Pathogenesis of Chronic Cardiorenal Syndrome: Is There a Role for Oxidative Stress?

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Oxidative Stress and Its Impact on Cellular Damage
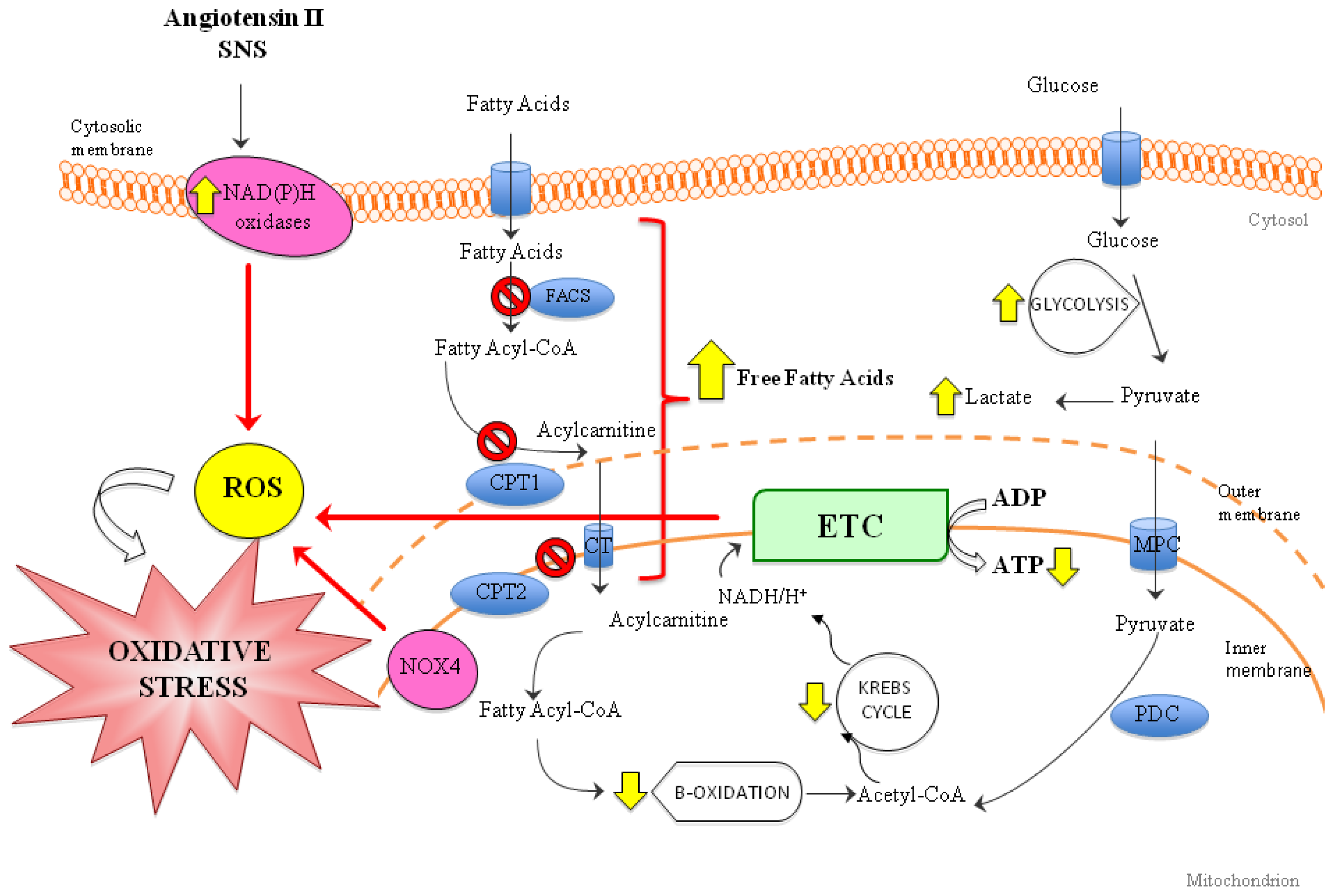
3. Role of Oxidative Stress in Heart Failure (HF)
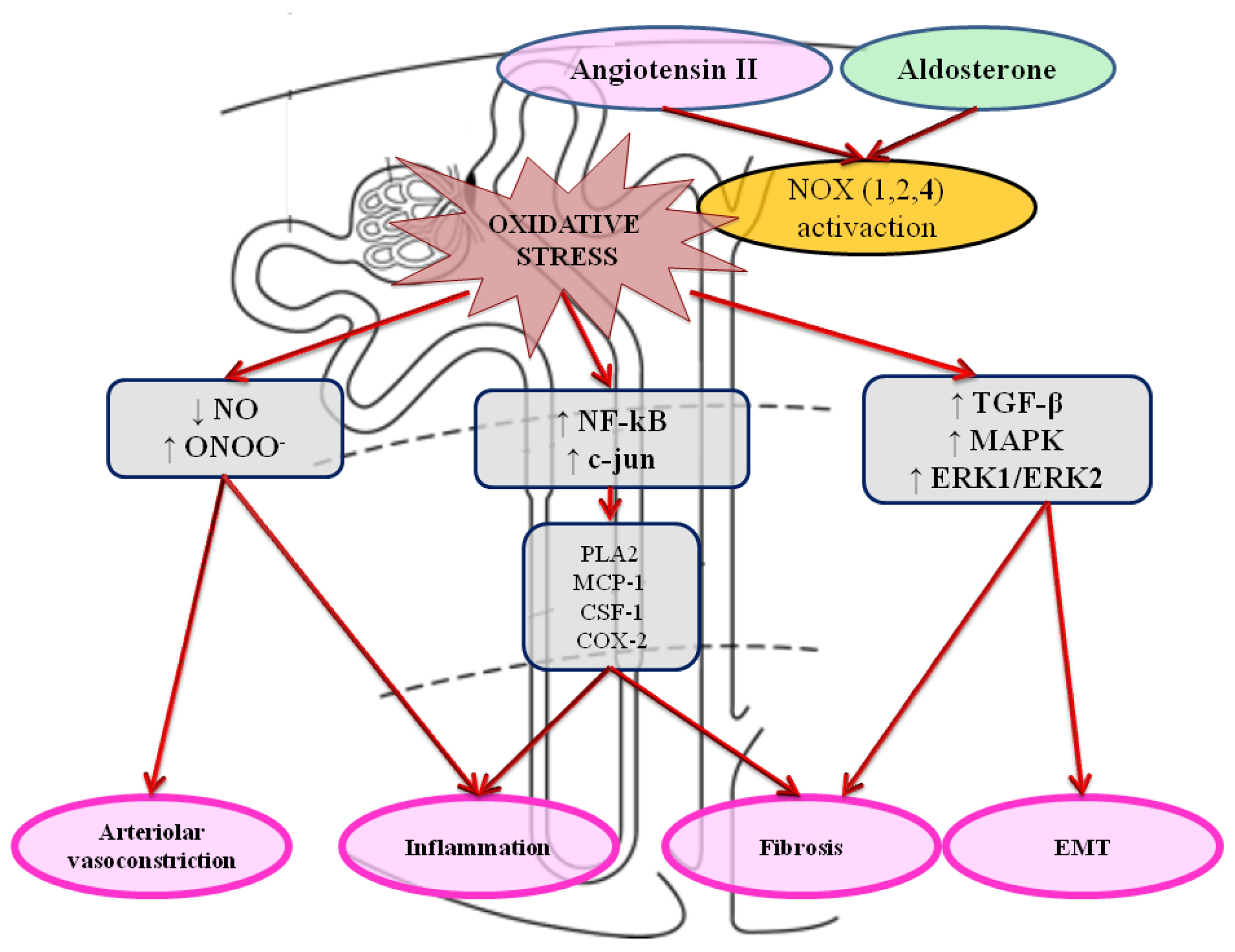
4. Role of Oxidative Stress in Kidney Damage and Failure
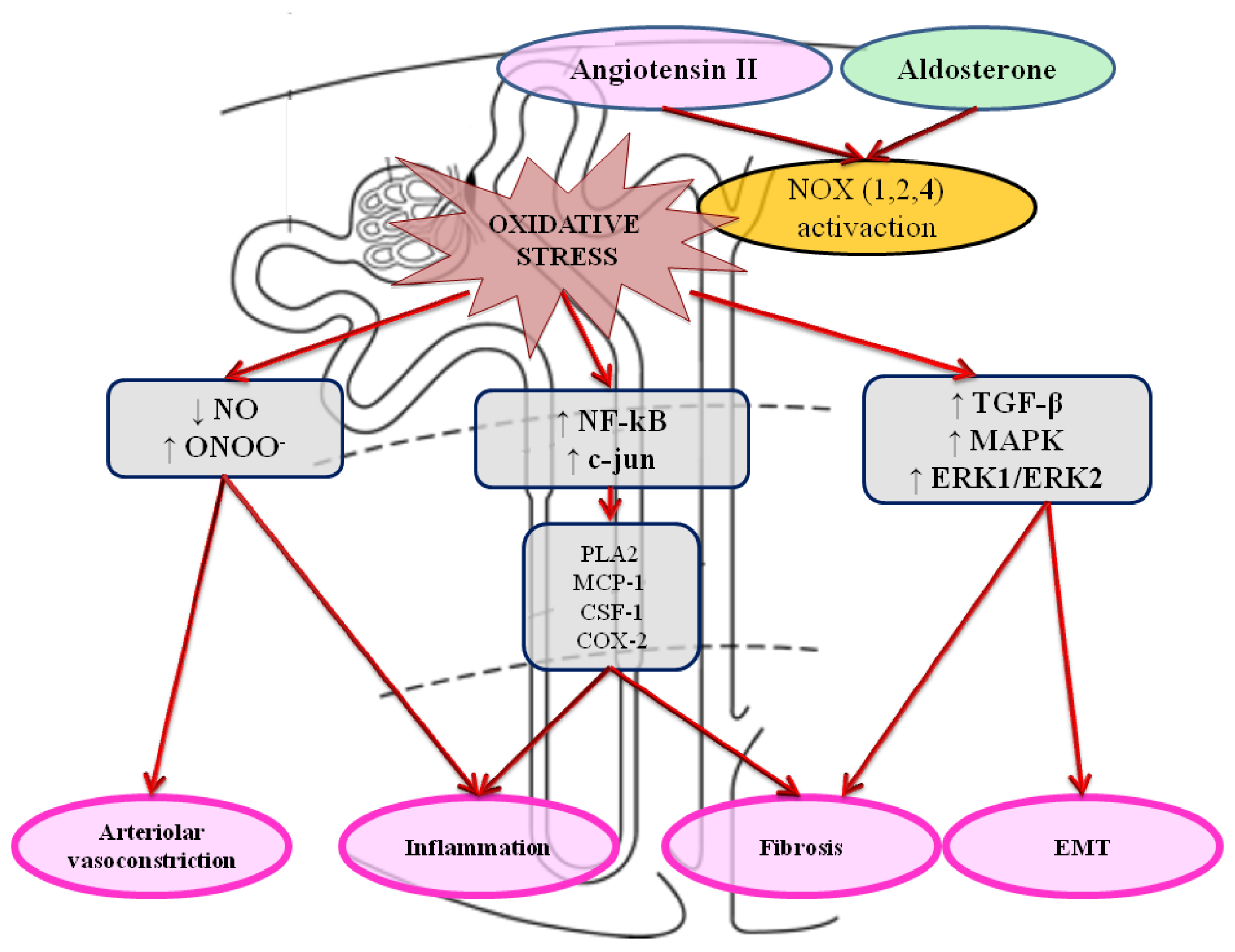
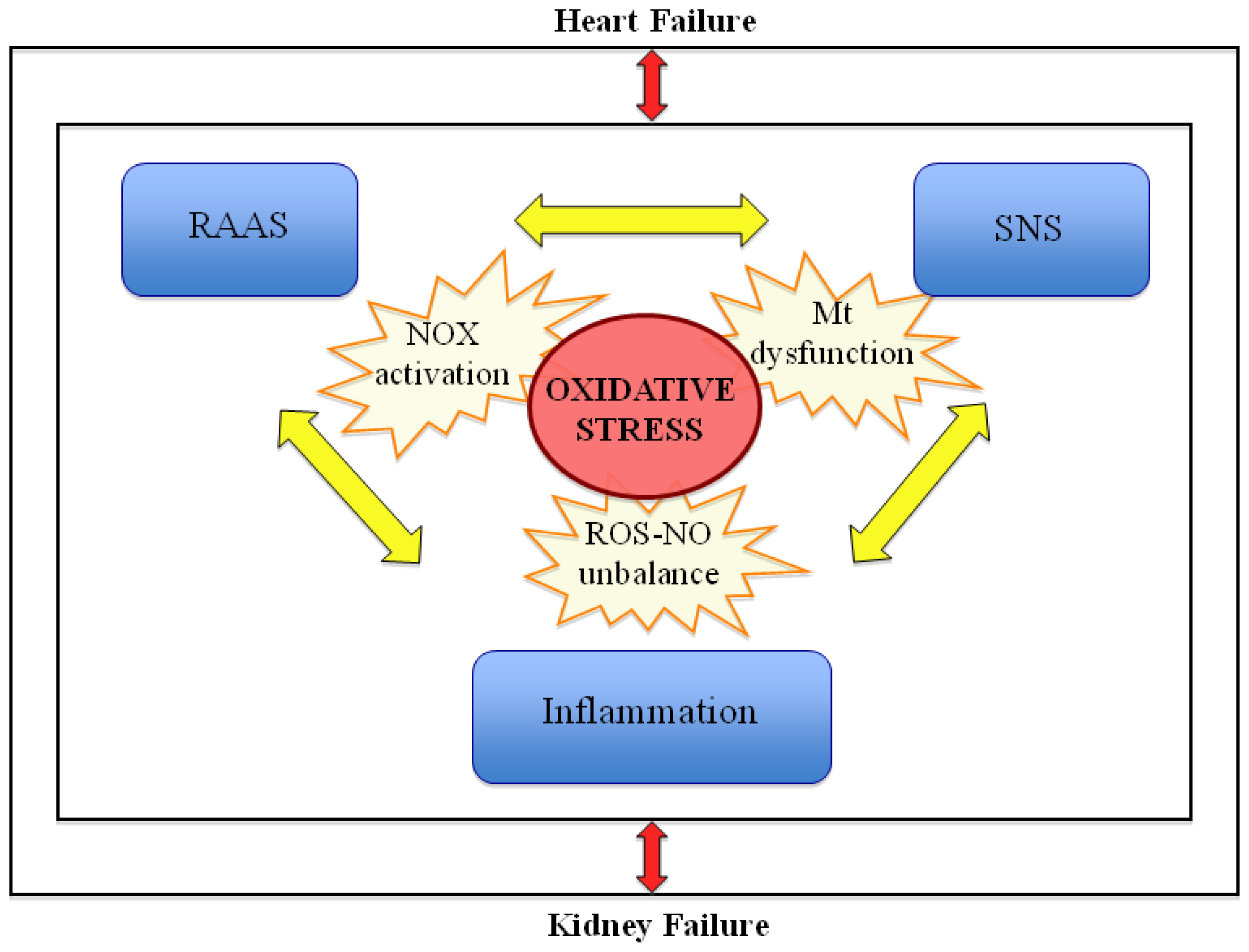
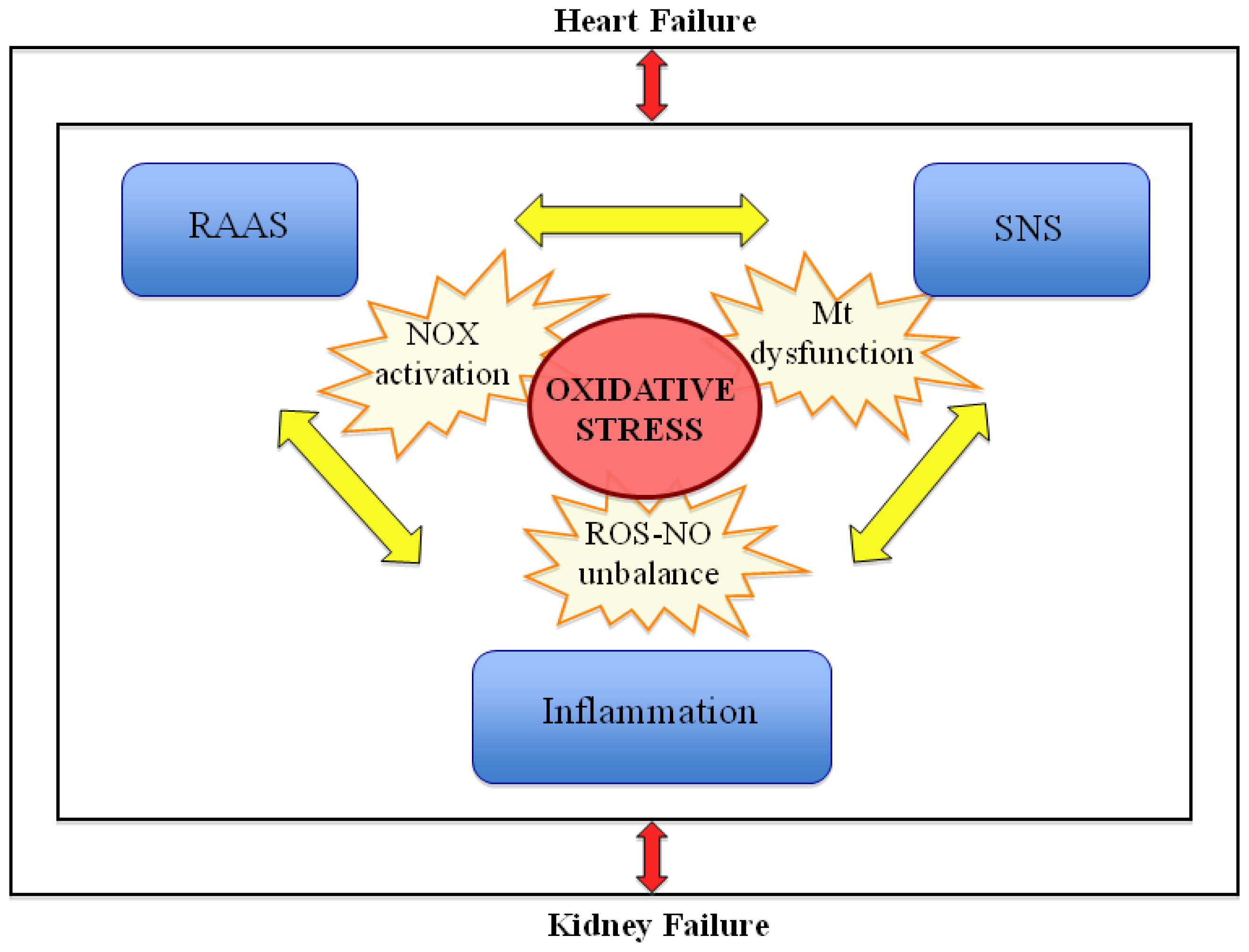
5. Implications of Increased Oxidative Stress in Chronic Cardiorenal Syndrome
6. Targeting Oxidative Stress to Treat CRS: Anything on the Horizon?
6.1. Drugs for HF with an Indirect Effect on Oxidative Stress
6.2. Oxidative Stress and NADPH Oxidase: Promising Therapies
6.3. Oxidative Stress and Mitochondria: Promising Therapies
7. Conclusions and Outlook
Acknowledgments
Conflicts of Interest
References
- Ronco, C.; House, A.A.; Haapio, M. Cardio-renal syndrome: Refining the definition of a complex symbiosis gone wrong. Intensiv. Care Med 2008, 34, 957–962. [Google Scholar]
- Ronco, C.; Ronco, F. Cardio-renal syndromes: A systematic approach for consensus definition and classification. Heart Fail Rev 2012, 17, 151–160. [Google Scholar]
- Ronco, C.; McCullough, P.; Anker, S.D.; Anand, I.; Aspromonte, N.; Bagshaw, S.M.; Bellomo, R.; Berl, T.; Bobek, I.; Cruz, D.N.; et al. Cardio-renal syndromes: Report from the consensus conference of the acute dialysis quality initiative. Eur. Heart J 2010, 31, 703–711. [Google Scholar]
- Guyton, A.C. The surprising kidney-fluid mechanism for pressure control—its infinite gain! Hypertension 1990, 16, 725–730. [Google Scholar]
- Lindner, A.; Charra, B.; Sherrard, D.J.; Scribner, B.H. Accelerated atherosclerosis in prolonged maintenance hemodialysis. N. Engl. J. Med 1974, 290, 697–701. [Google Scholar]
- Oh, J.; Wunsch, R.; Turzer, M.; Bahner, M.; Raggi, P.; Querfeld, U.; Mehls, O.; Schaefer, F. Advanced coronary and carotid arteriopathy in young adults with childhood-onset chronic renal failure. Circulation 2002, 106, 100–105. [Google Scholar]
- Rambausek, M.; Ritz, E.; Mall, G.; Mehls, O.; Katus, H. Myocardial hypertrophy in rats with renal insufficiency. Kidney Int 1985, 28, 775–782. [Google Scholar]
- Törnig, J.; Gross, M.L.; Simonaviciene, A.; Mall, G.; Ritz, E.; Amann, K. Hypertrophy of intramyocardial arteriolar smooth muscle cells in experimental renal failure. J. Am. Soc. Nephrol 1999, 10, 77–83. [Google Scholar]
- Safar, M.E.; London, G.M.; Plante, G.E. Arterial stiffness and kidney function. Hypertension 2004, 43, 163–168. [Google Scholar]
- Goodman, W.G.; Goldin, J.; Kuizon, B.D.; Yoon, C.; Gales, B.; Sider, D.; Wang, Y.; Chung, J.; Emerick, A.; Greaser, L.; et al. Coronary-artery calcification in young adults with end-stage renal disease who are undergoing dialysis. N. Engl. J. Med 2000, 342, 1478–1483. [Google Scholar]
- Sies, H. Oxidative stress: Oxidants and antioxidants. Exp. Physiol 1997, 82, 291–295. [Google Scholar]
- Hafstad, A.D.; Nabeebaccus, A.A.; Shah, A.M. Novel aspects of ROS signalling in heart failure. Basic Res. Cardiol 2013, 108, 359. [Google Scholar]
- Schramm, A.; Matusik, P.; Osmenda, G.; Guzik, T.J. Targeting NADPH oxidases in vascular pharmacology. Vasc. Pharmacol 2012, 56, 216–231. [Google Scholar]
- Sirker, A.; Zhang, M.; Shah, A.M. NADPH oxidases in cardiovascular disease: Insights from in vivo models and clinical studies. Basic Res. Cardiol 2011, 106, 735–747. [Google Scholar]
- Modlinger, P.S.; Wilcox, C.S.; Aslam, S. Nitric oxide, oxidative stress, and progression of chronic renal failure. WB Saunders. Semin. Nephrol 2004, 24, 354–365. [Google Scholar]
- Maack, C.; Böhm, M. Targeting mitochondrial oxidative stress in heart failure. Throttling the afterburner. J. Am. Coll. Cardiol 2011, 58, 83–86. [Google Scholar]
- Matsushima, S.; Ide, T.; Yamato, M.; Matsusaka, H.; Hattori, F.; Ikeuchi, M.; Kubota, T.; Sunagawa, K.; Hasegawa, Y.; Kurihara, T.; et al. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2006, 113, 1779–1786. [Google Scholar]
- Osterholt, M.; Nguyen, T.D.; Schwarzer, M.; Doenst, T. Alterations in mitochondrial function in cardiac hypertrophy and heart failure. Heart Fail Rev 2013, 18, 645–656. [Google Scholar]
