-1 promoter polymorphisms in human disease. Free Radic. Biol. Med 2004, 37, 1097–1104. [Google Scholar]
- Kappas, A.; Drummond, G.S. Control of heme and cytochrome P-450 metabolism by inorganic metals, organometals and synthetic metalloporphyrins. Environ. Health Perspect 1984, 57, 301–306. [Google Scholar]
- Sasaki, T.; Takahashi, T.; Maeshima, K.; Shimizu, H.; Toda, Y.; Morimatsu, H.; Takeuchi, M.; Yokoyama, M.; Akagi, R.; Morita, K. Heme arginate pretreatment attenuates pulmonary NF-kappaB and AP-1 activation induced by hemorrhagic shock via heme oxygenase-1 induction. Med. Chem 2006, 2, 271–274. [Google Scholar]
- Tenhunen, R.; Mustajoki, P. Acute porphyria: Treatment with heme. Semin. Liver Dis 1998, 18, 53–55. [Google Scholar]
- Barbagallo, I.; Galvano, F.; Frigiola, A.; Cappello, F.; Riccioni, G.; Murabito, P.; D’Orazio, N.; Torella, M.; Gazzolo, D.; Li Volti, G. Potential therapeutic effects of natural heme oxygenase-1 inducers in cardiovascular diseases. Antioxid. Redox Signal 2013, 18, 507–521. [Google Scholar]
- Motterlini, R.; Foresti, R.; Bassi, R.; Green, C.J. Curcumin, an antioxidant and anti-inflammatory agent, induces heme oxygenase-1 and protects endothelial cells against oxidative stress. Free Radic. Biol. Med 2000, 28, 1303–1312. [Google Scholar]
- Balogun, E.; Hoque, M.; Gong, P.; Killeen, E.; Green, C.J.; Foresti, R.; Alam, J.; Motterlini, R. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem. J 2003, 371, 887–895. [Google Scholar]
- McNally, S.J.; Harrison, E.M.; Ross, J.A.; Garden, O.J.; Wigmore, S.J. Curcumin induces heme oxygenase 1 through generation of reactive oxygen species, p38 activation and phosphatase inhibition. Int. J. Mol. Med 2007, 19, 165–172. [Google Scholar]
- Lin, H.C.; Cheng, T.H.; Chen, Y.C.; Juan, S.H. Mechanism of heme oxygenase-1 gene induction by quercetin in rat aortic smooth muscle cells. Pharmacology 2004, 71, 107–112. [Google Scholar]
- Yao, P.; Nussler, A.; Liu, L.; Hao, L.; Song, F.; Schirmeier, A.; Nussler, N. Quercetin protects human hepatocytes from ethanol-derived oxidative stress by inducing heme oxygenase-1 via the MAPK/Nrf2 pathways. J. Hepatol 2007, 47, 253–261. [Google Scholar]
- Wannamethee, S.G.; Lowe, G.D.; Rumley, A.; Bruckdorfer, K.R.; Whincup, P.H. Associations of vitamin C status, fruit and vegetable intakes, and markers of inflammation and hemostasis. Am. J. Clin. Nutr 2006, 83, 567–574, ; quiz 726–567.. [Google Scholar]
- Homma, S.; Azuma, A.; Taniguchi, H.; Ogura, T.; Mochiduki, Y.; Sugiyama, Y.; Nakata, K.; Yoshimura, K.; Takeuchi, M.; Kudoh, S.; et al. Efficacy of inhaled N-acetylcysteine monotherapy in patients with early stage idiopathic pulmonary fibrosis. Respirology 2012, 17, 467–477. [Google Scholar]
- Demedts, M.; Behr, J.; Buhl, R.; Costabel, U.; Dekhuijzen, R.; Jansen, H.M.; MacNee, W.; Thomeer, M.; Wallaert, B.; Laurent, F.; et al. High-dose acetylcysteine in idiopathic pulmonary fibrosis. N. Engl. J. Med 2005, 353, 2229–2242. [Google Scholar]
- Tomioka, H.; Kuwata, Y.; Imanaka, K.; Hashimoto, K.; Ohnishi, H.; Tada, K.; Sakamoto, H.; Iwasaki, H. A pilot study of aerosolized N-acetylcysteine for idiopathic pulmonary fibrosis. Respirology 2005, 10, 449–455. [Google Scholar]
