Neurodegeneration and Neuroprotection in Diabetic Retinopathy


{kind=link}
Abstract
:1. Introduction
2. Neurodegenerative Factors and Potential Therapeutic Targets
3. Hyperglycemia
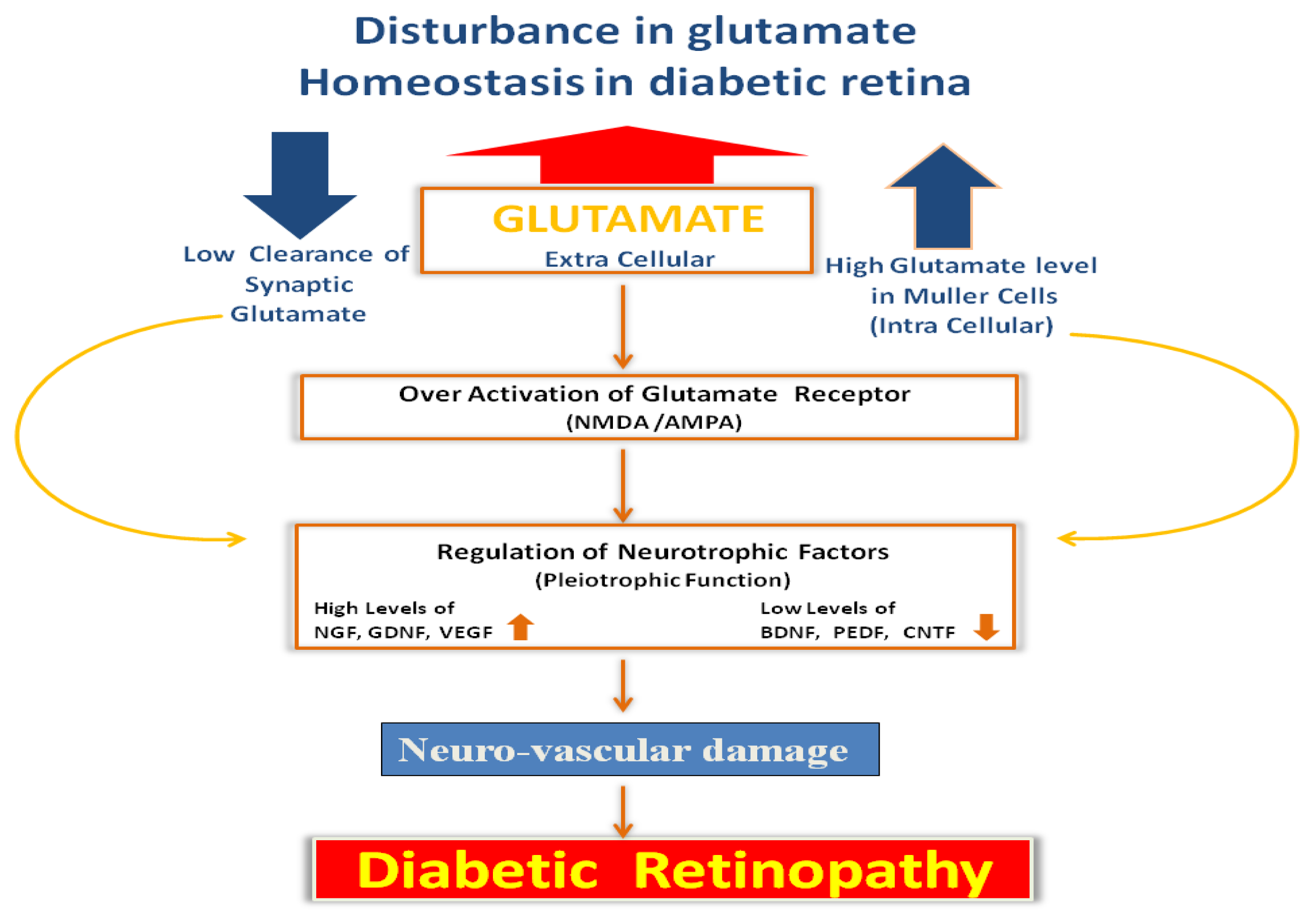
4. Glutamate Excitotoxicity
5. Nutrients and Vitamins
6. Metabolites of Tryptophan Pathway
7. Neurotrophic Factors
8. Conclusions
Acknowledgement
Conflict of Interest
References
- World Health Organization. Diabetes Fact Sheet Number 312; WHO: Geneva, Switzerland. Available online: http://www.who.int/mediacentre/factsheets/fs312/en (accessed on 30 August 2011).