- Graziewicz, M.A.; Day, B.J.; Copeland, W.C. The mitochondrial DNA polymerase as a target of oxidative damage. Nucleic Acids Res 2002, 30, 2817–2824. [Google Scholar]
- Chen, L.; Knowlton, A.A. Mitochondrial dynamics in heart failure. Congest. Heart Fail 2010, 17, 257–261. [Google Scholar]
- Stanley, W.C.; Chandler, M.P. Energy metabolism in the normal and failing heart: Potential for therapeutic interventions. Heart Fail Rev 2002, 7, 115–130. [Google Scholar]
- Beer, M.; Seyfarth, T.; Sandstede, J.; Landschütz, W.; Lipke, C.; Köstler, H.; von Kienlin, M.; Harre, K.; Hahn, D.; Neubauer, S. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J. Am. Coll. Cardiol 2002, 40, 1267–1274. [Google Scholar]
- Conway, M.A.; Allis, J.; Ouwerkerk, R.; Niioka, T.; Rajagopalan, B.; Radda, G.K. Detection of low phosphocreatine to ATP ratio in failing hypertrophied human myocardium by 31P magnetic resonance spectroscopy. Lancet 1991, 338, 973–976. [Google Scholar]
- Tian, R.; Nascimben, L.; Kaddurah-Daouk, R.; Ingwall, J.S. Depletion of energy reserve via the creatine kinase reaction during the evolution of heart failure in cardiomyopathic hamsters. J. Mol. Cell. Cardiol 1996, 28, 755–765. [Google Scholar]
- Kato, T.; Niizuma, S.; Inuzuka, Y.; Kawashima, T.; Okuda, J.; Tamaki, Y.; Iwanaga, Y.; Narazaki, M.; Matsuda, T.; Soga, T.; et al. Analysis of metabolic remodeling in compensated left ventricular hypertrophy and heart failure. Circ. Heart Fail 2010, 3, 420–430. [Google Scholar]
- Lei, B.; Lionetti, V.; Young, M.E.; Chandler, M.P.; d’Agostino, C.; Kang, E.; Altarejos, M.; Matsuo, K.; Hintze, T.H.; Stanley, W.C. Paradoxical downregulation of the glucose oxidation pathway despite enhanced flux in severe heart failure. J. Mol. Cell. Cardiol 2004, 36, 567–576. [Google Scholar]
- Katz, A.M.; Konstam, M.A. Heart Failure: Pathophysiology, Molecular Biology, and Clinical Management; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2009. [Google Scholar]
- Rosca, M.G.; Vazquez, E.J.; Kerner, J.; Parland, W.; Chandler, M.P.; Stanley, W.; Sabbah, H.N.; Hoppel, C.L. Cardiac mitochondria in heart failure: Decrease in respirasomes and oxidative phosphorylation. Cardiovasc. Res 2008, 80, 30–39. [Google Scholar]
- Scheubel, R.J.; Tostlebe, M.; Simm, A.; Rohrbach, S.; Prondzinsky, R.; Gellerich, F.N.; Silber, R.E.; Holtz, J. Dysfunction of mitochondrial respiratory chain complex I in human failing myocardium is not due to disturbed mitochondrial gene expression. J. Am. Coll. Cardiol 2002, 40, 2174–2181. [Google Scholar]
- Lemieux, H.; Semsroth, S.; Antretter, H.; Höfer, D.; Gnaiger, E. Mitochondrial respiratory control and early defects of oxidative phosphorylation in the failing human heart. Int. J. Biochem. Cell Biol 2011, 43, 1729–1738. [Google Scholar]
- Buchwald, A.; Till, H.; Unterberg, C.; Oberschmidt, R.; Figulla, H.R.; Wiegand, V. Alterations of the mitochondrial respiratory chain in human dilated cardiomyopathy. Eur. Heart J 1990, 11, 509–516. [Google Scholar]
- Marin-Garcia, J.; Goldenthal, M.J.; Moe, G.W. Mitochondrial pathology in cardiac failure. Cardiovasc. Res 2001, 49, 17–26. [Google Scholar]
- Rosca, M.G.; Hoppel, C.L. New aspects of impaired mitochondrial function in heart failure. J. Bioenerg. Biomembr 2009, 41, 107–112. [Google Scholar]
- Kimura, S.; Zhang, G.X.; Nishiyama, A.; Shokoji, T.; Yao, L.; Fan, Y.Y.; Rahman, M.; Suzuki, T.; Maeta, H.; Abe, Y. Role of NAD (P) H oxidase-and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension 2005, 45, 860–866. [Google Scholar]
- Dai, D.F.; Johnson, S.C.; Villarin, J.J.; Chin, M.T.; Nieves-Cintrón, M.; Chen, T.; Marcinek, D.J.; Dorn, G.W., II; Kang, Y.J.; Prolla, T.A.; et al. Mitochondrial oxidative stress mediates angiotensin II–induced cardiac hypertrophy and Gαq overexpression–induced heart failure. Novelty and significance. Circ. Res 2011, 108, 837–846. [Google Scholar]
- Marín-García, J.; Akhmedov, A.T.; Moe, G.W. Mitochondria in heart failure: The emerging role of mitochondrial dynamics. Heart Fail Rev 2013, 18, 439–456. [Google Scholar]
- Rosca, M.G.; Hoppel, C.L. Mitochondria in heart failure. Cardiovasc. Res 2010, 88, 40–50. [Google Scholar]
- Siwik, D.A.; Pagano, P.J.; Colucci, W.S. Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am. J. Physiol. Cell Physiol 2001, 280, C53–C60. [Google Scholar]
