Phylogeography and Genetic Differentiation among Populations of the Moon Turban Snail Lunella granulata Gmelin, 1791 (Gastropoda: Turbinidae)

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Genetic Diversity of L. granulata
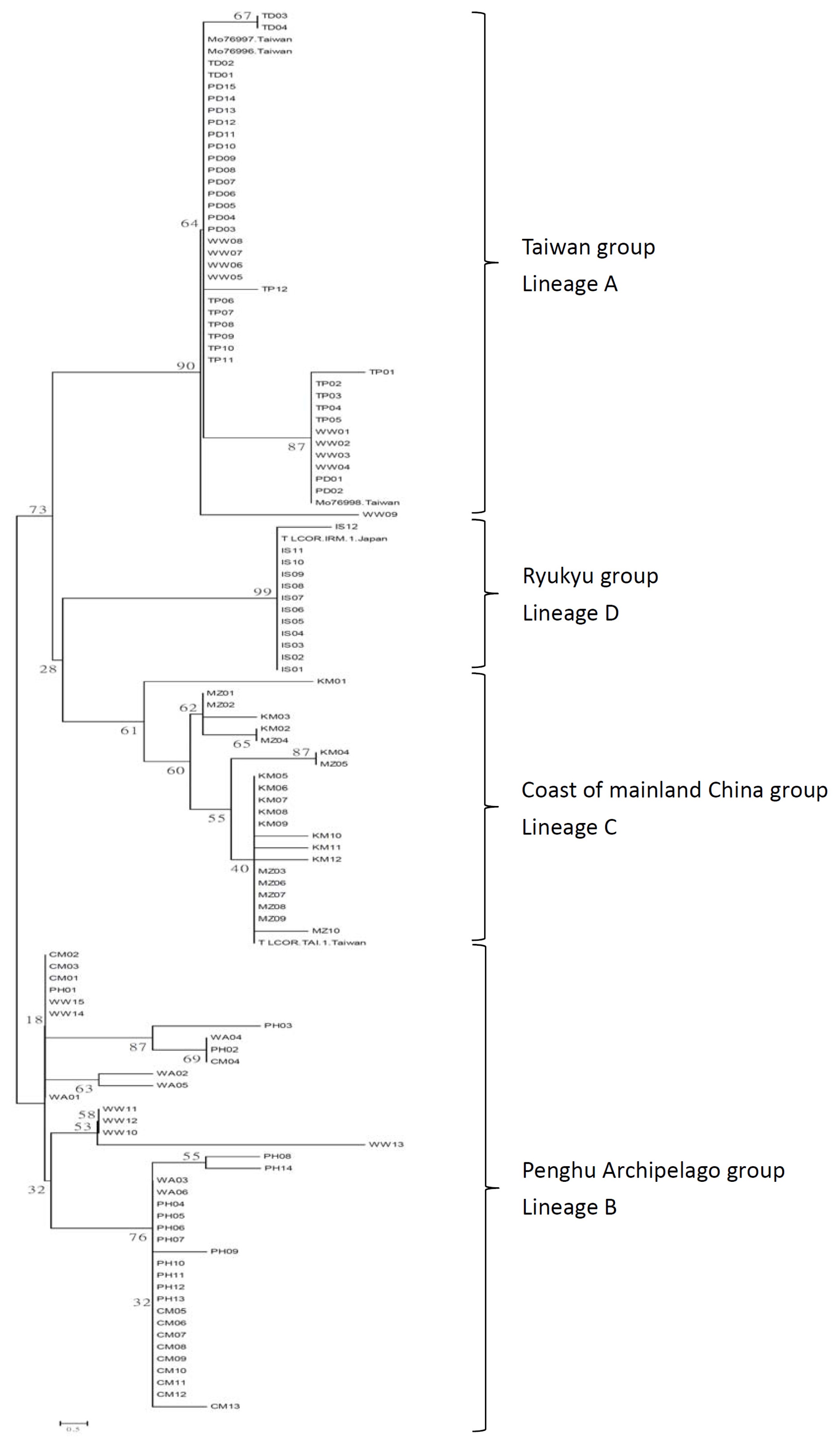
2.1.2. Phylogenetic Analysis
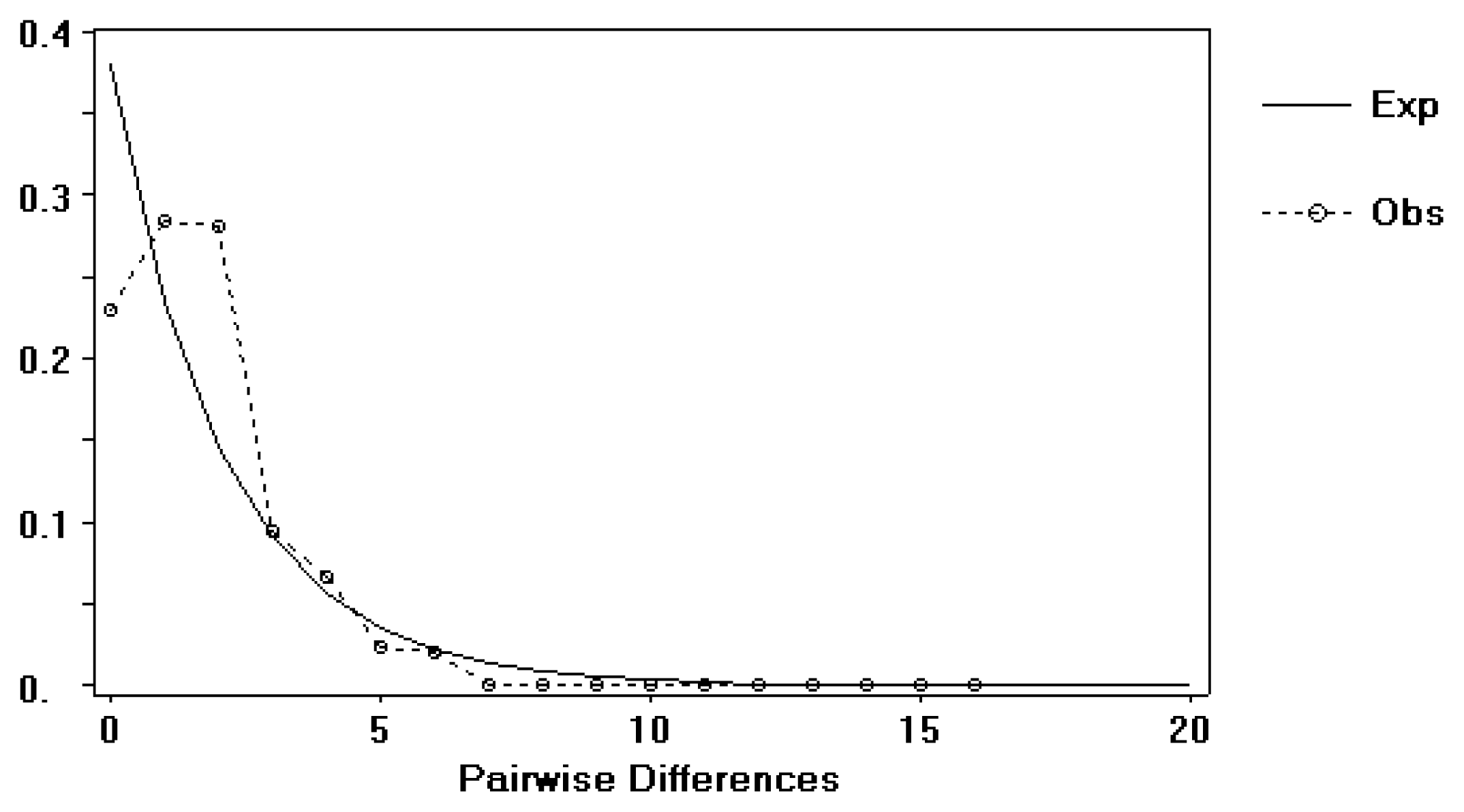
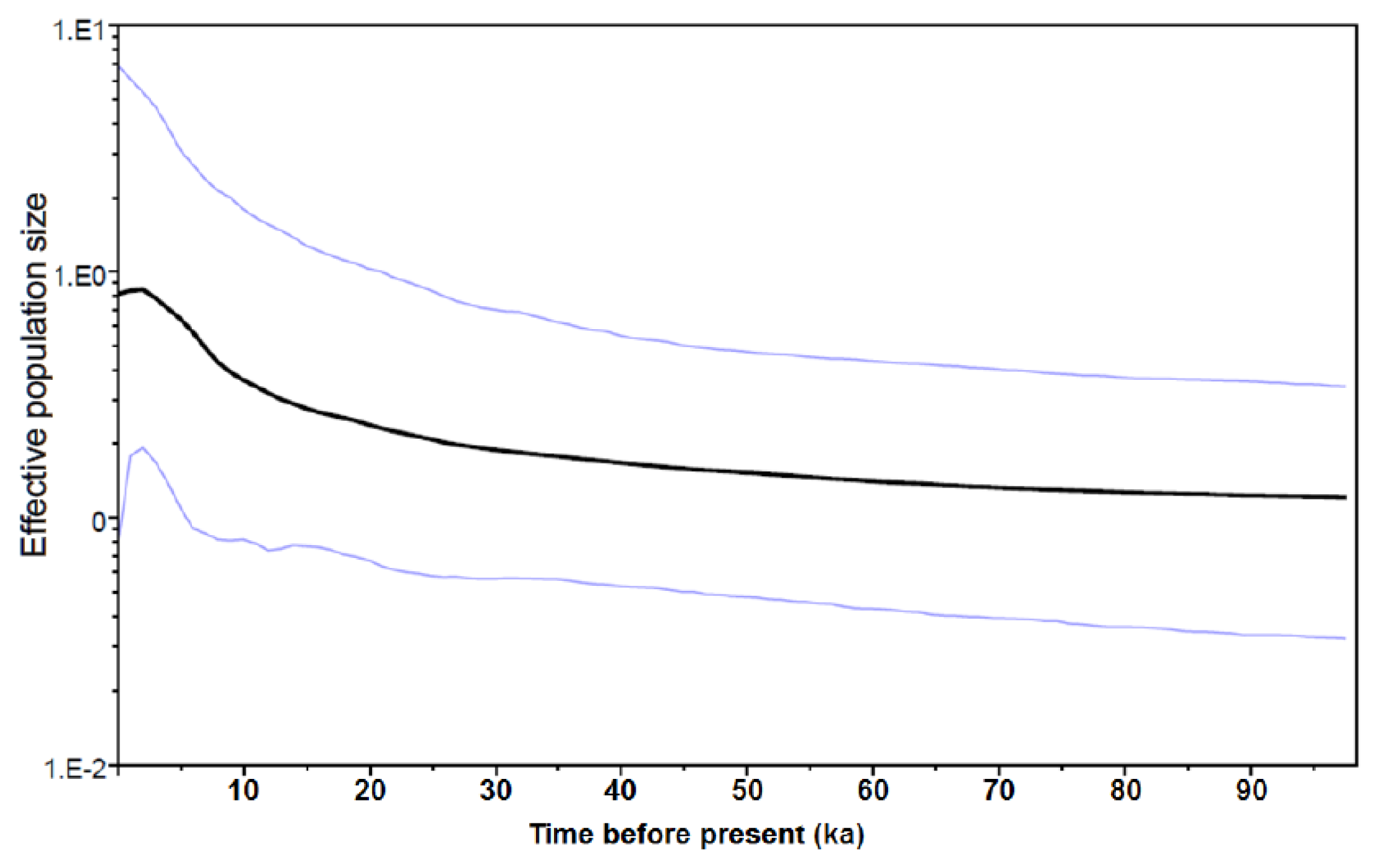
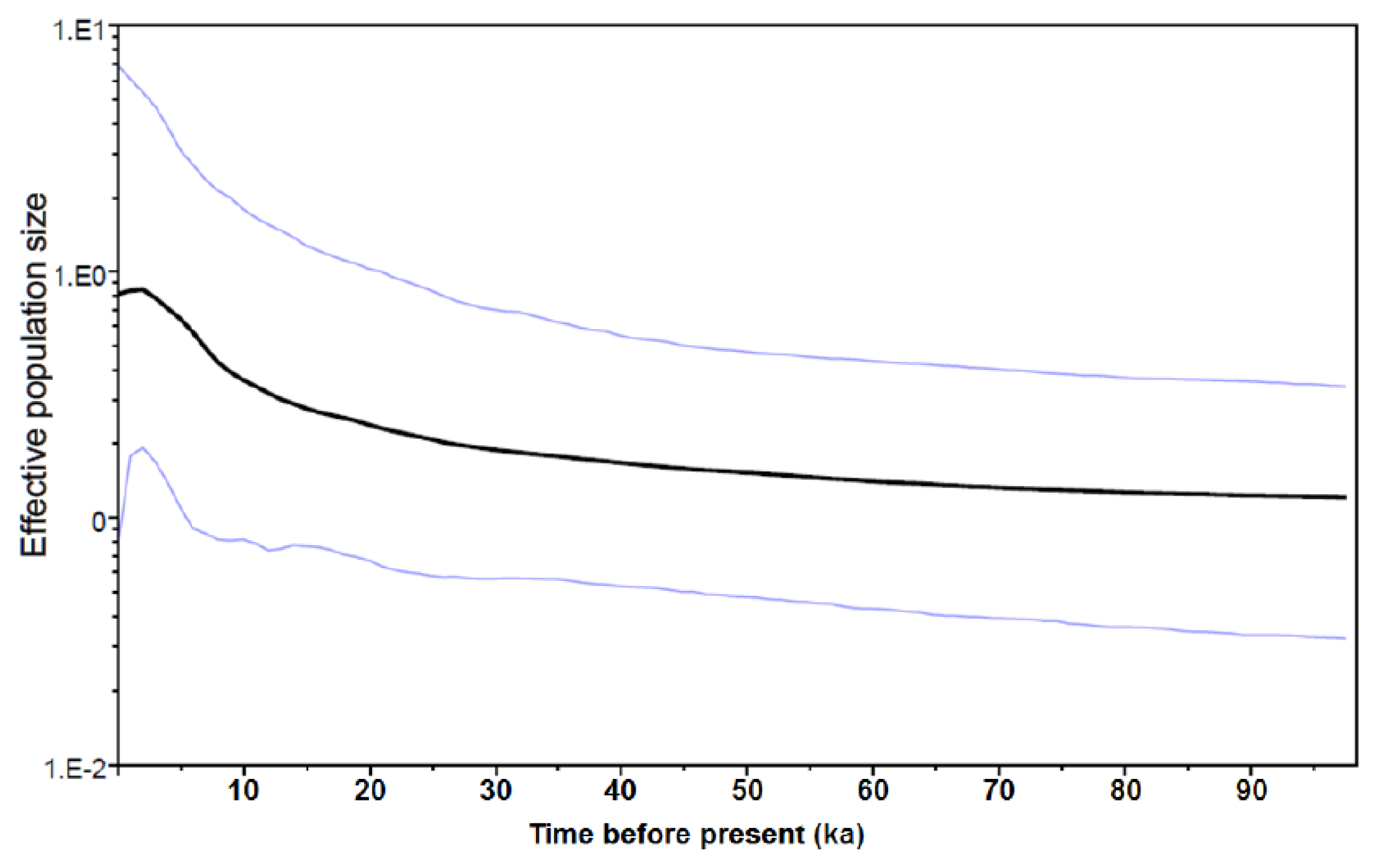
2.1.3. Historical Demography
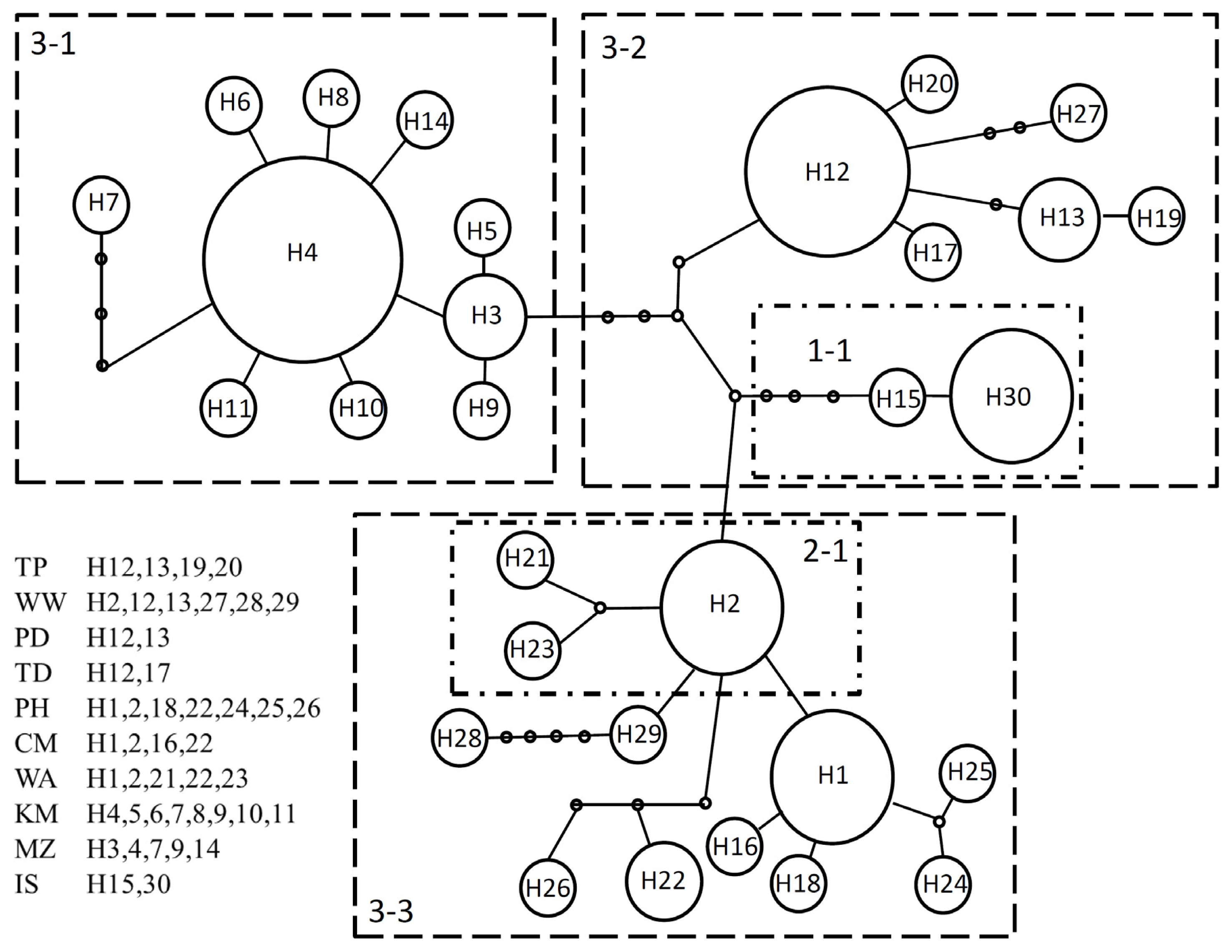
2.1.4. Population Differentiation
2.2. Discussion
2.2.1. Historical Demography and Genetic Diversity
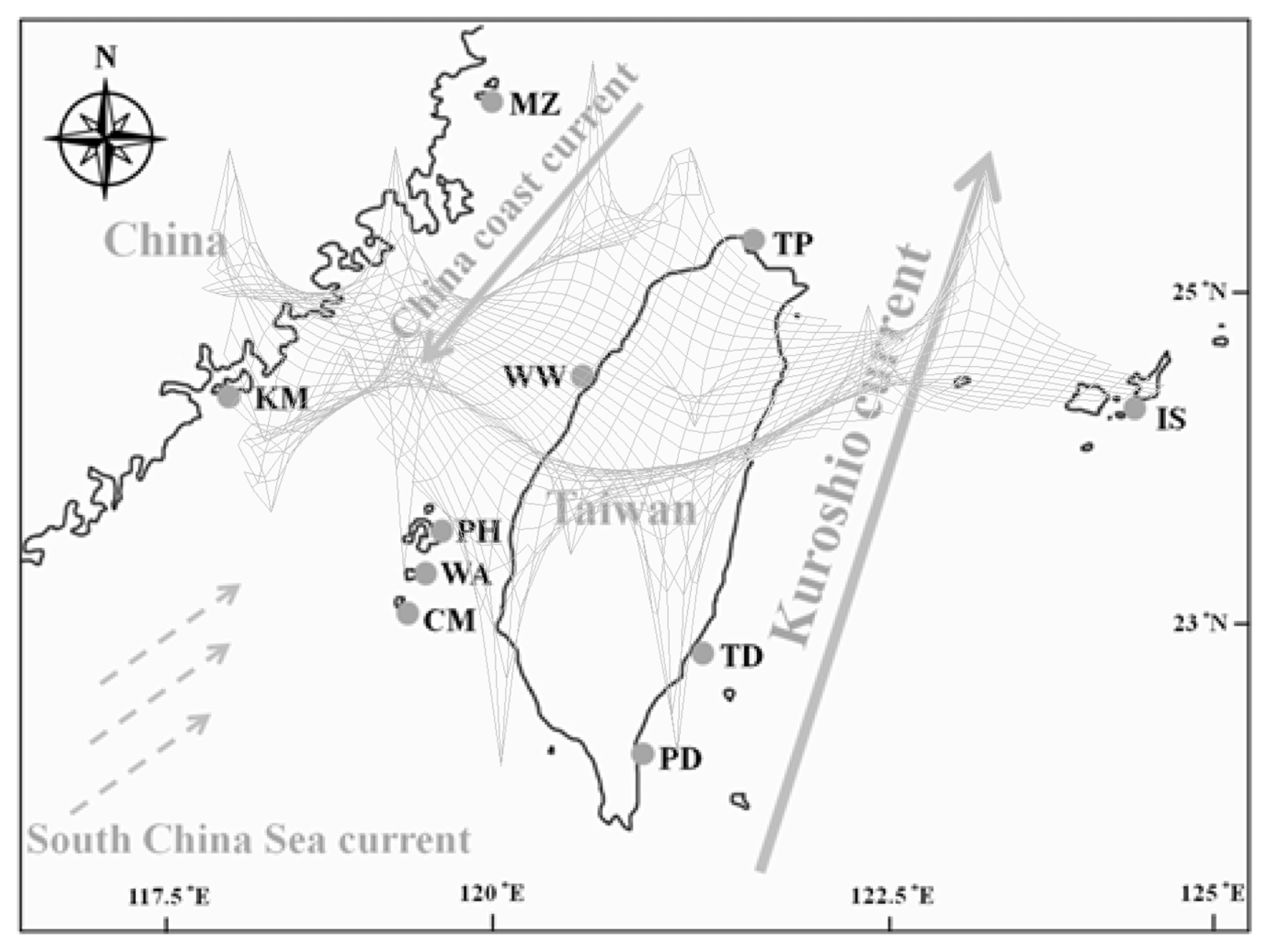
2.2.2. Phylogeographic Patterns
2.2.3. Population Genetic Structure and Historical Demography
3. Experimental Section
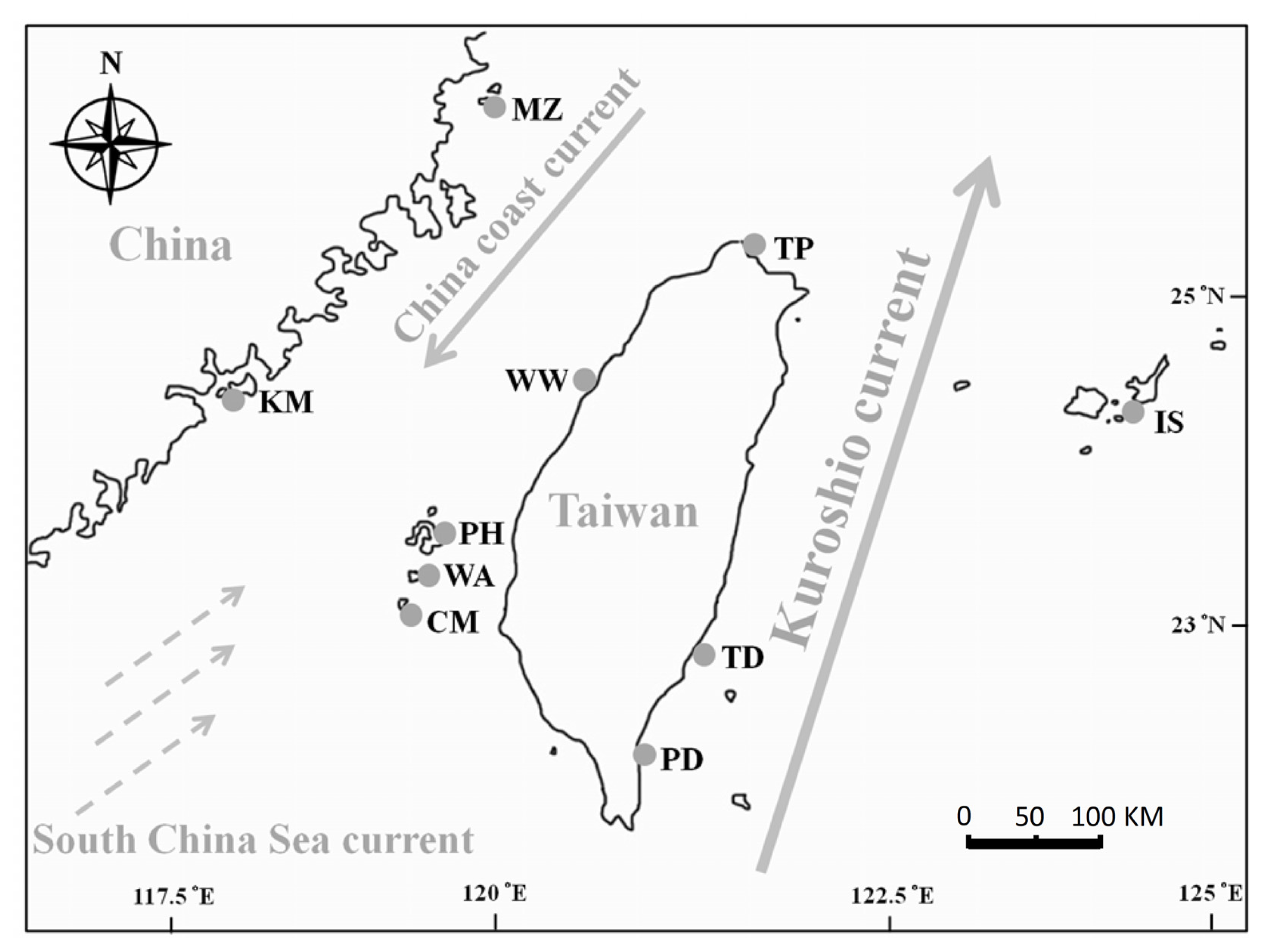
3.1. Sampling
3.2. DNA Extraction, Amplification, and Sequencing
3.3. Genetic Diversity
3.4. Historical Demography
3.5. Population Differentiation
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Hillis, D.M.; Mable, B.K.; Moritz, C. Molecular Systematics, 2nd Ed ed; Sinauer: Sunderland, MA, USA, 1996. [Google Scholar]
- Avise, J.C. The history and purview of phylogeography: A personal reflection. Mol. Ecol 1998, 7, 371–379. [Google Scholar]
- Nakano, T.; Takahashi, K.; Ozawa, T. Description of an endangered new species of Lunella. (Gastropoda: Turbinidae) from the Ogasawara Islands. Venus 2007, 66, 1–10. [Google Scholar]
- Williams, S.T.; Apte, D.; Ozawa, T.; Kagilis, F.; Nakano, T. Speciation and dispersal along continental coastlines and island arcs in the Indo-West Pacific turbinid gastropod genus Lunella. Evolution 2011, 65, 1752–1771. [Google Scholar]
- Williams, S.T.; Hall, A.; Kuklinski, P. Unraveling cryptic diversity in the Indo-West Pacific gastropod genus Lunella. (Turbinidae) using elliptic Fourier analysis. Am. Malacol. Bull 2012, 30, 189–206. [Google Scholar]