- Lieth, E.; Barber, A.J.; Xu, B.; Dice, C.; Ratz, M.J.; Tanase, D.; Strother, J.M. Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy. Penn State Retina Research Group. Diabetes 1998, 47, 815–820. [Google Scholar]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Invest 1998, 102, 783–791. [Google Scholar]
- Peng, P.H.; Lin, H.S.; Lin, S. Nerve fibre layer thinning in patients with preclinical retinopathy. Can. J. Ophthalmol 2009, 44, 417–422. [Google Scholar]
- Bearse, M.A., Jr; Han, Y.; Schneck, M.E.; Adams, A.J. Retinal function in normal and diabetic eyes mapped with the slow flash multifocal electroretinogram. Invest. Ophthalmol. Vis. Sci. 2004, 45, 296–304. [Google Scholar]
- Fletcher, E.L.; Phipps, J.A.; Ward, M.M.; Puthussery, T.; Wilkinson-Berka, J.L. Neuronal and glial cell abnormality as predictors of progression of diabetic retinopathy. Curr. Pharm. Des 2007, 13, 2699–2712. [Google Scholar]
- Mohr, S.; Xi, X.; Tang, J.; Kern, T.S. Caspase activation in retinas of diabetic and galactosemic mice and diabetic patients. Diabetes 2002, 51, 1172–1179. [Google Scholar]
- Metea, M.R.; Newman, E.A. Signalling within the neurovascular unit in the mammalian retina. Exp. Physiol 2007, 92, 635–640. [Google Scholar]
- Kusari, J.; Zhou, S.; Padillo, E.; Clarke, K.G.; Gil, D.W. Effect of memantine on neuroretinal function and retinal vascular changes of streptozotocin-induced diabetic rats. Invest. Ophthalmol. Vis. Sci 2007, 48, 5152–5159. [Google Scholar]
- Kusari, J.; Zhou, S.X.; Padillo, E.; Clarke, K.G.; Gil, D.W. Inhibition of vitreoretinal VEGF elevation and blood-retinal barrier breakdown in streptozotocin-induced diabetic rats by brimonidine. Invest. Ophthalmol. Vis. Sci 2010, 51, 1044–1051. [Google Scholar]
- Feng, Y.; Wang, Y.; Stock, O.; Pfister, F.; Tanimoto, N.; Seeliger, M.W.; Hillebrands, J.L.; Hoffmann, S.; Wolburg, H.; Gretz, N.; et al. Vasoregression linked to neuronal damage in the rat with defect of polycystin-2. PLoS One 2009, 6, e7328. [Google Scholar]
- Feng, Y.; Wang, Y.; Li, L.; Wu, L.; Hoffmann, S.; Gretz, N.; Hammes, H.P. Gene expression profiling of vasoregression in the retina–involvement of microglial cells. PLoS One 2011, 6, e16865. [Google Scholar]
- Kern, T.S.; Du, Y.; Miller, C.M.; Hatala, D.A.; Levin, L.A. Overexpression of Bcl-2 in vascular endothelium inhibits the microvascular lesions of diabetic retinopathy. Am. J. Pathol 2010, 176, 2550–2558. [Google Scholar]
- Martin, P.M.; Roon, P.; van Ells, T.K.; Ganapathy, V.; Smith, S.B. Death of retinal neurons in streptozotocin-induced diabetic mice. Invest. Ophthalmol. Vis. Sci 2004, 45, 3330–3336. [Google Scholar]
- Harrower, A.D.; Clarke, B.F. Diabetic retinopathy with normal glucose tolerance. Br. J. Ophthalmol 1976, 60, 459–463. [Google Scholar]
- Barnes, A.J.; Kohner, E.M.; Johnston, D.G.; Alberti, K.G. Severe retinopathy and mild carbohydrate intolerance: Possible role of insulin deficiency and elevated circulating growth hormone. Lancet 1985, 1, 1465–1468. [Google Scholar]