- Spinale, F.G.; Coker, M.L.; Thomas, C.V.; Walker, J.D.; Mukherjee, R.; Hebbar, L. Time-dependent changes in matrix metalloproteinase activity and expression during the progression of congestive heart failure relation to ventricular and myocyte function. Circ. Res 1998, 82, 482–495. [Google Scholar]
- Danial, N.N.; Korsmeyer, S.J. Cell death: Critical control points. Cell 2004, 116, 205–219. [Google Scholar]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001, 412, 95–99. [Google Scholar]
- Kwon, S.H.; Pimentel, D.R.; Remondino, A.; Sawyer, D.B.; Colucci, W.S. H2O2 regulates cardiac myocyte phenotype via concentration-dependent activation of distinct kinase pathways. J. Mol. Cell. Cardiol 2003, 35, 615–621. [Google Scholar]
- Chaanine, A.H.; Jeong, D.; Liang, L.; Chemaly, E.R.; Fish, K.; Gordon, R.E.; Hajjar, R.J. JNK modulates FOXO3a for the expression of the mitochondrial death and mitophagy marker BNIP3 in pathological hypertrophy and in heart failure. Cell Death Dis 2012, 3, 265. [Google Scholar]
- Chiong, M.; Wang, Z.V.; Pedrozo, Z.; Cao, D.J.; Troncoso, R.; Ibacache, M.; Criollo, A.; Nemchenko, A.; Hill, J.A.; Lavandero, S. Cardiomyocyte death: Mechanisms and translational implications. Cell Death Dis 2011, 2, e244. [Google Scholar]
- Vacek, T.P.; Vacek, J.C.; Tyagi, S.C. Mitochondrial mitophagic mechanisms of myocardial matrix metabolism and remodelling. Arch. Physiol. Biochem 2012, 118, 31–42. [Google Scholar]
- Gorin, Y.; Ricono, J.; Wagner, B.; Kim, N.; Bhandari, B.; Choudhury, G.; Abboud, H. Angiotensin II-induced ERK1/ERK2 activation and protein synthesis are redox-dependent in glomerular mesangial cells. Biochem. J 2004, 381, 231–239. [Google Scholar]
- Hannken, T.; Schroeder, R.; Stahl, R.A.; Wolf, G. Angiotensin II-mediated expression of p27Kip1 and induction of cellular hypertrophy in renal tubular cells depend on the generation of oxygen radicals1. Kidney Int 1998, 54, 1923–1933. [Google Scholar]
- Lodha, S.; Dani, D.; Mehta, R.; Bhaskaran, M.; Reddy, K.; Ding, G.; Singhal, P.C. Angiotensin II-induced mesangial cell apoptosis: Role of oxidative stress. Mol. Med 2002, 8, 830–840. [Google Scholar]
- López, B.; Salom, M.G.; Arregui, B.; Valero, F.; Fenoy, F.J. Role of superoxide in modulating the renal effects of angiotensin II. Hypertension 2003, 42, 1150–1156. [Google Scholar]
- Miyata, K.; Rahman, M.; Shokoji, T.; Nagai, Y.; Zhang, G.X.; Sun, G.P.; Kimura, S.; Yukimura, T.; Kiyomoto, H.; Kohno, M.; et al. Aldosterone stimulates reactive oxygen species production through activation of NADPH oxidase in rat mesangial cells. J. Am. Soc. Nephrol 2005, 16, 2906–2912. [Google Scholar]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev 2007, 87, 245–313. [Google Scholar]
- Nagase, M. Activation of the aldosterone/mineralocorticoid receptor system in chronic kidney disease and metabolic syndrome. Clin. Exp. Nephrol 2010, 14, 303–314. [Google Scholar]
- Zou, A.P.; Li, N.; Cowley, A.W. Production and actions of superoxide in the renal medulla. Hypertension 2001, 37, 547–553. [Google Scholar]
- Chabrashvili, T.; Tojo, A.; Onozato, M.L.; Kitiyakara, C.; Quinn, M.T.; Fujita, T.; Welch, W.J.; Wilcox, C.S. Expression and cellular localization of classic NADPH oxidase subunits in the spontaneously hypertensive rat kidney. Hypertension 2002, 39, 269–274. [Google Scholar]
- Geiszt, M.; Kopp, J.B.; Várnai, P.; Leto, T.L. Identification of renox, an NAD (P) H oxidase in kidney. Proc. Natl. Acad. Sci. USA 2000, 97, 8010–8014. [Google Scholar]
- Shiose, A.; Kuroda, J.; Tsuruya, K.; Hirai, M.; Hirakata, H.; Naito, S.; Hattori, M.; Sakaki, Y.; Sumimoto, H. A novel superoxide-producing NAD (P) H oxidase in kidney. J. Biol. Chem 2001, 276, 1417–1423. [Google Scholar]
- Cheng, G.; Cao, Z.; Xu, X.; Meir, E.G.V.; Lambeth, J.D. Homologs of gp91phox: Cloning and tissue expression of Nox3, Nox4, and Nox5. Gene 2001, 269, 131–140. [Google Scholar]
- Vaziri, N.D.; Dicus, M.; Ho, N.D.; Boroujerdi-Rad, L.; Sindhu, R.K. Oxidative stress and dysregulation of superoxide dismutase and NADPH oxidase in renal insufficiency. Kidney Int 2003, 63, 179–185. [Google Scholar]