- Amio, M. A comparative embryology of marine gastropods, with ecological consi derations. J. Shimonoseki Univ. Fish 1963, 12, 229–358. [Google Scholar]
- Hellberg, M.E. Dependence of gene flow on geographic distance in two solitary corals with different larval dispersal capabilities. Evolution 1996, 50, 1167–1175. [Google Scholar]
- Kojima, S.; Sagawa, S.; Hayashi, I. Genetic differentiation among populations of the Japanese turban shell Turbo (Batillus) cornutus corresponding to warm currents. Mar. Ecol. Prog. Ser 1997, 150, 149–155. [Google Scholar]
- Kojima, S.; Sagawa, R.; Hayashi, I. Stability of the courses of the warm coastal currents along the Kyushu Island suggested by the population structure of the Japanese turban shell, Turbo (Batillus) cornutus. J. Oceanogr 2000, 56, 601–604. [Google Scholar]
- Palumbi, S.R. Molecular biogeography of the Pacific. Coral Reefs 1997, 16, 47–52. [Google Scholar]
- Briggs, J.S. Coincident biogeographic patterns: Indo-West Pacific Ocean. Evolution 1999, 53, 326–335. [Google Scholar]
- Kyle, C.J.; Boulding, E.G. Comparative population genetic structure of marine gastropods (Littorina. spp.) with and without pelagic larval dispersal. Mar. Biol 2000, 137, 835–845. [Google Scholar]
- Gruenthal, K.M.; Burton, R.S. Genetic structure of natural populations of the California black abalone (Haliotis. cracherodii Leach, 1814), a candidate for endangered species status. J. Exp. Mar. Biol. Ecol 2008, 355, 47–58. [Google Scholar]
- Palumbi, S.R.; Wilson, A.C. Mitochondrial DNA diversity in the sea urchins Stronglyocentrotus. purpuratus and S. droebachiensis. Evolution 1990, 44, 403–415. [Google Scholar]
- Arndt, A.; Smith, J. Genetic diversity and population structure in two species of sea cucumber: Different patterns according to mode of development. Mol. Ecol 1998, 7, 1053–1064. [Google Scholar]