- Warboys, C.M.; Fraser, P.A. Hyperglycemia attenuates acute permeability response to advanced glycation end products in retinal microvasculature. Microvasc. Res 2010, 80, 174–176. [Google Scholar]
- Ismail-Beigi, F.; Craven, T.; Banerji, M.A.; Basile, J.; Calles, J.; Cohen, R.M.; Cuddihy, R.; Cushman, W.C.; Genuth, S.; Grimm, R.H., Jr; et al. Effect of intensive treatment of hyperglycaemia on microvascular outcomes in type 2 diabetes: An analysis of the ACCORD randomised trial. Lancet 2010, 376, 419–430. [Google Scholar]
- Patel, A.; MacMahon, S.; Chalmers, J.; Neal, B.; Billot, L.; Woodward, M.; Marre, M.; Cooper, M.; Glasziou, P.; Grobbee, D.; et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med 2008, 358, 2560–2572. [Google Scholar]
- Ola, M.S.; Nawaz, M.I.; Siddiquei, M.M.; Al-Amro, S.; Abu El-Asrar, A.M. Recent advances in understanding the biochemical and molecular mechanism of diabetic retinopathy. J. Diabetes Complicat 2012, 26, 56–64. [Google Scholar]
- Abu El-Asrar, A.M.; Al-Mezaine, H.S.; Ola, M.S. Pathophysiology and management of diabetic retinopathy. Expert Rev. Ophthalmol 2009, 4, 627–647. [Google Scholar]
- Ola, M.S.; Berkich, D.A.; Xu, Y.; King, M.T.; Gardner, T.W.; Simpson, I.; LaNoue, K.F. Analysis of glucose metabolism in diabetic rat retinas. Am. J. Physiol. Endocrinol. Metab 2006, 290, E1057–E1067. [Google Scholar]
- Diederen, R.M.; La Heij, E.C.; Deutz, N.E.; Kijlstra, A.; Kessels, A.G.; van Eijk, H.M.; Liem, A.T.; Dieudonné, S.; Hendrikse, F. Increased glutamate levels in the vitreous of patients with retinal detachment. Exp. Eye Res 2006, 83, 45–50. [Google Scholar]
- Yu, X.H.; Zhang, H.; Wang, Y.H.; Liu, L.J.; Teng, Y.; Liu, P. Time-dependent reduction of glutamine synthetase in retina of diabetic rats. Exp. Eye Res 2009, 89, 967–971. [Google Scholar]
- Ola, M.S.; Hosoya, K.; LaNoue, K.F. Regulation of glutamate metabolism by hydrocortisone and branched chain keto acids in cultured rat retinal Müller cells (TR-MUL). Neurochem. Int 2011, 59, 656–663. [Google Scholar]
- Zhang, Y.; Bhavnani, B.R. Glutamate-induced apoptosis in neuronal cells is mediated via caspase-dependent and independent mechanisms involving calpain and caspase-3 proteases as well as apoptosis inducing factor (AIF) and this process is inhibited by equine estrogens. BMC Neurosci 2006, 7, 49. [Google Scholar]
- Hutson, S.M.; Berkich, D.; Drown, P.; Xu, B.; Aschner, M.; LaNoue, K.F. Role of branched-chain aminotransferase isoenzymes and gabapentin in neurotransmitter metabolism. J. Neurochem 1998, 71, 863–874. [Google Scholar]
- Sweatt, A.J.; Garcia-Espinosa, M.A.; Wallin, R.; Hutson, S.M. Branched-chain amino acids and neurotransmitter metabolism: Expression of cytosolic branched-chain aminotransferase (BCATc) in the cerebellum and hippocampus. J. Comp. Neurol 2004, 477, 360–370. [Google Scholar]