- Gorin, Y.; Ricono, J.M.; Kim, N.H.; Bhandari, B.; Choudhury, G.G.; Abboud, H.E. Nox4 mediates angiotensin II-induced activation of Akt/protein kinase B in mesangial cells. Am. J. Physiol. Ren. Physiol 2003, 285, F219–F229. [Google Scholar]
- Wilcox, C.S. Redox regulation of the afferent arteriole and tubuloglomerular feedback. Acta Physiol. Scand 2003, 179, 217–223. [Google Scholar]
- Raij, L. Nitric oxide and cardiovascular and renal effects. Osteoarthr. Cartil 2008, 16, S21–S26. [Google Scholar]
- Liaudet, L.; Soriano, F.G.; Szabó, C. Biology of nitric oxide signaling. Crit. Care Med 2000, 28, N37–N52. [Google Scholar]
- Schnackenberg, C.G. Physiological and pathophysiological roles of oxygen radicals in the renal microvasculature. Am. J. Physiol. Regul. Integr. Comp. Physiol 2002, 282, R335–R342. [Google Scholar]
- Rhyu, D.Y.; Yang, Y.; Ha, H.; Lee, G.T.; Song, J.S.; Uh, S.T.; Lee, H.B. Role of reactive oxygen species in TGF-β1-induced mitogen-activated protein kinase activation and epithelial-mesenchymal transition in renal tubular epithelial cells. J. Am. Soc. Nephrol 2005, 16, 667–675. [Google Scholar]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial–mesenchymal transitions. Nat. Rev. Mol. Cell Biol 2006, 7, 131–142. [Google Scholar]
- Wolf, G.; Wenzel, U.; Hannken, T.; Stahl, R.A. Angiotensin II induces p27Kip1 expression in renal tubules in vivo: Role of reactive oxygen species. J. Mol. Med 2001, 79, 382–389. [Google Scholar]
- Napoli, C.; Casamassimi, A.; Crudele, V.; Infante, T.; Abbondanza, C. Kidney and heart interactions during cardiorenal syndrome: A molecular and clinical pathogenic framework. Future Cardiol 2011, 7, 485–497. [Google Scholar]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig 2009, 119, 1420–1428. [Google Scholar]
- Yoshikawa, M.; Hishikawa, K.; Marumo, T.; Fujita, T. Inhibition of histone deacetylase activity suppresses epithelial-to-mesenchymal transition induced by TGF-β1 in human renal epithelial cells. J. Am. Soc. Nephrol 2007, 18, 58–65. [Google Scholar]
- Ng, Y.Y.; Huang, T.P.; Yang, W.C.; Chen, Z.P.; Yang, A.H.; Mu, W.; Nikolic-Paterson, D.J.; Atkins, R.C.; Lan, H.Y. Tubular epithelial-myofibroblast transdifferentiation in progressive tubulointerstitial fibrosis in 5/6 nephrectomized rats. Kidney Int 1998, 54, 864–876. [Google Scholar]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig 2002, 110, 341–350. [Google Scholar]
- Yang, J.; Liu, Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am. J. Pathol 2001, 159, 1465–1475. [Google Scholar]
- Böttinger, E.P.; Bitzer, M. TGF-beta signaling in renal disease. J. Am. Soc. Nephrol 2002, 13, 2600–2610. [Google Scholar]
- Dorsam, G.; Taher, M.M.; Valerie, K.C.; Kuemmerle, N.B.; Chan, J.C.; Franson, R.C. Diphenyleneiodium chloride blocks inflammatory cytokine-induced up-regulation of group IIA phospholipase A2 in rat mesangial cells. J. Pharmacol. Exp. Ther 2000, 292, 271–279. [Google Scholar]
- Cui, X.L.; Douglas, J.G. Arachidonic acid activates c-jun N-terminal kinase through NADPH oxidase in rabbit proximal tubular epithelial cells. Proc. Natl. Acad. Sci. USA 1997, 94, 3771–3776. [Google Scholar]
- Satriano, J.A.; Shuldiner, M.; Hora, K.; Xing, Y.; Shan, Z.; Schlondorff, D. Oxygen radicals as second messengers for expression of the monocyte chemoattractant protein, JE/MCP-1, and the monocyte colony-stimulating factor, CSF-1, in response to tumor necrosis factor-alpha and immunoglobulin G. Evidence for involvement of reduced nicotinamide adenine dinucleotide phosphate (NADPH)-dependent oxidase. J. Clin. Investig 1993, 92, 1564–1571. [Google Scholar]
- Feng, L.; Xia, Y.; Garcia, G.E.; Hwang, D.; Wilson, C.B. Involvement of reactive oxygen intermediates in cyclooxygenase-2 expression induced by interleukin-1, tumor necrosis factor-alpha, and lipopolysaccharide. J. Clin. Investig 1995, 95, 1669–1675. [Google Scholar]
- Di Castro, S.; Scarpino, S.; Marchitti, S.; Bianchi, F.; Stanzione, R.; Cotugno, M.; Sironi, L.; Gelosa, P.; Duranti, E.; Ruco, L.; et al. Differential modulation of UCP2 in kidneys of stroke-prone spontaneously hypertensive rats under high salt/low potassium diet. Hypertension 2013, 61, 534–541. [Google Scholar]
- Mattiasson, G.; Sullivan, P.G. The emerging functions of UCP2 in health, disease, and therapeutics. Antioxid. Redox Signal 2006, 8, 1–38. [Google Scholar]