- Behr, J.; Demedts, M.; Buhl, R.; Costabel, U.; Dekhuijzen, R.P.; Jansen, H.M.; MacNee, W.; Thomeer, M.; Wallaert, B.; Laurent, F.; et al. Lung function in idiopathic pulmonary fibrosis—Extended analyses of the IFIGENIA trial. Respir. Res 2009, 10, 101. [Google Scholar]
- Crestani, B.; Besnard, V.; Boczkowski, J. Signalling pathways from NADPH oxidase-4 to idiopathic pulmonary fibrosis. Int. J. Biochem. Cell Biol 2011, 43, 1086–1089. [Google Scholar]
- Atkuri, K.R.; Mantovani, J.J.; Herzenberg, L.A.; Herzenberg, L.A. N-Acetylcysteine—A safe antidote for cysteine/glutathione deficiency. Curr. Opin. Pharmacol 2007, 7, 355–359. [Google Scholar]
- Raghu, G.; Anstrom, K.J.; King, T.E., Jr; Lasky, J.A.; Martinez, F.J. Idiopathic Pulmonary Fibrosis Clinical Research, N. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med 2012, 366, 1968–1977. [Google Scholar]
- Schagen, S.K.; Zampeli, V.A.; Makrantonaki, E.; Zouboulis, C.C. Discovering the link between nutrition and skin aging. Dermato-Endocrinol 2012, 4, 298–307. [Google Scholar]
- Placzek, M.; Gaube, S.; Kerkmann, U.; Gilbertz, K.P.; Herzinger, T.; Haen, E.; Przybilla, B. Ultraviolet B-induced DNA damage in human epidermis is modified by the antioxidants ascorbic acid and D-alpha-tocopherol. J. Investig. Dermatol 2005, 124, 304–307. [Google Scholar]
- Morganti, P.; Bruno, C.; Guarneri, F.; Cardillo, A.; Del Ciotto, P.; Valenzano, F. Role of topical and nutritional supplement to modify the oxidative stress. Int. J. Cosmet. Sci 2002, 24, 331–339. [Google Scholar]
- Eberlein-Konig, B.; Ring, J. Relevance of vitamins C and E in cutaneous photoprotection. J. Cosmet. Dermatol 2005, 4, 4–9. [Google Scholar]
- Stahl, W.; Heinrich, U.; Jungmann, H.; Sies, H.; Tronnier, H. Carotenoids and carotenoids plus vitamin E protect against ultraviolet light-induced erythema in humans. Am. J. Clin. Nutr 2000, 71, 795–798. [Google Scholar]
- Lee, J.; Jiang, S.; Levine, N.; Watson, R.R. Carotenoid supplementation reduces erythema in human skin after simulated solar radiation exposure. Proc. Soc. Exp. Biol. Med 2000, 223, 170–174. [Google Scholar]
- Heinrich, U.; Gartner, C.; Wiebusch, M.; Eichler, O.; Sies, H.; Tronnier, H.; Stahl, W. Supplementation with beta-carotene or a similar amount of mixed carotenoids protects humans from UV-induced erythema. J. Nutr 2003, 133, 98–101. [Google Scholar]
- Telfer, A.; Dhami, S.; Bishop, S.M.; Phillips, D.; Barber, J. beta-Carotene quenches singlet oxygen formed by isolated photosystem II reaction centers. Biochemistry 1994, 33, 14469–14474. [Google Scholar]
- Bose, B.; Chatterjee, S.N. UVA-induced peroxidation of lipid in the dried film state. J. Photochem. Photobiol. B 1994, 23, 119–123. [Google Scholar]
- Eicker, J.; Kurten, V.; Wild, S.; Riss, G.; Goralczyk, R.; Krutmann, J.; Berneburg, M. Betacarotene supplementation protects from photoaging-associated mitochondrial DNA mutation. Photochem. Photobiol. Sci 2003, 2, 655–659. [Google Scholar]
- Manach, C.; Scalbert, A.; Morand, C.; Remesy, C.; Jimenez, L. Polyphenols: Food sources and bioavailability. Am. J. Clin. Nutr 2004, 79, 727–747. [Google Scholar]