- Xu, Y.; Ola, M.S.; Berkich, D.A.; Gardner, T.W.; Barber, A.J.; Palmieri, F.; Hutson, S.M.; LaNoue, K.F. Energy sources for glutamate neurotransmission in the retina: Absence of the aspartate/glutamate carrier produces reliance on glycolysis in glia. J. Neurochem 2007, 101, 120–131. [Google Scholar]
- Lieth, E.; LaNoue, K.F.; Berkich, D.A.; Xu, B.; Ratz, M.; Taylor, C.; Hutson, S.M. Nitrogen shuttling between neurons and glial cells during glutamate synthesis. J. Neurochem 2001, 76, 1712–1723. [Google Scholar]
- Hutson, S.M.; Lieth, E.; LaNoue, K.F. Function of leucine in excitatory neurotransmitter metabolism in the central nervous system. J. Nutr 2001, 131, 846S–850S. [Google Scholar]
- Weber, M.; Bonaventure, N.; Sahel, J.A. Protective role of excitatory amino acid antagonists in experimental retinal ischemia. Graefes. Arch. Clin. Exp. Ophthalmol 1995, 233, 360–365. [Google Scholar]
- Vorwerk, C.K.; Lipton, S.A.; Zurakowski, D.; Hyman, B.T.; Sabel, B.A.; Dreyer, E.B. Chronic low-dose glutamate is toxic to retinal ganglion cells. Toxicity blocked by memantine. Invest. Ophthalmol. Vis. Sci 1996, 37, 1618–1624. [Google Scholar]
- Smith, S.B.; Duplantier, J.; Dun, Y.; Mysona, B.; Roon, P.; Martin, P.M.; Ganapathy, V. In vivo protection against retinal neurodegeneration by sigma receptor 1 ligand (+)-pentazocine. Invest. Ophthalmol. Vis. Sci 2008, 49, 4154–4161. [Google Scholar]
- Lieberman, T.W.; Podos, S.M.; Hartstein, J. Acute glaucoma, ectopia lentis and homocystinuria. Am. J. Ophthalmol 1966, 61, 252–255. [Google Scholar]
- Brazionis, L.; Rowley, K., Sr; Itsiopoulos, C.; Harper, C.A.; O’Dea, K. Homocysteine and diabetic retinopathy. Diabetes Care 2008, 31, 50–56. [Google Scholar]
- Wijekoon, E.P.; Brosnan, M.E.; Brosnan, J.T. Homocysteine metabolism in diabetes. Biochem. Soc. Trans 2007, 5, 1175–1179. [Google Scholar]
- Coral, K.; Angayarkanni, N.; Gomathy, N.; Bharathselvi, M.; Pukhraj, R.; Rupak, R. Homocysteine levels in the vitreous of proliferative diabetic retinopathy and rhegmatogenous retinal detachment: Its modulating role on lysyl oxidase. Invest. Ophthalmol. Vis. Sci 2009, 50, 3607–3612. [Google Scholar]
- Neugebauer, S.; Baba, T.; Kurokawa, K.; Watanabe, T. Defective homocysteine metabolism as a risk factor for diabetic retinopathy. Lancet 1997, 349, 473–474. [Google Scholar]
- Naggar, H.; Ola, M.S.; Moore, P.; Huang, W.; Bridges, C.C.; Ganapathy, V.; Smith, S.B. Downregulation of reduced-folate transporter by glucose in cultured RPE cells and in RPE of diabetic mice. Invest. Ophthalmol. Vis. Sci 2002, 43, 556–563. [Google Scholar]
- Wright, A.D.; Martin, N.; Dodson, P.M. Homocysteine, folates, and the eye. Eye (Lond. ) 2008, 22, 989–993. [Google Scholar]
- Satyanarayana, A.; Balakrishna, N.; Pitla, S.; Reddy, P.Y.; Mudili, S.; Lopamudra, P.; Suryanarayana, P.; Viswanath, K.; Ayyagari, R.; Reddy, G.B. Status of B-vitamins and homocysteine in diabetic retinopathy: Association with vitamin-B12 deficiency and hyperhomocysteinemia. PLoS One 2011, 6, e26747. [Google Scholar]