- Yoshida, T.; Kato, K.; Fujimaki, T.; Yokoi, K.; Oguri, M.; Watanabe, S.; Metoki, N.; Yoshida, H.; Satoh, K.; Aoyagi, Y.; et al. Association of genetic variants with chronic kidney disease in Japanese individuals. Clin. J. Am. Soc. Nephrol 2009, 4, 883–890. [Google Scholar]
- Metra, M.; Davison, B.; Bettari, L.; Sun, H.; Edwards, C.; Lazzarini, V.; Piovanelli, B.; Carubelli, V.; Bugatti, S.; Lombardi, C.; et al. Is worsening renal function an ominous prognostic sign in patients with acute heart failure? The role of congestion and its interaction with renal function. Circ. Heart Fail 2012, 5, 54–62. [Google Scholar]
- Shlipak, M.G.; Fried, L.F.; Crump, C.; Bleyer, A.J.; Manolio, T.A.; Tracy, R.P.; Furberg, C.D.; Psaty, B.M. Elevations of inflammatory and procoagulant biomarkers in elderly persons with renal insufficiency. Circulation 2003, 107, 87–92. [Google Scholar]
- Stuveling, E.M.; Hillege, H.L.; Bakker, S.J.; Gans, R.O.; de Jong, P.E.; de Zeeuw, D. C-reactive protein is associated with renal function abnormalities in a non-diabetic population. Kidney Int 2003, 63, 654–661. [Google Scholar]
- Ikizler, T.A.; Morrow, J.D.; Roberts, L.J.; Evanson, J.A.; Becker, B.; Hakim, R.M.; Shyr, Y.; Himmelfarb, J. Plasma F2-isoprostane levels are elevated in chronic hemodialysis patients. Clin. Nephrol 2002, 58, 190–197. [Google Scholar]
- Dounousi, E.; Papavasiliou, E.; Makedou, A.; Ioannou, K.; Katopodis, K.P.; Tselepis, A.; Siamopoulos, K.C.; Tsakiris, D. Oxidative stress is progressively enhanced with advancing stages of CKD. Am. J. Kidney Dis 2006, 48, 752–760. [Google Scholar]
- Oberg, B.P.; McMenamin, E.; Lucas, F.L.; McMonagle, E.; Morrow, J.; Ikizler, T.A.; Himmelfarb, J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int 2004, 65, 1009–1016. [Google Scholar]
- Nerpin, E.; Helmersson-Karlqvist, J.; Risérus, U.; Sundström, J.; Larsson, A.; Jobs, E.; Basu, S.; Ingelsson, E.; Arnlöv, J. Inflammation, oxidative stress, glomerular filtration rate, and albuminuria in elderly men: A cross-sectional study. BMC Res. Notes 2012, 5, 537. [Google Scholar]
- El Nahas, M. Cardio-Kidney-Damage: A unifying concept. Kidney Int 2010, 78, 14–18. [Google Scholar]
- Chaney, E.; Shaw, A. Pathophysiology of fluid retention in heart failure. Contrib. Nephrol 2010, 164, 46–53. [Google Scholar]
- Colombo, P.C.; Ganda, A.; Lin, J.; Onat, D.; Harxhi, A.; Iyasere, J.E.; Uriel, N.; Cotter, G. Inflammatory activation: Cardiac, renal, and cardio-renal interactions in patients with the cardiorenal syndrome. Heart Fail Rev 2012, 17, 177–190. [Google Scholar]
- Bongartz, L.G.; Cramer, M.J.; Doevendans, P.A.; Joles, J.A.; Braam, B. The severe cardiorenal syndrome: ‘Guyton revisited’. Eur. Heart J 2005, 26, 11–17. [Google Scholar]
- Bongartz, L.G.; Braam, B.; Gaillard, C.A.; Cramer, M.J.; Goldschmeding, R.; Verhaar, M.C.; Doevendans, P.A.; Joles, J.A. Target organ cross talk in cardiorenal syndrome: Animal models. Am. J. Physiol. Ren. Physiol 2012, 303, F1253–F1263. [Google Scholar]
- Griendling, K.K.; Minieri, C.A.; Ollerenshaw, J.D.; Alexander, R.W. Angiotensin II stimulates NADH and NADPH oxidase activity in cultured vascular smooth muscle cells. Circ. Res 1994, 74, 1141–1148. [Google Scholar]
- Ushio-Fukai, M.; Zafari, A.M.; Fukui, T.; Ishizaka, N.; Griendling, K.K. p22phox is a critical component of the superoxide-generating NADH/NADPH oxidase system and regulates angiotensin II-induced hypertrophy in vascular smooth muscle cells. J. Biol. Chem 1996, 271, 23317–23321. [Google Scholar]
- Chabrashvili, T.; Kitiyakara, C.; Blau, J.; Karber, A.; Aslam, S.; Welch, W.J.; Wilcox, C.S. Effects of ANG II type 1 and 2 receptors on oxidative stress, renal NADPH oxidase, and SOD expression. Am. J. Physiol. Regul. Integr. Comp. Physiol 2003, 285, R117–R124. [Google Scholar]
- Nakagami, H.; Takemoto, M.; Liao, J.K. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. J. Mol. Cell. Cardiol 2003, 35, 851–859. [Google Scholar]
- Heymes, C.; Bendall, J.K.; Ratajczak, P.; Cave, A.C.; Samuel, J.L.; Hasenfuss, G.; Shah, A.M. Increased myocardial NADPH oxidase activity in human heart failure. J. Am. Coll. Cardiol 2003, 41, 2164–2171. [Google Scholar]