- Nichols, J.A.; Katiyar, S.K. Skin photoprotection by natural polyphenols: Anti-inflammatory, antioxidant and DNA repair mechanisms. Arch. Dermatol. Res 2010, 302, 71–83. [Google Scholar]
- Agarwal, R.; Katiyar, S.K.; Khan, S.G.; Mukhtar, H. Protection against ultraviolet B radiation-induced effects in the skin of SKH-1 hairless mice by a polyphenolic fraction isolated from green tea. Photochem. Photobiol 1993, 58, 695–700. [Google Scholar]
- Katiyar, S.K.; Elmets, C.A.; Agarwal, R.; Mukhtar, H. Protection against ultraviolet-B radiation-induced local and systemic suppression of contact hypersensitivity and edema responses in C3H/HeN mice by green tea polyphenols. Photochem. Photobiol 1995, 62, 855–861. [Google Scholar]
- Elmets, C.A.; Singh, D.; Tubesing, K.; Matsui, M.; Katiyar, S.; Mukhtar, H. Cutaneous photoprotection from ultraviolet injury by green tea polyphenols. J. Am. Acad. Dermatol 2001, 44, 425–432. [Google Scholar]
- Katiyar, S.K.; Elmets, C.A. Green tea polyphenolic antioxidants and skin photoprotection (Review). Int. J. Oncol 2001, 18, 1307–1313. [Google Scholar]
- Van Laethem, A.; Claerhout, S.; Garmyn, M.; Agostinis, P. The sunburn cell: Regulation of death and survival of the keratinocyte. Int. J. Biochem. Cell Biol 2005, 37, 1547–1553. [Google Scholar]
- Lambert, J.D.; Hong, J.; Yang, G.Y.; Liao, J.; Yang, C.S. Inhibition of carcinogenesis by polyphenols: Evidence from laboratory investigations. Am. J. Clin. Nutr 2005, 81, 284S–291S. [Google Scholar]
- Shindo, Y.; Witt, E.; Han, D.; Epstein, W.; Packer, L. Enzymic and non-enzymic antioxidants in epidermis and dermis of human skin. J. Investig. Dermatol 1994, 102, 122–124. [Google Scholar]
- Kwong, L.K.; Kamzalov, S.; Rebrin, I.; Bayne, A.C.; Jana, C.K.; Morris, P.; Forster, M.J.; Sohal, R.S. Effects of coenzyme Q(10) administration on its tissue concentrations, mitochondrial oxidant generation, and oxidative stress in the rat. Free Radic. Biol. Med 2002, 33, 627–638. [Google Scholar]
- Sohal, R.S.; Kamzalov, S.; Sumien, N.; Ferguson, M.; Rebrin, I.; Heinrich, K.R.; Forster, M.J. Effect of coenzyme Q10 intake on endogenous coenzyme Q content, mitochondrial electron transport chain, antioxidative defenses, and life span of mice. Free Radic. Biol. Med 2006, 40, 480–487. [Google Scholar]
- Prahl, S.; Kueper, T.; Biernoth, T.; Wohrmann, Y.; Munster, A.; Furstenau, M.; Schmidt, M.; Schulze, C.; Wittern, K.P.; Wenck, H.; et al. Aging skin is functionally anaerobic: Importance of coenzyme Q10 for anti aging skin care. BioFactors 2008, 32, 245–255. [Google Scholar]
- Blatt, T.; Lenz, H.; Koop, U.; Jaspers, S.; Weber, T.; Mummert, C.; Wittern, K.P.; Stab, F.; Wenck, H. Stimulation of skin’s energy metabolism provides multiple benefits for mature human skin. BioFactors 2005, 25, 179–185. [Google Scholar]
- Blatt, T.; Littarru, G.P. Biochemical rationale and experimental data on the antiaging properties of CoQ(10) at skin level. BioFactors 2011, 37, 381–385. [Google Scholar]
- Makrantonaki, E.; Zouboulis, C.C. Skin alterations and diseases in advanced age. Drug Discov. Today Dis. Mech 2008, 5, 153–162. [Google Scholar]