- Moore, P.; El-sherbeny, A.; Roon, P.; Schoenlein, P.V.; Ganapathy, V.; Smith, S.B. Apoptotic cell death in the mouse retinal ganglion cell layer is induced in vivo by the excitatory amino acid homocysteine. Exp. Eye Res 2001, 73, 45–57. [Google Scholar]
- Ganapathy, P.S.; Perry, R.L.; Tawfik, A.; Smith, R.M.; Perry, E.; Roon, P.; Bozard, B.R.; Ha, Y.; Smith, S.B. Homocysteine-mediated modulation of mitochondrial dynamics in retinal ganglion cells. Invest. Ophthalmol. Vis. Sci 2011, 52, 5551–5558. [Google Scholar]
- Ganapathy, P.S.; White, R.E.; Ha, Y.; Bozard, B.R.; McNeil, P.L.; Caldwell, R.W.; Kumar, S.; Black, S.M.; Smith, S.B. The role of N-methyl-d-aspartate receptor activation in homocysteine-induced death of retinal ganglion cells. Invest. Ophthalmol. Vis. Sci 2011, 52, 5515–5524. [Google Scholar]
- Lipton, S.A.; Kim, W.K.; Choi, Y.B.; Kumar, S.; D’Emilia, D.M.; Rayudu, P.V.; Arnelle, D.R.; Stamler, J.S. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-d-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928. [Google Scholar]
- Zieminska, E.; Matyja, E.; Kozlowska, H.; Stafiej, A.; Lazarewicz, J.W. Excitotoxic neuronal injury in acute homocysteine neurotoxicity: Role of calcium and mitochondrial alterations. Neurochem. Int 2006, 48, 491–497. [Google Scholar]
- Ziemińska, E.; Stafiej, A.; Łazarewicz, J.W. Role of group I metabotropic glutamate receptors and NMDA receptors in homocysteine-evoked acute neurodegeneration of cultured cerebellar granule neurones. Neurochem. Int 2003, 43, 481–492. [Google Scholar]
- Maler, J.M.; Seifert, W.; Hüther, G.; Wiltfang, J.; Rüther, E.; Kornhuber, J.; Bleich, S. Homocysteine induces cell death of rat astrocytes in vitro. Neurosci. Lett 2003, 347, 85–88. [Google Scholar]
- Poloschek, C.M.; Fowler, B.; Unsold, R.; Lorenz, B. Disturbed visual system function in methionine synthase deficiency. Graefes. Arch. Clin. Exp. Ophthalmol 2005, 243, 497–500. [Google Scholar]
- Luchowska, E.; Luchowski, P.; Paczek, R.; Ziembowicz, A.; Kocki, T.; Turski, W.A.; Wielosz, M.; Lazarewicz, J.; Urbanska, E.M. Dual effect of DL-homocysteine and S-adenosylhomocysteine on brain synthesis of the glutamate receptor antagonist, kynurenic acid. J. Neurosci. Res 2005, 79, 375–382. [Google Scholar]
- Chmiel-Perzyńska, I.; Perzyński, A.; Wielosz, M.; Urbańska, E.M. Hyperglycemia enhances the inhibitory effect of mitochondrial toxins and d,l-homocysteine on the brain production of kynurenic acid. Pharmacol. Rep 2007, 59, 268–273. [Google Scholar]
- Munipally, P.K.; Agraharm, S.G.; Valavala, V.K.; Gundae, S.; Turlapati, N.R. Evaluation of indoleamine 2,3-dioxygenase expression and kynurenine pathway metabolites levels in serum samples of diabetic retinopathy patients. Arch. Physiol. Biochem 2011, 117, 254–258. [Google Scholar]