- Tojo, A.; Onozato, M.L.; Kobayashi, N.; Goto, A.; Matsuoka, H.; Fujita, T. Angiotensin II and oxidative stress in Dahl Salt-sensitive rat with heart failure. Hypertension 2002, 40, 834–839. [Google Scholar]
- Hornig, B.; Landmesser, U.; Kohler, C.; Ahlersmann, D.; Spiekermann, S.; Christoph, A.; Tatge, H.; Drexler, H. Comparative effect of ACE inhibition and angiotensin II type 1 receptor antagonism on bioavailability of nitric oxide in patients with coronary artery disease role of superoxide dismutase. Circulation 2001, 103, 799–805. [Google Scholar]
- Ruiz-Ortega, M.; Lorenzo, O.; Egido, J. Angiotensin III increases MCP-1 and activates NF-kappaB and AP-1 in cultured mesangial and mononuclear cells. Kidney Int 2000, 57, 2285–2298. [Google Scholar]
- Pueyo, M.E.; Gonzalez, W.; Nicoletti, A.; Savoie, F.; Arnal, J.F.; Michel, J.B. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-κB activation induced by intracellular oxidative stress. Arterioscler. Thromb. Vasc. Biol 2000, 20, 645–651. [Google Scholar]
- Klein, I.H.; Ligtenberg, G.; Oey, P.L.; Koomans, H.A.; Blankestijn, P.J. Enalapril and losartan reduce sympathetic hyperactivity in patients with chronic renal failure. J. Am. Soc. Nephrol 2003, 14, 425–430. [Google Scholar]
- Jackson, G.; Gibbs, C.R.; Davies, M.K.; Lip, G.Y.H. ABC of heart failure: Pathophysiology. BMJ 2000, 320, 167–170. [Google Scholar]
- Amin, J.K.; Xiao, L.; Pimental, D.R.; Pagano, P.J.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Reactive oxygen species mediate alpha-adrenergic receptor-stimulated hypertrophy in adult rat ventricular myocytes. J. Mol. Cell. Cardiol 2001, 33, 131–139. [Google Scholar]
- Bleeke, T.; Zhang, H.; Madamanchi, N.; Patterson, C.; Faber, J.E. Catecholamine-induced vascular wall growth is dependent on generation of reactive oxygen species. Circ. Res 2004, 94, 37–45. [Google Scholar]
- Bianchi, P.; Séguélas, M.H.; Parini, A.; Cambon, C. Activation of pro-apoptotic cascade by dopamine in renal epithelial cells is fully dependent on hydrogen peroxide generation by monoamine oxidases. J. Am. Soc. Nephrol 2003, 14, 855–862. [Google Scholar]
- Liao, J.; Keiser, J.A.; Scales, W.E.; Kunkel, S.L.; Kluger, M.J. Role of epinephrine in TNF and IL-6 production from isolated perfused rat liver. Am. J. Physiol. Regul. Integr. Comp. Physiol 1995, 268, R896–R901. [Google Scholar]
- Oddis, C.V.; Simmons, R.L.; Hattler, B.G.; Finkel, M.S. cAMP enhances inducible nitric oxide synthase mRNA stability in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol 1995, 269, H2044–H2050. [Google Scholar]
- Arici, M.; Walls, J. End-stage renal disease, atherosclerosis, and cardiovascular mortality: Is C-reactive protein the missing link? Kidney Int 2001, 59, 407–414. [Google Scholar]
- Zebrack, J.S.; Anderson, J.L.; Beddhu, S.; Horne, B.D.; Bair, T.L.; Cheung, A.; Muhlestein, J.B. Do associations with C-reactive protein and extent of coronary artery disease account for the increased cardiovascular risk of renal insufficiency? J. Am. Coll. Cardiol 2003, 42, 57–63. [Google Scholar]
- Irish, A. Cardiovascular disease, fibrinogen and the acute phase response: Associations with lipids and blood pressure in patients with chronic renal disease. Atherosclerosis 1998, 137, 133–139. [Google Scholar]
- Testa, M.; Yeh, M.; Lee, P.; Fanelli, R.; Loperfido, F.; Berman, J.W.; LeJemtel, T.H. Circulating levels of cytokines and their endogenous modulators in patients with mild to severe congestive heart failure due to coronary artery disease or hypertension. J. Am. Coll. Cardiol 1996, 28, 964–971. [Google Scholar]
- Sanchez-Lozada, L.G.; Tapia, E.; Johnson, R.J.; Rodriguez-Iturbe, B.; Herrera-Acosta, J. Glomerular hemodynamic changes associated with arteriolar lesions and tubulointerstitial inflammation. Kidney Int 2003, 64, S9–S14. [Google Scholar]
- Niijima, A.; Hori, T.; Aou, S.; Oomura, Y. The effects of interleukin-1β on the activity of adrenal, splenic and renal sympathetic nerves in the rat. J. Auton. Nerv. Syst 1991, 36, 183–192. [Google Scholar]
- Wassmann, S.; Stumpf, M.; Strehlow, K.; Schmid, A.; Schieffer, B.; Böhm, M.; Nickenig, G. Interleukin-6 induces oxidative stress and endothelial dysfunction by overexpression of the angiotensin II type 1 receptor. Circ. Res 2004, 94, 534–541. [Google Scholar]