- Schizas, N.V.; Street, G.T.; Coull, B.C.; Chandler, G.T.; Quattro, J.M. Molecular population structure of the marine benthic copepod Microarthridion littorale along the southeastern and Gulf coasts of the USA. Mar. Biol 1999, 135, 399–405. [Google Scholar]
- Palumbi, S.R. Genetic divergence, reproductive isolation, and marine speciation. Annu. Rev. Ecol. Syst 1994, 25, 547–572. [Google Scholar]
- Benzie, J.A.H.; Williams, S.T. Genetic structure of giant clam (Tridacna. maxima) populations in the West Pacific is not consistent with dispersal by present-day ocean currents. Evolution 1997, 51, 768–783. [Google Scholar]
- Brown, G.G.; Gadaleta, G.; Pepe, G.; Saccone, C.; Sbisa, E. Structural conservation and variation in the D-loop containing region of vertebrate mitochondrial DNA. J. Mol. Biol 1986, 192, 503–511. [Google Scholar]
- Wilding, C.S.; Mill, P.J.; Grahame, J. Partial sequence of the mitochondrial genome of Littorina saxatilis: Relevance to gas-tropod phylogenetics. J. Mol. Evol 1999, 48, 348–359. [Google Scholar]
- Grant, W.S.; Bowen, B.W. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. J. Hered 1998, 89, 415–426. [Google Scholar]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000. [Google Scholar]
- Fu, Y. Statistical tests of neutrality of mutations against population growth, hitchhiking, and background selection. Genetics 1997, 147, 915–925. [Google Scholar]
- Pons, O.; Petit, R.J. Measuring and testing genetic differentiation with ordered vs. unordered alleles. Genetics 1996, 144, 1237–1245. [Google Scholar]
- González-Wangüemert, M.; Froufe, E.; Pérez-Ruzafa, A.; Alexandrino, P. Phylogeographical history of the white seabream Diplodus. sargus (Sparidae): Implications for insularity. Mar. Biol. Res 2011, 7, 250–260. [Google Scholar]
- Díaz-Ferguson, E.; Haney, R.; Wares, J.; Silliman, B. Population genetics of a trochid gastropod broadens picture of caribbean sea connectivity. PLoS One 2010, 5, e12675. [Google Scholar]