- Zwilling, D.; Huang, S.Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Guidetti, P.; Wu, H.Q.; Lee, J.; Truong, J.; Andrews-Zwilling, Y.; Hsieh, E.W.; et al. Kynurenine 3-monooxygenase inhibition in blood ameliorates neurodegeneration. Cell 2011, 145, 863–874. [Google Scholar]
- Park, K.S.; Kim, S.S.; Kim, J.C.; Kim, H.C.; Im, Y.S.; Ahn, C.W.; Lee, H.K. Serum and tear levels of nerve growth factor in diabetic retinopathy patients. Am. J. Ophthalmol 2008, 145, 432–437. [Google Scholar]
- Mattson, M.P.; Maudsley, S.; Martin, B. BDNF and 5-HT: A dynamic duo in age-related neuronal plasticity and neurodegenerative disorders. Trends Neurosci 2004, 27, 589–594. [Google Scholar]
- Carmeliet, P.; Tessier-Lavigne, M. Common mechanisms of nerve and blood vessel wiring. Nature 2005, 436, 193–200. [Google Scholar]
- Nagahara, A.H.; Tuszynski, M.H. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat. Rev. Drug. Discov 2011, 10, 209–219. [Google Scholar]
- Eichmann, A.; Makinen, T.; Alitalo, K. Neural guidance molecules regulate vascular remodeling and vessel navigation. Genes Dev 2005, 19, 1013–1021. [Google Scholar]
- Suchting, S.; Bicknell, R.; Eichmann, A. Neuronal clues to vascular guidance. Exp. Cell. Res 2006, 312, 668–675. [Google Scholar]
- Yoshii, A.; Constantine-Paton, M. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev. Neurobiol 2010, 70, 304–322. [Google Scholar]
- Cohen-Cory, S.; Kidane, A.H.; Shirkey, N.J.; Marshak, S. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev. Neurobiol 2010, 70, 271–288. [Google Scholar]
- Rohrer, B.; Korenbrot, J.I.; LaVail, M.M.; Reichardt, L.F.; Xu, B. Role of neurotrophin receptor TrkB in the maturation of rod photoreceptors and establishment of synaptic transmission to the inner retina. J. Neurosci 1999, 19, 8919–8930. [Google Scholar]
- Chen, H.; Weber, A.J. BDNF enhances retinal ganglion cell survival in cats with optic nerve damage. Invest. Ophthalmol. Vis. Sci 2001, 42, 966–974. [Google Scholar]
- Lom, B.; Cogen, J.; Sanchez, A.L.; Vu, T.; Cohen-Cory, S. Local and target-derived brain-derived neurotrophic factor exert opposing effects on the dendritic arborization of retinal ganglion cells in vivo. J. Neurosci 2002, 22, 7639–7649. [Google Scholar]
- Pinzón-Duarte, G.; Arango-González, B.; Guenther, E.; Kohler, K. Effects of brain-derived neurotrophic factor on cell survival, differentiation and patterning of neuronal connections and Müller glia cells in the developing retina. Eur. J. Neurosci 2004, 19, 1475–1484. [Google Scholar]
- Hu, Y.; Cho, S.; Goldberg, J.L. Neurotrophic effect of a novel TrkB agonist on retinal ganglion cells. Invest. Ophthalmol. Vis. Sci 2010, 51, 1747–1754. [Google Scholar]
- Sánchez-Migallón, M.C.; Nadal-Nicolás, F.M.; Jiménez-López, M.; Sobrado-Calvo, P.; Vidal-Sanz, M.; Agudo-Barriuso, M. Brain derived neurotrophic factor maintains Brn3a expression in axotomized rat retinal ganglion cells. Exp. Eye Res 2011, 92, 260–267. [Google Scholar]
- Dai, M.; Xia, X.-B.; Xiong, S.-Q. BDNF regulates GLAST and glutamine synthetase in mouse retinal Müller cells. J. Cell. Physiol 2012, 227, 596–603. [Google Scholar]