- Han, R.; Liu, L.; Li, J.; Du, G.; Chen, J. Functions, applications and production of 2-O-D-glucopyranosyl-L-ascorbic acid. Appl. Microbiol. Biotech 2012, 95, 313–320. [Google Scholar]
- Pawar, R.S.; Tamta, H.; Ma, J.; Krynitsky, A.J.; Grundel, E.; Wamer, W.G.; Rader, J.I. Updates on chemical and biological research on botanical ingredients in dietary supplements. Anal. Bioanal. Chem. 2013. [Google Scholar] [CrossRef]
- Ristow, M.; Zarse, K.; Oberbach, A.; Kloting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Bluher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar]
- Stanner, S.A.; Hughes, J.; Kelly, C.N.; Buttriss, J. A review of the epidemiological evidence for the ‘antioxidant hypothesis’. Public Health Nutr 2004, 7, 407–422. [Google Scholar]
- Marik, P.E.; Flemmer, M. Do dietary supplements have beneficial health effects in industrialized nations: What is the evidence? J. Parenteral Enteral Nutr 2012, 36, 159–168. [Google Scholar]
- Mursu, J.; Robien, K.; Harnack, L.J.; Park, K.; Jacobs, D.R., Jr. Dietary supplements and mortality rate in older women: The Iowa Women’s Health Study. Arch. Intern. Med 2011, 171, 1625–1633. [Google Scholar]
- Sedlak, T.W.; Saleh, M.; Higginson, D.S.; Paul, B.D.; Juluri, K.R.; Snyder, S.H. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. USA 2009, 106, 5171–5176. [Google Scholar]
- Dekker, D.; Dorresteijn, M.J.; Pijnenburg, M.; Heemskerk, S.; Rasing-Hoogveld, A.; Burger, D.M.; Wagener, F.A.; Smits, P. The bilirubin-increasing drug atazanavir improves endothelial function in patients with type 2 diabetes mellitus. Arterioscler. Thromb. Vasc. Biol 2011, 31, 458–463. [Google Scholar]
- Scheffler, I.E. Mitochondria make a come back. Advan. Drug Delivery Rev 2001, 49, 3–26. [Google Scholar]
- Davis, R.E.; Williams, M. Mitochondrial function and dysfunction: An update. J. Pharmacol. Exp. Ther 2012, 342, 598–607. [Google Scholar]
- Shults, C.W.; Oakes, D.; Kieburtz, K.; Beal, M.F.; Haas, R.; Plumb, S.; Juncos, J.L.; Nutt, J.; Shoulson, I.; Carter, J.; et al. Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol 2002, 59, 1541–1550. [Google Scholar]
- Shults, C.W.; Flint Beal, M.; Song, D.; Fontaine, D. Pilot trial of high dosages of coenzyme Q10 in patients with Parkinson’s disease. Exp. Neurol 2004, 188, 491–494. [Google Scholar]
- Ernster, L.; Dallner, G. Biochemical, physiological and medical aspects of ubiquinone function. Biochim. Biophys 1995, 1271, 195–204. [Google Scholar]
- Zhang, Y.; Aberg, F.; Appelkvist, E.L.; Dallner, G.; Ernster, L. Uptake of dietary coenzyme Q supplement is limited in rats. J. Nutr 1995, 125, 446–453. [Google Scholar]
- Matthews, R.T.; Yang, L.; Browne, S.; Baik, M.; Beal, M.F. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc. Natl. Acad. Sci. USA 1998, 95, 8892–8897. [Google Scholar]
- Cocheme, H.M.; Kelso, G.F.; James, A.M.; Ross, M.F.; Trnka, J.; Mahendiran, T.; Asin-Cayuela, J.; Blaikie, F.H.; Manas, A.R.; Porteous, C.M.; et al. Mitochondrial targeting of quinones: Therapeutic implications. Mitochondrion 2007, 7, S94–S102. [Google Scholar]