- Chandler, E.A.; McDowell, J.R.; Graves, J.E. Genetically monomorphic invasive populations of the rapa whelk, Rapana venosa. Mol. Ecol 2008, 17, 4079–4091. [Google Scholar]
- Kochzius, M.; Nuryanto, A. Strong genetic population structure in the boring giant clam, Tridacna crocea, across the Indo-Malay Archipelago: Implications related to evolutionary processes and connectivity. Mol. Ecol 2008, 17, 3775–3787. [Google Scholar]
- Kurihara, T. Spatiotemporal variations in rocky intertidal malacofauna throughout Japan in the 1970s and 1980s. Mar. Biol 2007, 153, 61–70. [Google Scholar]
- Tsoi, K.H.; Chan, T.Y.; Chu, K.H. Molecular population structure of the kuruma shrimp Penaeus japonicus species complex in western Pacific. Mar. Biol 2007, 150, 1345–1364. [Google Scholar]
- Vo, S.T. The hermatypic Scleractinia of South Vietnam. In the Marine Biology of the South China Sea III; Morton, B., Ed.; Hong Kong University Press: Hong Kong, China, 1998; pp. 11–21. [Google Scholar]
- Niino, H.; Emery, K.O. Sediments of shallow portions of East China Sea and South China Sea. Geol. Soc. Am. Bull 1961, 72, 731–762. [Google Scholar]
- Chu, T.Y. A study on the water exchange between Pacific Ocean and the South China Sea. Acta Ocean. Taiwan 1972, 2, 11–24. [Google Scholar]
- Farris, A.; Wimbush, M. Wind-induced Kuroshio intrusion into the South China Sea. J. Oceanogr 1996, 52, 771–784. [Google Scholar]
- Jan, S.; Wang, J.; Chern, C.S.; Chao, S.Y. Seasonal variation of the circulation in the Taiwan Strait. J. Mar. Syst 2002, 35, 249–268. [Google Scholar]
- Zhang, J.B.; Cai, Z.P.; Huang, L.M. Population genetic structure of crimson snapper Lutjanus erythropterus in East Asia, revealed by analysis of the mitochondrial control region. ICES J. Mar. Sci 2006, 63, 693–704. [Google Scholar]
- Zhao, F.; Dong, Y.; Zhuang, P.; Zhang, T.; Zhang, L.Z.; Shi, Z.H. Genetic diversity of silver pomfret (Pampus argenteus) in the Southern Yellow and East China Seas. Biochem. Syst. Ecol 2011, 39, 145–150. [Google Scholar]