- Nakagawa, T.; Ono-Kishino, M.; Sugaru, E.; Yamanaka, M.; Taiji, M.; Noguchi, H. Brain-derived neurotrophic factor (BDNF) regulates glucose and energy metabolism in diabetic mice. Diabetes Metab. Res. Rev 2002, 18, 185–191. [Google Scholar]
- Krabbe, K.S.; Nielsen, A.R.; Krogh-Madsen, R.; Plomgaard, P.; Rasmussen, P.; Erikstrup, C.; Fischer, C.P.; Lindegaard, B.; Petersen, A.M.; Taudorf, S.; et al. Brain-derived neurotrophic factor (BDNF) and type 2 diabetes. Diabetologia 2007, 50, 431–438. [Google Scholar]
- Yamanaka, M.; Itakura, Y.; Ono-Kishino, M.; Tsuchida, A.; Nakagawa, T.; Taiji, M. Intermittent administration of brain-derived neurotrophic factor (BDNF) ameliorates glucose metabolism and prevents pancreatic exhaustion in diabetic mice. J. Biosci. Bioeng 2008, 105, 395–402. [Google Scholar]
- Seki, M.; Tanaka, T.; Nawa, H.; Usui, T.; Fukuchi, T.; Ikeda, K.; Abe, H.; Takei, N. Involvement of brain-derived neurotrophic factor in early retinal neuropathy of streptozotocin-induced diabetes in rats: Therapeutic potential of brain-derived neurotrophic factor for dopaminergic amacrine cells. Diabetes 2004, 53, 2412–2419. [Google Scholar]
- Sasaki, M.; Ozawa, Y.; Kurihara, T.; Kubota, S.; Yuki, K.; Noda, K.; Kobayashi, S.; Ishida, S.; Tsubota, K. Neurodegenerative influence of oxidative stress in the retina of a murine model of diabetes. Diabetologia 2010, 53, 971–979. [Google Scholar]
- Barhwal, K.; Hota, S.K.; Prasad, D.; Singh, S.B.; Ilavazhagan, G. Hypoxia-induced deactivation of NGF-mediated ERK1/2 signaling in hippocampal cells: Neuroprotection by acetyl-l-carnitine. J. Neurosci. Res 2008, 86, 2705–2721. [Google Scholar]
- Harada, C.; Harada, T.; Quah, H.M.; Maekawa, F.; Yoshida, K.; Ohno, S.; Wada, K.; Parada, L.F.; Tanaka, K. Potential role of glial cell line-derived neurotrophic factor receptors in Müller glial cells during light induced retinal degeneration. Neuroscience 2003, 122, 229–235. [Google Scholar]
- Azadi, S.; Johnson, L.E.; Paquet-Durand, F.; Perez, M.T.; Zhang, Y.; Ekström, P.A.; van Veen, T. CNTF + BDNF treatment and neuroprotective pathways in the rd1 mouse retina. Brain Res 2007, 1129, 116–129. [Google Scholar]
- Barnstable, C.J.; Tombran-Tink, J. Neuroprotective and antiangiogenic actions of PEDF in the eye: Molecular targets and therapeutic potential. Prog. Retin. Eye Res 2004, 23, 561–577. [Google Scholar]
- Saint-Geniez, M.; Maharaj, A.S.R.; Walshe, T.E.; Tucker, B.A.; Sekiyama, E.; Kurihara, T.; Darland, D.C.; Young, M.J.; D’Amore, P.A. Endogenous VEGF Is Required for Visual Function: Evidence for a Survival Role on Müller Cells and Photoreceptors. PLoS One 2008, 3, e3554. [Google Scholar]
- Li, Y.; Zhang, F.; Nagai, N.; Tang, Z.; Zhang, S.; Scotney, P.; Lennartsson, J.; Zhu, C.; Qu, Y.; Fang, C.; et al. VEGF-B inhibits apoptosis via VEGFR-1-mediated suppression of the expression of BH3-only protein genes in mice and rats. J. Clin. Invest 2008, 118, 913–923. [Google Scholar]