- Tsutamoto, T.; Hisanaga, T.; Wada, A.; Maeda, K.; Ohnishi, M.; Fukai, D.; Mabuchi, N.; Sawaki, M.; Kinoshita, M. Interleukin-6 spillover in the peripheral circulation increases with the severity of heart failure, and the high plasma level of interleukin-6 is an important prognostic predictor in patients with congestive heart failure. J. Am. Coll. Cardiol 1998, 31, 391–398. [Google Scholar]
- Anker, S.D.; Egerer, K.R.; Volk, H.D.; Kox, W.J.; Poole-Wilson, P.A.; Coats, A.J. Elevated soluble CD14 receptors and altered cytokines in chronic heart failure. Am. J. Cardiol 1997, 79, 1426–1430. [Google Scholar]
- Kawai, M.; Naruse, K.; Komatsu, S.; Kobayashi, S.; Nagino, M.; Nimura, Y.; Sokabe, M. Mechanical stress-dependent secretion of interleukin 6 by endothelial cells after portal vein embolization: Clinical and experimental studies. J. Hepatol 2002, 37, 240–246. [Google Scholar]
- McMurray, J.J. CONSENSUS to EMPHASIS: The overwhelming evidence which makes blockade of the renin–angiotensin–aldosterone system the cornerstone of therapy for systolic heart failure. Eur. J. Heart Fail 2011, 13, 929–936. [Google Scholar]
- Kim, Y.S.; Greenberg, B. Update on renin-angiotensin-aldosterone blockade in heart failure. Curr. Treat. Options Cardiovasc. Med 2009, 11, 455–466. [Google Scholar]
- Konstam, M.A.; Rousseau, M.F.; Kronenberg, M.W.; Udelson, J.E.; Melin, J.; Stewart, D.; Dolan, N.; Edens, T.R.; Ahn, S.; Kinan, D.; et al. Effects of the angiotensin converting enzyme inhibitor enalapril on the long-term progression of left ventricular dysfunction in patients with heart failure. SOLVD Investigators. Circulation 1992, 86, 431–438. [Google Scholar]
- Brenner, B.M.; Cooper, M.E.; de Zeeuw, D.; Grunfeld, J.P.; Keane, W.F.; Kurokawa, K.; McGill, J.B.; Mitch, W.E.; Parving, H.H.; Remuzzi, G.; et al. The losartan renal protection study—rationale, study design and baseline characteristics of RENAAL (Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan). J. Renin Angiotensin Aldosterone Syst 2000, 1, 328–335. [Google Scholar]
- Granger, C.B.; McMurray, J.J.; Yusuf, S.; Held, P.; Michelson, E.L.; Olofsson, B.; Ostergren, J.; Pfeffer, M.A.; Swedberg, K. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: The CHARM-Alternative trial. Lancet 2003, 362, 772–776. [Google Scholar]
- Parving, H.H.; Lehnert, H.; Bröchner-Mortensen, J.; Gomis, R.; Andersen, S.; Arner, P. The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. N. Engl. J. Med 2001, 345, 870–878. [Google Scholar]
- Lewis, E.J.; Hunsicker, L.G.; Clarke, W.R.; Berl, T.; Pohl, M.A.; Lewis, J.B.; Ritz, E.; Atkins, R.C.; Rohde, R.; Raz, I. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N. Engl. J. Med 2001, 345, 851–860. [Google Scholar]
- Maschio, G.; Alberti, D.; Janin, G.; Locatelli, F.; Mann, J.F.; Motolese, M.; Ponticelli, C.; Ritz, E.; Zucchelli, P. Effect of the angiotensin-converting–enzyme inhibitor benazepril on the progression of chronic renal insufficiency. N. Engl. J. Med 1996, 334, 939–945. [Google Scholar]
- Hou, F.F.; Zhang, X.; Zhang, G.H.; Xie, D.; Chen, P.Y.; Zhang, W.R.; Jiang, J.P.; Liang, M.; Wang, G.B.; Liu, Z.R.; et al. Efficacy and safety of benazepril for advanced chronic renal insufficiency. N. Engl. J. Med 2006, 354, 131–140. [Google Scholar]
- Pitt, B.; Remme, W.; Zannad, F.; Neaton, J.; Martinez, F.; Roniker, B.; Bittman, R.; Hurley, S.; Kleiman, J.; Gatlin, M. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N. Engl. J. Med 2003, 348, 1309–1321. [Google Scholar]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. N. Engl. J. Med 1999, 341, 709–717. [Google Scholar]
- Eichhorn, E.J.; Heesch, C.M.; Barnett, J.H.; Alvarez, L.G.; Fass, S.M.; Grayburn, P.A.; Hatfield, B.A.; Marcoux, L.G.; Malloy, C.R. Effect of metoprolol on myocardial function and energetics in patients with nonischemic dilated cardiomyopathy: A randomized, double-blind, placebo-controlled study. J. Am. Coll. Cardiol 1994, 24, 1310–1320. [Google Scholar]
- Sawyer, D.B.; Colucci, W.S. Mitochondrial oxidative stress in heart failure “oxygen wastage” revisited. Circ. Res 2000, 86, 119–120. [Google Scholar]
- Guzik, T.J.; Griendling, K.K. NADPH oxidases: Molecular understanding finally reaching the clinical level? Antioxid. Redox Signal 2009, 11, 2365–2370. [Google Scholar]