- Hewitt, G.M. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar]
- Hewitt, G.M. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc 1996, 58, 247–276. [Google Scholar]
- Liu, M.; Gao, T.X.; Sakurai, Y.; Jia, N.; Zhao, L.L.; Du, X.; Jiang, Q.; Lu, Z.C. Mitochondrial DNA control region diversity and population structure of Pacific herring (Clupea pallasii) in the Yellow Sea and the Sea of Japan. Chin. J. Oceanol. Limnol 2011, 29, 317–325. [Google Scholar]
- Xiao, J.; Cordes, J.F.; Moss, J.A.; Reece, K.S. Genetic diversity in U.S. hatchery stocks of Crassostrea ariakensis (Fujita, 1913) and comparison with natural populations in Asia. J. Shellfish Res 2011, 30, 751–760. [Google Scholar]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotech 1994, 3, 294–299. [Google Scholar]
- Simon, C.; Frati, F.; Beckenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, and phylogenetic utility of mitochondrial gene sequences and a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am 1994, 87, 651–701. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol 2011, 28, 2731, –2739.. [Google Scholar]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009. [Google Scholar] [CrossRef]
- Nei, M.; Tajima, F. Maximum likelihood estimation of the number of nucleotide substitutions from restriction sites data. Genetics 1983, 105, 207–217. [Google Scholar]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol 2000, 9, 1657–1660. [Google Scholar]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Rand, D.M. Neutrality tests of molecular markers and the connections between DNA polymorphism, demography, and conservation biology. Conserv. Biol 1996, 10, 665–671. [Google Scholar]
- Harpending, H. Signature of ancient population growth in a low resolution mitochondrial DNA mismatch distribution. Hum. Biol 1994, 66, 591–600. [Google Scholar]