- Nishijima, K.; Ng, Y.; Zhong, L.; Bradley, J.; Schubert, W.; Jo, N.; Akita, J.; Samuelsson, S.J.; Robinson, G.S.; Adamis, A.P.; et al. Vascular Endothelial Growth Factor-A Is a Survival Factor for Retinal Neurons and a Critical Neuroprotectant during the Adaptive Response to Ischemic Injury. Am. J. Pathol 2007, 171, 53–67. [Google Scholar]
- Barber, A.J.; Antonetti, D.A.; Kern, T.S.; Reiter, C.E.; Soans, R.S.; Krady, J.K.; Levison, S.W.; Gardner, T.W.; Bronson, S.K. The Ins2Akita mouse as a model of early retinal complications in diabetes. Invest. Ophthalmol. Vis. Sci 2005, 46, 2210–2218. [Google Scholar]
- Barber, A.J.; Nakamura, M.; Wolpert, E.B.; Reiter, C.E.; Seigel, G.M.; Antonetti, D.A.; Gardner, T.W. Insulin rescues retinal neurons from apoptosis by a phosphatidylinositol 3-kinase/Akt-mediated mechanism that reduces the activation of caspase-3. J. Biol. Chem 2001, 276, 32814–32821. [Google Scholar]
- Reiter, C.E.; Wu, X.; Sandirasegarane, L.; Nakamura, M.; Gilbert, K.A.; Singh, R.S.; Fort, P.E.; Antonetti, D.A.; Gardner, T.W. Diabetes reduces basal retinal insulin receptor signaling: Reversal with systemic and local insulin. Diabetes 2006, 55, 1148–1156. [Google Scholar]
- Kondo, T.; Kahn, C.R. Altered insulin signaling in retinal tissue in diabetic states. J. Biol. Chem 2004, 279, 37997–38006. [Google Scholar]
- Yi, X.; Schubert, M.; Peachey, N.S.; Suzuma, K.; Burks, D.J.; Kushner, J.A.; Suzuma, I.; Cahill, C.; Flint, C.L.; Dow, M.A.; et al. Insulin receptor substrate 2 is essential for maturation and survival of photoreceptor cells. J. Neurosci 2005, 25, 1240–1248. [Google Scholar]
- Reiter, C.E.; Sandirasegarane, L.; Wolpert, E.B.; Klinger, M.; Simpson, I.A.; Barber, A.J.; Antonetti, D.A.; Kester, M.; Gardner, T.W. Characterization of insulin signaling in rat retina in vivo and ex vivo. Am. J. Physiol. Endocrinol. Metab 2003, 285, E763–E774. [Google Scholar]
- Misra, G.P.; Singh, R.S.; Aleman, T.S.; Jacobson, S.G.; Gardner, T.W.; Lowe, T.L. Subconjunctivally implantable hydrogels with degradable and thermoresponsive properties for sustained release of insulin to the retina. Biomaterials 2009, 30, 6541–6547. [Google Scholar]
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ola, M.S.; Nawaz, M.I.; Khan, H.A.; Alhomida, A.S. Neurodegeneration and Neuroprotection in Diabetic Retinopathy. Int. J. Mol. Sci. 2013, 14, 2559-2572. https://doi.org/10.3390/ijms14022559
Ola MS, Nawaz MI, Khan HA, Alhomida AS. Neurodegeneration and Neuroprotection in Diabetic Retinopathy. International Journal of Molecular Sciences. 2013; 14(2):2559-2572. https://doi.org/10.3390/ijms14022559
Chicago/Turabian StyleOla, Mohammad Shamsul, Mohd Imtiaz Nawaz, Haseeb A. Khan, and Abdullah S. Alhomida. 2013. "Neurodegeneration and Neuroprotection in Diabetic Retinopathy" International Journal of Molecular Sciences 14, no. 2: 2559-2572. https://doi.org/10.3390/ijms14022559