- Guzik, T.J.; Harrison, D.G. Vascular NADPH oxidases as drug targets for novel antioxidant strategies. Drug Discov. Today 2006, 11, 524–533. [Google Scholar]
- Wind, S.; Beuerlein, K.; Eucker, T.; Muller, H.; Scheurer, P.; Armitage, M.E.; Ho, H.; Schmidt, H.H.; Wingler, K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br. J. Pharmacol 2010, 161, 885–898. [Google Scholar]
- Stolk, J.; Hiltermann, T.J.; Dijkman, J.H.; Verhoeven, A.J. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am. J. Respir. Cell Mol. Biol 1994, 11, 95–102. [Google Scholar]
- Cayatte, A.J.; Rupin, A.; Oliver-Krasinski, J.; Maitland, K.; Sansilvestri-Morel, P.; Boussard, M.F.; Wierzbicki, M.; Verbeuren, T.J.; Cohen, R.A. S17834, a new inhibitor of cell adhesion and atherosclerosis that targets NADPH oxidase. Arterioscler. Thromb. Vasc. Biol 2001, 21, 1577–1584. [Google Scholar]
- Xu, S.; Jiang, B.; Hou, X.; Shi, C.; Bachschmid, M.M.; Zang, M.; Verbeuren, T.J.; Cohen, R.A. High-fat diet increases and the polyphenol, S17834, decreases acetylation of the sirtuin-1-dependent lysine-382 on p53 and apoptotic signaling in atherosclerotic lesion-prone aortic endothelium of normal mice. J. Cardiovasc. Pharmacol 2011, 58, 263–271. [Google Scholar]
- Rey, F.E.; Cifuentes, M.E.; Kiarash, A.; Quinn, M.T.; Pagano, P.J. Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O(2)(−) and systolic blood pressure in mice. Circ. Res 2001, 89, 408–414. [Google Scholar]
- Dai, D.F.; Hsieh, E.J.; Chen, T.; Menendez, L.G.; Basisty, N.B.; Tsai, L.; Beyer, R.P.; Crispin, D.A.; Shulman, N.J.; Szeto, H.H.; et al. Global proteomics and pathway analysis of pressure-overload-induced heart failure and its attenuation by mitochondrial-targeted peptides. Circ. Heart Fail 2013, 6, 1067–1076. [Google Scholar]
- Bayeva, M.; Gheorghiade, M.; Ardehali, H. Mitochondria as a therapeutic target in heart failure. J. Am. Coll. Cardiol 2013, 61, 599–610. [Google Scholar]
- McQueen, M.J.; Lonn, E.; Gerstein, H.C.; Bosch, J.; Yusuf, S.; The, HOPE. (Heart Outcomes Prevention Evaluation) Study and its consequences. Scand. J. Clin. Lab. Investig 2005, 65, 143–156. [Google Scholar]
- Collins, R.; Armitage, J.; Parish, S.; Sleight, P.; Peto, R. MRC/BHF Heart Protection Study of antioxidant vitamin supplementation in 20536 high-risk individuals: A randomised placebo-controlled trial. Lancet 2002, 360, 23–33. [Google Scholar]
- Smith, R.A.; Porteous, C.M.; Gane, A.M.; Murphy, M.P. Delivery of bioactive molecules to mitochondria in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 5407–5412. [Google Scholar]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.; Murphy, M.P.; Sammut, I.A. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J 2005, 19, 1088–1095. [Google Scholar]
- Neuzil, J.; Widén, C.; Gellert, N.; Swettenham, E.; Zobalova, R.; Dong, L.F.; Wang, X.F.; Lidebjer, C.; Dalen, H.; Headrick, J.P.; et al. Mitochondria transmit apoptosis signalling in cardiomyocyte-like cells and isolated hearts exposed to experimental ischemia-reperfusion injury. Redox Rep 2007, 12, 148–162. [Google Scholar]
- Parajuli, N.; Campbell, L.H.; Marine, A.; Brockbank, K.G.; MacMillan-Crow, L.A. MitoQ blunts mitochondrial and renal damage during cold preservation of porcine kidneys. PLoS One 2012, 7, e48590. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rubattu, S.; Mennuni, S.; Testa, M.; Mennuni, M.; Pierelli, G.; Pagliaro, B.; Gabriele, E.; Coluccia, R.; Autore, C.; Volpe, M. Pathogenesis of Chronic Cardiorenal Syndrome: Is There a Role for Oxidative Stress? Int. J. Mol. Sci. 2013, 14, 23011-23032. https://doi.org/10.3390/ijms141123011
Rubattu S, Mennuni S, Testa M, Mennuni M, Pierelli G, Pagliaro B, Gabriele E, Coluccia R, Autore C, Volpe M. Pathogenesis of Chronic Cardiorenal Syndrome: Is There a Role for Oxidative Stress? International Journal of Molecular Sciences. 2013; 14(11):23011-23032. https://doi.org/10.3390/ijms141123011
Chicago/Turabian StyleRubattu, Speranza, Silvia Mennuni, Marco Testa, Mara Mennuni, Giorgia Pierelli, Beniamino Pagliaro, Erica Gabriele, Roberta Coluccia, Camillo Autore, and Massimo Volpe. 2013. "Pathogenesis of Chronic Cardiorenal Syndrome: Is There a Role for Oxidative Stress?" International Journal of Molecular Sciences 14, no. 11: 23011-23032. https://doi.org/10.3390/ijms141123011