- Rogers, A.R.; Harpending, H. Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol 1992, 9, 552–569. [Google Scholar]
- Drummond, A.J.; Rambaut, A.; Shapiro, B.; Pybus, O.G. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol 2005, 22, 1185–1192. [Google Scholar]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol 2007, 7, 214. [Google Scholar]
- Donald, K.M.; Kennedy, M.; Spencer, H.G. Cladogenesis as the result of long-distance rafting events in Pacific topshells (Gastropoda: Trochoidea). Evolution 2005, 59, 1701–1711. [Google Scholar]
- Hellberg, M.E.; Vacquier, V.D. Rapid evolution of fertilization selectivity and lysine cDNA sequences in teguline gastropods. Mol. Biol. Evol 1999, 16, 839–848. [Google Scholar]
- Marko, P.B. Fossil calibration of molecular clocks and the divergence times of geminate species pairs separated by the Isthmus of Panama. Mol. Biol. Evol 2002, 19, 2005–2021. [Google Scholar]
- Dupanloup, I.; Schneidera, S.; Excoffier, N.L. A simulated annealing approach to define the genetic structure of populations. Mol. Ecol 2002, 11, 2571–2581. [Google Scholar]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs toperform population genetics analyses under Linux and Windows. Mol. Ecol. Res 2010, 10, 564–567. [Google Scholar]
- Beerli, P.; Felsenstein, J. Maximum likelihood estimation of a migration matrix and effective population size in n subpopulations by using a coalescent approach. Proc. Natl. Acad. Sci. USA 2001, 98, 4563–4568. [Google Scholar]
- Miller, M.P. Alleles in space: Computer software for the joint analysis of inter-individual spatial and genetic information. J. Hered 2005, 96, 722–724. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regions | Population | Site code | Coordinates | N | Nh | h | π (10−2) | Tajima’s D | Fu’s Fs | R2 | SSD | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Taiwan Island (Lineage A) | 48 | 9 | 0.659 | 0.18 | −1.246 | −0.694 | 0.069 | 0.538 * | ||||