- Kelso, G.F.; Porteous, C.M.; Coulter, C.V.; Hughes, G.; Porteous, W.K.; Ledgerwood, E.C.; Smith, R.A.; Murphy, M.P. Selective targeting of a redox-active ubiquinone to mitochondria within cells: Antioxidant and antiapoptotic properties. J. Biol. Chem 2001, 276, 4588–4596. [Google Scholar]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J 2005, 19, 1088–1095. [Google Scholar]
- Murphy, M.P.; Smith, R.A. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Ann. Rev. Pharmacol. Toxicol 2007, 47, 629–656. [Google Scholar]
- Skulachev, V.P. A biochemical approach to the problem of aging: “Megaproject” on membrane-penetrating ions. The first results and prospects. Biochemistry (Mosc.) 2007, 72, 1385–1396. [Google Scholar]
- Antonenko, Y.N.; Roginsky, V.A.; Pashkovskaya, A.A.; Rokitskaya, T.I.; Kotova, E.A.; Zaspa, A.A.; Chernyak, B.V.; Skulachev, V.P. Protective effects of mitochondria-targeted antioxidant SkQ in aqueous and lipid membrane environments. J. Membr. Biol 2008, 222, 141–149. [Google Scholar]
- Distelmaier, F.; Valsecchi, F.; Forkink, M.; van Emst-de Vries, S.; Swarts, H.G.; Rodenburg, R.J.; Verwiel, E.T.; Smeitink, J.A.; Willems, P.H.; Koopman, W.J. Trolox-sensitive reactive oxygen species regulate mitochondrial morphology, oxidative phosphorylation and cytosolic calcium handling in healthy cells. Antioxid. Redox Signal 2012, 17, 1657–1669. [Google Scholar]
- Marrache, S.; Dhar, S. Engineering of blended nanoparticle platform for delivery of mitochondria-acting therapeutics. Proc. Natl. Acad. Sci. USA 2012, 109, 16288–16293. [Google Scholar]
- Bindu, S.; Pal, C.; Dey, S.; Goyal, M.; Alam, A.; Iqbal, M.S.; Dutta, S.; Sarkar, S.; Kumar, R.; Maity, P.; et al. Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J. Biol. Chem 2011, 286, 39387–39402. [Google Scholar]
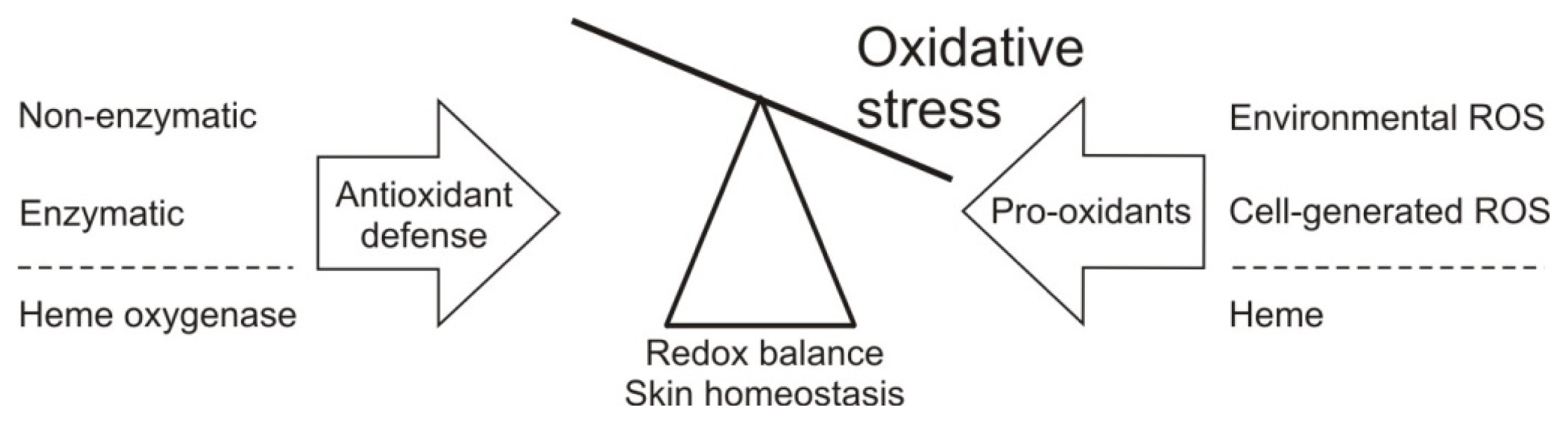

{kind=link}
{kind=link}
| Antioxidants | Examples | Target |
|---|---|---|
| Enzymatic | Superoxide dismutase | Superoxide |
| Catalase | Hydrogen peroxide | |
| Glutathione peroxidase | Hydrogen peroxide, lipid peroxides | |
| Peroxiredoxin | Hydrogen peroxide | |
| Heme oxygenase | Heme | |
| Non-enzymatic | Bilirubin | Lipid peroxides |
| Vitamin C | Superoxide, hydroxyl radical, reactive nitrogen species, trace metals | |
| Vitamin E | Lipid peroxides | |
| Metal-binding proteins | Ferritin | Free iron |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wagener, F.A.D.T.G.; Carels, C.E.; Lundvig, D.M.S. Targeting the Redox Balance in Inflammatory Skin Conditions. Int. J. Mol. Sci. 2013, 14, 9126-9167. https://doi.org/10.3390/ijms14059126
Wagener FADTG, Carels CE, Lundvig DMS. Targeting the Redox Balance in Inflammatory Skin Conditions. International Journal of Molecular Sciences. 2013; 14(5):9126-9167. https://doi.org/10.3390/ijms14059126
Chicago/Turabian StyleWagener, Frank A. D. T. G., Carine E. Carels, and Ditte M. S. Lundvig. 2013. "Targeting the Redox Balance in Inflammatory Skin Conditions" International Journal of Molecular Sciences 14, no. 5: 9126-9167. https://doi.org/10.3390/ijms14059126
APA StyleWagener, F. A. D. T. G., Carels, C. E., & Lundvig, D. M. S. (2013). Targeting the Redox Balance in Inflammatory Skin Conditions. International Journal of Molecular Sciences, 14(5), 9126-9167. https://doi.org/10.3390/ijms14059126