| Taipei | TP | 25.29°N 121.57°E | 12 | 4 | H12,13,19,20 | 0.682 | 0.11 | 0.184 | 0.097 | 0.162 | 0.040 | |
| Miaoli | WW | 24.60°N 120.73°E | 15 | 6 | H2,12,13,27,28,29 | 0.848 | 0.35 | 0.051 | 1.325 | 0.149 | 0.024 | |
| Pintung | PD | 22.13°N 120.88°E | 16 | 2 | H12,13 | 0.325 | 0.04 | 0.200 | 1.738 | 0.162 | 0.309 | |
| Taitung | TD | 22.83°N 121.18°E | 5 | 2 | H12,17 | 0.600 | 0.05 | 1.224 | 0.626 | 0.300 | 0.090 | |
| Penghu Archipelago (Lineage B) | 33 | 10 | 0.686 | 0.18 | −1.296 | −2.383 * | 0.067 | 0.038 | ||||
| Penghu | PH | 22.53°N 119.62°E | 14 | 7 | H1,2,18,22,24,25,26 | 0.692 | 0.19 | −1.237 | −1.557 | 0.089 | 0.585 * | |
| Qimei | CM | 23.22°N 119.44°E | 13 | 4 | H1,2,16,22 | 0.603 | 0.12 | −0.754 | 0.438 | 0.157 | 0.484 * | |
| Wangan | WA | 23.34°N 119.49°E | 6 | 5 | H1,2,21,22,23 | 0.933 | 0.26 | −0.399 | −1.121 | 0.167 | 0.076 | |
| Coast of mainland China (Lineage C) | 23 | 10 | 0.771 | 0.13 | −1.876 * | −5.060 * | 0.076 | 0.003 | ||||
| Kinmen | KM | 24.45°N 118.38°E | 12 | 8 | H4,5,6,7,8,9,10,11 | 0.848 | 0.17 | −1.942 * | −3.762 * | 0.093 | 0.004 | |
| Matsu | MZ | 26.21°N 119.98°E | 11 | 5 | H3,4,7,9,14 | 0.709 | 0.09 | −1.219 | −1.684 | 0.135 | 0.002 | |
| Ryukyu (Lineage D) | 13 | 2 | 0.154 | 0.01 | −1.149 | −0.537 | 0.266 | 0.023 | ||||
| Ishigaki | IS | 24.36°N 124.11°E | 13 | 2 | H15,30 | 0.154 | 0.01 | −1.149 | −0.537 | 0.266 | 0.023 | |
| Total | 117 | 30 | 0.899 | 0.46 | −1.128 | 01506.888 ** | 0.056 | 0.020 * | ||||
| Source of variation | Variance components | Percentage of variation |
|---|---|---|
| Among geographic districts | 2.603 *** | 71.31 |
| Among populations within geographic district | 0.127 ** | 3.49 |
| Within populations | 0.92 *** | 25.2 |
| Number of groups | Groupings | ΦCT % | Variance among groups |
|---|---|---|---|
| 2 | (TP, WW, PD, TD, PH, CM, WA, KM, MZ) (IS) | 0.346 | 34.61 |
| 3 | (TP, WW, PD, TD) (PH, CM, WA, IS) (KM, MZ) | 0.533 | 53.28 |
| 4 | (TP, WW, PD, TD) (PH, CM, WA) (KM, MZ) (IS) | 0.713 * | 71.31 |
| 5 | (TP, WW) (PD, TD) (PH, CM, WA) (KM, MZ) (IS) | 0.699 * | 69.86 |
| 6 | (TP, WW) (PD, TD) (PH, CM) (WA) (KM, MZ) (IS) | 0.698 * | 69.81 |
| 7 | (TP, WW) (PD) (TD) (PH, CM, WA) (KM) (MZ) (IS) | 0.671 * | 67.06 |
| Lineage | θ | Taiwan | Ryukyu | China | Penghu |
|---|---|---|---|---|---|
| Taiwan | 0.0058 (0.0045, 0.0077) | - | 0 | 0 | 0.761 (0.14, 2.68) |
| Ryukyu | 0.0059 (0.0037, 0.0101) | 0 | - | 0 | 0.765 (0.114, 3.474) |
| China | 0.0064 (0.0045, 0.0093) | 0 | 0.297 (0.022, 1.574) | - | 0 |
| Penghu | 0.0084 (0.0066, 0.011) | 0 | 0 | 0 | - |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chiu, Y.-W.; Bor, H.; Tan, M.-S.; Lin, H.-D.; Jean, C.-T. Phylogeography and Genetic Differentiation among Populations of the Moon Turban Snail Lunella granulata Gmelin, 1791 (Gastropoda: Turbinidae). Int. J. Mol. Sci. 2013, 14, 9062-9079. https://doi.org/10.3390/ijms14059062
Chiu Y-W, Bor H, Tan M-S, Lin H-D, Jean C-T. Phylogeography and Genetic Differentiation among Populations of the Moon Turban Snail Lunella granulata Gmelin, 1791 (Gastropoda: Turbinidae). International Journal of Molecular Sciences. 2013; 14(5):9062-9079. https://doi.org/10.3390/ijms14059062
Chicago/Turabian StyleChiu, Yuh-Wen, Hor Bor, Mian-Shin Tan, Hung-Du Lin, and Chuen-Tan Jean. 2013. "Phylogeography and Genetic Differentiation among Populations of the Moon Turban Snail Lunella granulata Gmelin, 1791 (Gastropoda: Turbinidae)" International Journal of Molecular Sciences 14, no. 5: 9062-9079. https://doi.org/10.3390/ijms14059062
APA StyleChiu, Y.-W., Bor, H., Tan, M.-S., Lin, H.-D., & Jean, C.-T. (2013). Phylogeography and Genetic Differentiation among Populations of the Moon Turban Snail Lunella granulata Gmelin, 1791 (Gastropoda: Turbinidae). International Journal of Molecular Sciences, 14(5), 9062-9079. https://doi.org/10.3390/ijms14059062